

Abstract
The emerging literature implicates a role for glia/cytokines in persistent pain. However, the mechanisms by which these non-neural elements contribute to CNS activity-dependent plasticity and pain are unclear. Using a trigeminal model of inflammatory hyperalgesia, here we provide evidence that demonstrates a mechanism by which glia interact with neurons, leading to activity-dependent plasticity and hyperalgesia. In response to masseter inflammation, there was an upregulation of glial fibrillary acidic proteins (GFAPs), a marker of astroglia, and interleukin-1β (IL-1β), a prototype proinflammatory cytokine, in the region of the trigeminal nucleus specifically related to the processing of deep orofacial input. The activated astroglia exhibited hypertrophy and an increased level of connexin 43, an astroglial gap junction protein. The upregulated IL-1β was selectively localized to astrocytes but not to microglia and neurons. Local anesthesia of the masseter nerve prevented the increase in GFAP and IL-1β after inflammation, and substance P, a prototype neurotransmitter of primary afferents, induced similar increases in GFAP and IL-1β, which was blocked by a nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester. Injection of IL-1 receptor antagonist and fluorocitrate, a glial inhibitor, attenuated hyperalgesia and NMDA receptor phosphorylation after inflammation. In vitro application of IL-1β induced NR1 phosphorylation, which was blocked by an IL-1 receptor antagonist, a PKC inhibitor (chelerythrine), an IP3 receptor inhibitor (2-aminoethoxydiphenylborate), and inhibitors of phospholipase C [1-[6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione] and phospholipase A2 (arachidonyltrifluoromethyl ketone). These findings provide evidence of astroglial activation by tissue injury, concomitant IL-1β induction, and the coupling of NMDA receptor phosphorylation through IL-1 receptor signaling.
Keywords: GFAP, IL-1β, NMDA receptor, gap junction, inflammation, rat
Introduction
The emerging literature strongly implicates a role for glia and proinflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α, in the genesis or maintenance of persistent pain (Watkins and Maier, 2005). Independent of release of cytokines from the peripheral site of injury, inflammatory cytokines are induced in the CNS (Lee et al., 2004; Raghavendra et al., 2004; McMahon et al., 2005). It has become clear that CNS cytokines, in association with activated glia, may influence or modulate neuronal function (Srinivasan et al., 2004). Evidence suggests that glia are intimately involved in the active control of neuronal activity (Araque et al., 1999; Frerking, 2004). Through still unknown mechanisms, glia can be activated after injury and release chemical mediators that modulate neuronal activity and synaptic strength. What is most intriguing is that IL-1β signaling may facilitate NMDA receptor (NMDAR) activation in neurons (Viviani et al., 2003; Yang et al., 2005). Despite the increasing amount of evidence for the involvement of glia and related chemicals in persistent pain conditions, very few studies have addressed the mechanisms by which these non-neural elements contribute to CNS activity-dependent neuronal plasticity and behavioral hyperalgesia (Tsuda et al., 2003; Milligan et al., 2004; Tanga et al., 2005; Hains and Waxman, 2006; Zhuang et al., 2006).
Some of the most prevalent and debilitating persistent pain conditions are associated with deep tissues, such as muscles, joints, and viscera. The most common persistent orofacial pain condition, temporomandibular joint disorder (TMJD), affects the musculoskeletal and joint tissues, is often not successfully treated and afflicts ∼10% of the population (Dworkin and LeResche, 1992). The etiology and pathology of these disorders remain unclear. Recent studies demonstrate that, in addition to the medullary dorsal horn, the homolog of the spinal dorsal horn involved in initial pain processing, orofacial injury activates a distinct region in the spinal trigeminal complex, the subnuclei interpolaris/caudalis (Vi/Vc) transition zone (Strassman and Vos, 1993; Hathaway et al., 1995; Bereiter et al., 2000; Ro et al., 2003; Dubner and Ren, 2004; Sugiyo et al., 2005; Wang et al., 2006). Further analysis indicates that the ventral portion of the Vi/Vc transition zone plays an important role in the processing of orofacial deep inputs (Sugiyo et al., 2005; Wang et al., 2006).
In the present study, we have examined the cellular mechanisms of the trigeminal processing of deep orofacial input after inflammation with an emphasis on glial–cytokine–neuronal interactions in the trigeminal transition zone. We hypothesize that trigeminal glial activation and inflammatory cytokine release affect or facilitate neuronal plasticity through interactions with neuronal glutamate receptors and play a critical role in the development of inflammatory pain. We tested this hypothesis in a rat model of masseter muscle inflammation and pain hypersensitivity because TMJD often results in persistent pain associated with the muscles of mastication. Our findings provide new and important evidence of astroglial activation and its role in signal coupling between cytokine and NMDAR, leading to activity-dependent neuronal plasticity and chronic or persistent pain.
Materials and Methods
Animals.
Male Sprague Dawley rats were used (100–300 g; Harlan, Indianapolis, IN). The rats received complete Freund's adjuvant (CFA; 0.05 ml, 1:1 oil/saline suspension; Sigma, St. Louis, MO) into the bilateral or unilateral masseter muscle under brief halothane anesthesia. Saline-injected and naive rats were used as controls. The injections were made mainly into the larger caudal region of the muscle to ensure that CFA was confined to the muscle tissue. The selective inflammation of the masseter tissues was verified with Evans' blue extravasation as described previously (Wang et al., 2006). The experiments were approved by the Institutional Animal Care and Use Committee of the University of Maryland Dental School.
Brainstem tissue dissection.
It has been shown that orofacial input activates a distinct region in the spinal trigeminal complex, the Vi/Vc transition zone (Strassman and Vos, 1993, Sugiyo et al., 2005; Wang et al., 2006). In particular, the ventral portion of the Vi/Vc transition zone (see Fig. 1a) selectively receives deep orofacial inputs (Sugiyo et al., 2005; Wang et al., 2006). We analyzed the responses of neurons and glial cells in this region to inflammation of the masseter muscle. The rats were anesthetized with halothane and quickly decapitated. A block of caudal brainstem tissues was cut. The tissue block includes a 2 mm segment at the level of the obex. The full length of the trigeminal Vi/Vc transition zone is enclosed in this block. The tissue block is then turned coronally and the ventral portions of the transition zone were harvested by taking punches with a 15 gauge puncture needle.
Figure 1.
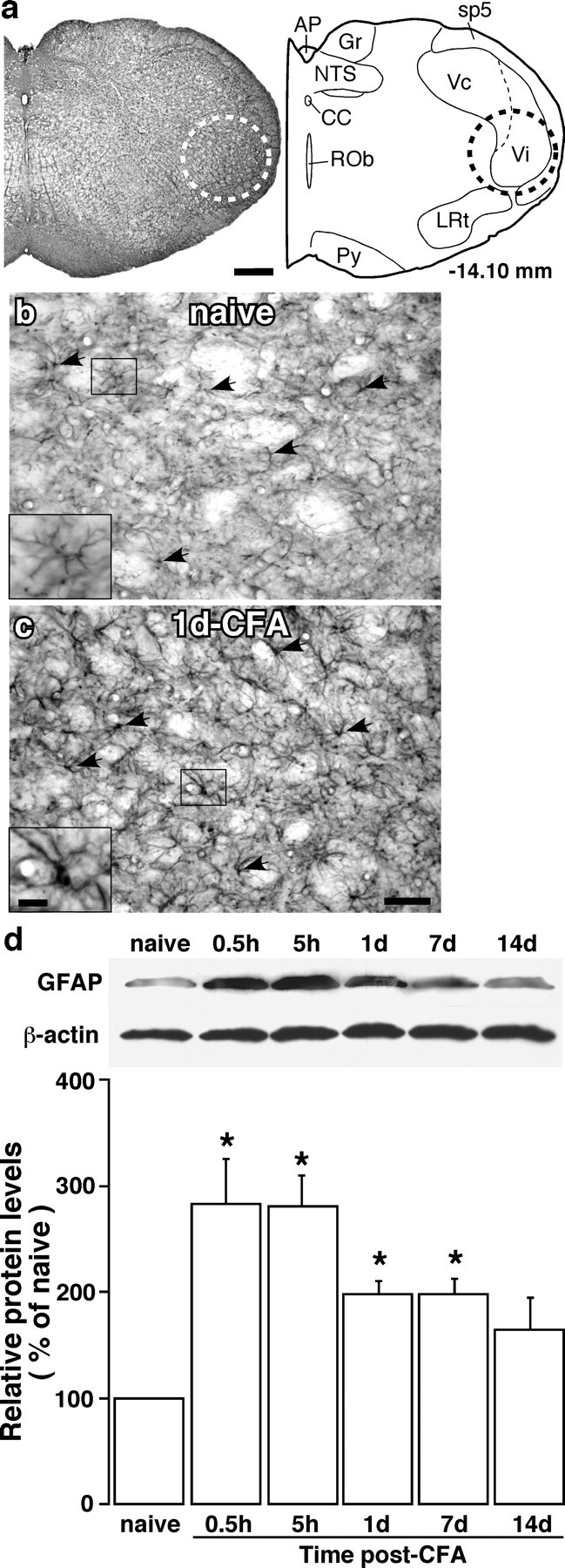
Upregulation of GFAP in astroglia after inflammation. a, Brainstem section (left) and its drawing (right) at a level ∼14.1 mm posterior to bregma illustrates the area analyzed (dashed circles) in the present study, which included the ventral portion of the Vi/Vc transition zone. AP, Area postrema; cc, central canal; Gr, gracile nucleus; LRt, lateral reticular nucleus; NTS, nucleus tractus solitarius; Py, pyramidal tract; Rob, nucleus raphe obscurus; sp5, spinal trigeminal tract. Scale bar, 0.4 mm. b, c, GFAP-like immunostaining in the ventral Vi/Vc transition zone in noninflamed (b; naive) and CFA-inflamed (c; 1 d post-CFA) rats. The insets (scale bar, 0.025 mm) at the bottom left corners were enlarged from small rectangles in b and c, respectively. Note the hypertrophy of astroglia in the inflamed rat (arrows, insets) compared with the naive rat. Scale bar, 0.1 mm. d, Western immunoblot illustrating masseter inflammation-induced increase in GFAP. The ventral portion of the Vi/Vc was punched out at various time points and total proteins were isolated. β-Actin was used as a loading control. An example of the blot is shown (top), and the relative protein levels are shown in the bottom histogram. *p < 0.05 vs naive rats. n = 4 for each time point. Error bars represent SEM.
Brainstem slices.
Adult male Sprague Dawley rats weighing 150–200 g (Harlan) were anesthetized with 2% halothane and decapitated. The caudal medulla was removed quickly and kept in cold artificial CSF consisting of the following (in mm): 124 NaCl, 4.4 KCl, 25 NaHCO3, 2.0 CaCl2, 1.0 MgSO4, 1.0 NaH2PO4, 10 d-glucose, pH 7.4, and bubbled with 95% O2 and 5% CO2. Transverse medullary slices (0.6 mm thick) at the Vi/Vc level were obtained. The slices were treated at room temperature with different drugs to test their effect on glial activation and receptor phosphorylation. The ventral Vi/Vc tissues were harvested and homogenized to extract proteins for Western blot analysis or postfixed with 10% paraformaldehyde for immunohistochemistry.
Western blot.
The tissues were homogenized in solubilization buffer (50 mm Tris HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 1 mm Na3VO4, 1 U/ml aprotinin, 20 μg/ml leupetin, 20 μg/ml pepstatin A). The homogenate was centrifuged at 20,200 × g for 10 min at 4°C. The supernatant was removed. The protein concentration is determined using a detergent-compatible protein assay with a bovine serum albumin standard. Each sample contains proteins from one animal. The proteins (50 μg) were separated on 7.5% SDS-PAGE and blotted on a nitrocellulose membrane (GE Healthcare, Piscataway, NJ). The blots were blocked with 5% milk in Tris-buffered saline (TBS) buffer and then incubated with the respective antibody. The membrane was washed with TBS and incubated with anti-goat or mouse IgG (1:3000; Santa Cruz Biotechnology, Santa Cruz, CA). The immunoreactivity was detected using enhanced chemiluminescence (ECL; GE Healthcare). The loading and blotting of the amount of protein was verified by reprobing the membrane with anti-β-actin antiserum (Sigma) and with Coomassie blue staining.
Immunohistochemistry.
Rats were deeply anesthetized with pentobarbital sodium (100 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer at pH 7.4. The brainstem was removed, postfixed, and transferred to 25% sucrose (w/v) for cryoprotection. Free-floating sections are incubated with relevant antibodies with 1–3% relevant normal sera. After washes in PBS, the sections were incubated with biotinylated secondary IgG (1:400; Vector Laboratories, Burlingame, CA) and then with avidin and biotinylated horseradish peroxidase complex (1:100; Vector Laboratories). The tissue sections were finally reacted with 0.05% diaminobenzidine dihydrochloride (Sigma) in 0.1 m phosphate buffer containing 0.003% hydrogen peroxide for 3–6 min. Control sections were processed with the same method except that the primary antisera are omitted or adsorbed by respective antigens. Double-labeling immunofluorescence was performed with cyanine 2 (Cy2) and Cy3 (1:500; Jackson ImmunoResearch, West Grove, PA) or Alexa Fluor 488 (1:500; Invitrogen, San Diego, CA) and Cy3 after incubation with respective primary antibodies.
Behavioral testing (Ren, 1999).
All behavioral tests were conducted under blind conditions. The rat was not restrained but habituated to stand on its hind paws and lean against the experimenter's hand who was wearing a regular leather work glove. The habituation required no more than normal petting of the rat, and it can be achieved within half an hour (Ren, 1999). A series of calibrated von Frey filaments were applied perpendicularly to the hairy skin above the masseter muscle site that received the CFA injection. The stimulation of the whisker pad was avoided. An active withdrawal of the head from the probing filament was defined as a response. Each von Frey filament was applied five times at intervals of a few seconds. The response frequencies [(number of responses per number of stimuli) × 100%] to a range of von Frey filament forces were determined and a stimulus–response frequency (S-R) curve plotted. After a nonlinear regression analysis, an EF50 value, defined as the von Frey filament force (g) that produces a 50% response frequency, was derived from the S-R curve. We used EF50 values as a measure of mechanical sensitivity. A leftward shift of the S-R curve, resulting in a reduction of EF50, occurred after inflammation (Guo et al., 2004; Sugiyo et al., 2005). This shift of the curve suggests the presence of mechanical hyperalgesia and allodynia because there was an increase in response to suprathreshold stimuli and a decreased response threshold for nocifensive behavior.
Antibodies.
The following antibodies were used: anti-glial fibrillary acidic proteins (GFAPs), anti-NeuN, anti-NR1, anti-P-ser896 NR1 (Millipore, Bedford, MA); anti-IL-1β (Endogen, Rockford, IL); anti-NR2A, anti-NR2B, anti-IL-1RI, anti-NK1R (Santa Cruz Biotechnology); anti-CD11b (clone OX-42; AbD Serotec, Raleigh, NC); anti-connexin 43 (Cx43), anti-β-actin (Sigma); anti-connexin 36 (Cx36; Invitrogen-Zymed, Carlsbad, CA); anti-NK1R (Calbiochem, La Jolla, CA).
Drug administration.
For microinjection, the guide cannula (C315G, 26 gauge; Plastics One, Roanoke, VA) was implanted ipsilateral to the side of inflammation under anesthesia with 50 mg/kg pentobarbital sodium injected intraperitoneally. Animals were securely placed into a stereotaxic device. A burr hole was drilled, and a guide cannula was lowered into the ventral Vi/Vc transition zone by referring to the rat brain atlas (Paxinos and Watson, 2005). The guide cannula was then secured with cranioplastic cement. To prevent clogging of the guide cannula, a dummy cannula (C315DC; Plastics One) was inserted and secured in place until the time of injection. The wound was cleaned with an antiseptic solution and closed with 4-0 silk suture. Animals were allowed to recover for 7 d before further experimentation. Drugs were injected into the Vi/Vc transition zone through a 33 gauge injection cannula (C315I; Plastics One) inserted through the tip of the guide cannula. The injection cannula was connected to a 0.01 ml Hamilton syringe by polyethylene-10 tubing. All injections (500 nl) were performed by delivering drug or vehicle solution slowly over a 2 min period. The sections of neural tissue were stained with cresyl violet for histological verification of the injection sites.
Osmotic pumps were used for delivering IL-1 receptor antagonist (IL-1ra; 12.5 pg per 500 nl/h) per manufacturer's instruction (model 2002, Alzet osmotic pumps; Durect, Cupertino, CA). The outlet of the pump is connected to PE60 tubing that is connected to a cannula implanted at the level of the obex. Drug vehicle (saline) was infused as a control. The infusion of the drug started at 3 d before CFA injection.
Drugs.
IL-1ra (kineret) was purchased from Amgen (Thousand Oaks, CA). The following agents were purchased from Biomol International (Plymouth Meeting, PA): 1-[6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (U73122; phospholipase C inhibitor), 1-(6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidine-dione (U73343; inactive analog of U73122), arachidonyltrifluoromethyl ketone (AACOCF3; phospholipase A2 inhibitor), and arachidonylmethylketone (AACOCH3; inactive analog of AACOCF3). The following agents were purchased from EMD Biosciences (La Jolla, CA) and Calbiochem: 2-aminoethoxydiphenylborate (2APB; inhibitor of IP3-mediated calcium release) and chelerythrine chloride (PKC inhibitor). The following drugs were also used: IL-1β (IL-1ra) and TNF-α (Peprotech, Rocky Hill, NJ); propentofylline (glial modulator), fluorocitrate (metabolic gliotoxin), substance P (SP), and calcitonin gene-related peptide (CGRP; Sigma); (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801, NMDAR channel blocker; Sigma-RBI, Natick, MA); NG-nitro-l-arginine methyl ester hydrochloride (l-NAME, nitric oxide synthase inhibitor; Tocris, Ellisville, MO); Nω-nitro-d-arginine methyl ester (d-NAME, inactive isoform of l-NAME; Rachem, Torrance, CA).
Data analysis.
Data are presented as means ± SEM. Statistical comparisons were made by the use of Student's t test or ANOVA with Fisher's protected least significant difference test for post hoc analysis. For Western blot analysis, the ECL-exposed films were digitized and densitometric quantification of immunoreactive bands was performed using U-SCAN-IT gel (version 4.3; Silk Scientific, Orem, UT). The relative protein levels were obtained by comparing the respective specific band to the β-actin control from the same membrane. The deduced ratios were further normalized to that of the naive rats on the same membrane and illustrated as the percentage of the naive controls. Raw data (ratios of the respective band over the β-actin band) were used for statistical comparisons. ANOVA and the unpaired two-tailed t test were used to determine significant differences. For the animals that are subject to repeated testing, ANOVA with repeated measures is used with time as a within animal effect. p < 0.05 is considered significant for all cases.
Results
Inflammation induces glial activation
We injected the inflammatory agent CFA into the masseter of the rat to produce muscle inflammation and hyperalgesia (Sugiyo et al., 2005; Watanabe et al., 2005). After CFA injection, rats exhibited an increased responsiveness and reduced response threshold to mechanical stimuli, characteristic of mechanical hyperalgesia (increased pain to a noxious stimulus) and allodynia (pain induced by a normally nonpainful stimulus) (Sugiyo et al., 2005; Watanabe et al., 2005).
We first examined whether masseter inflammation induced glial activation in the Vi/Vc transition zone. Using GFAP immunostaining, reactive astroglia were clearly seen in the Vi/Vc transition zone after CFA injection. Compared with naive rats (n = 4) (Fig. 1b), the activated astroglial cells typically exhibited hypertrophy with thicker processes and larger and densely stained cell bodies after inflammation (n = 4) (Fig. 1c). Injection of normal saline did not induce glial activation (n = 3). The upregulation of GFAP was quantified by Western blot. The tissues from the ventral Vi/Vc transition zone (Fig. 1a) were punched out, and total proteins were isolated. As shown in Figure 1d, the increase in GFAP levels was seen as early as at 0.5 h and lasted for ∼1 week after inflammation. The levels of CD11b (cluster of differentiation 11b), a marker of activated microglia, were also upregulated by masseter inflammation at 0.5 h after inflammation and lasted for approximately a week (data not shown). The glial activation by masseter inflammation correlates with the development of hyperalgesia (Sugiyo et al., 2005; Watanabe et al., 2005).
The activation of astroglia after inflammation was further indicated by activation of Cx43, the gap junction protein of astrocytes at 1 d after CFA-induced inflammation (Fig. 2). The increased Cx43 staining was associated with hypertrophied astrocytes, characteristic of reactive astrocytes (Fig. 2e–g). Double immunohistochemistry confirmed that the Cx43-like immunoreactivity (LI) colocalized with GFAP (a marker of astrocytes) but not with CD11b (a marker of microglia) neuron-specific nuclear protein (NeuN; a neuronal marker) and Cx36 (a neuronal gap junction protein) (Fig. 2f–k).
Figure 2.
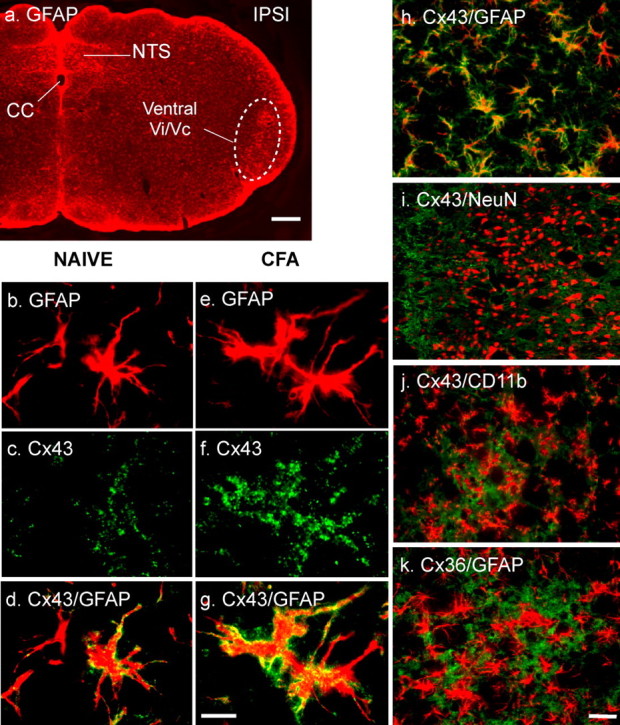
Connexin 43 (Cx43) is increased in reactive astrocytes in Vi/Vc after masseter inflammation. a, Immunofluorescent labeling of GFAP. An area of ventral Vi/Vc (dashed circle) is enlarged in e–k. cc, Central canal; NTS, nucleus tractus solitarius; IPSI, ipsilateral. b–g, Compared with the naive rat (b–d), CFA- (1 d; e–g) induced hypertrophy of astrocytes (b, e) and an increased staining intensity for Cx43 (c, f). d and g show double labeling of Cx43 with GFAP (yellow-orange). h–j, Double immunofluorescence labeling with antibodies against Cx43 (green) and GFAP (h; red), NeuN (i; red, neuronal marker), or CD11b (j; red, microglial marker). k, Immunofluorescence labeling of Cx36 (red, neuronal gap junction protein) with GFAP. Note the presence of overlapped staining in h (yellow-orange) and lack of overlap in staining in i–k, indicating that Cx43 is an astrocytic gap junction protein. Scale bars: a, 0.2 mm; b–k, 0.02 mm.
Inflammation induces an increase in proinflammatory cytokine IL-1β in astroglia
We next examined whether activation of glia by masseter inflammation is accompanied by an increase in inflammatory cytokine levels. Compared with the naive rats (Fig. 3a), the immunostaining for IL-1β (n = 4) was clearly increased in the Vi/Vc transition zone (Fig. 3b). The immunostaining for TNF-α one of the other prototype proinflammatory cytokines, did not show changes after inflammation (n = 4; data not shown). Western blot confirmed the increase of IL-1β in the Vi/Vc transition zone with a time course of 0.5 h to 7 d (Fig. 3c), similar to the time course of astrocyte hypertrophy after masseter inflammation. IL-1β was found in astroglia as shown by double immunofluorescence staining. Figure 4a–c shows colocalization of IL-1β and GFAP 1 d after inflammation, suggesting that activated astroglia is a source of IL-1β release. However, the IL-1β staining in the Vi/Vc transition zone did not colocalize with CD11b (Fig. 4d–f) and NeuN, a neuronal marker (Fig. 4g–i).
Figure 3.
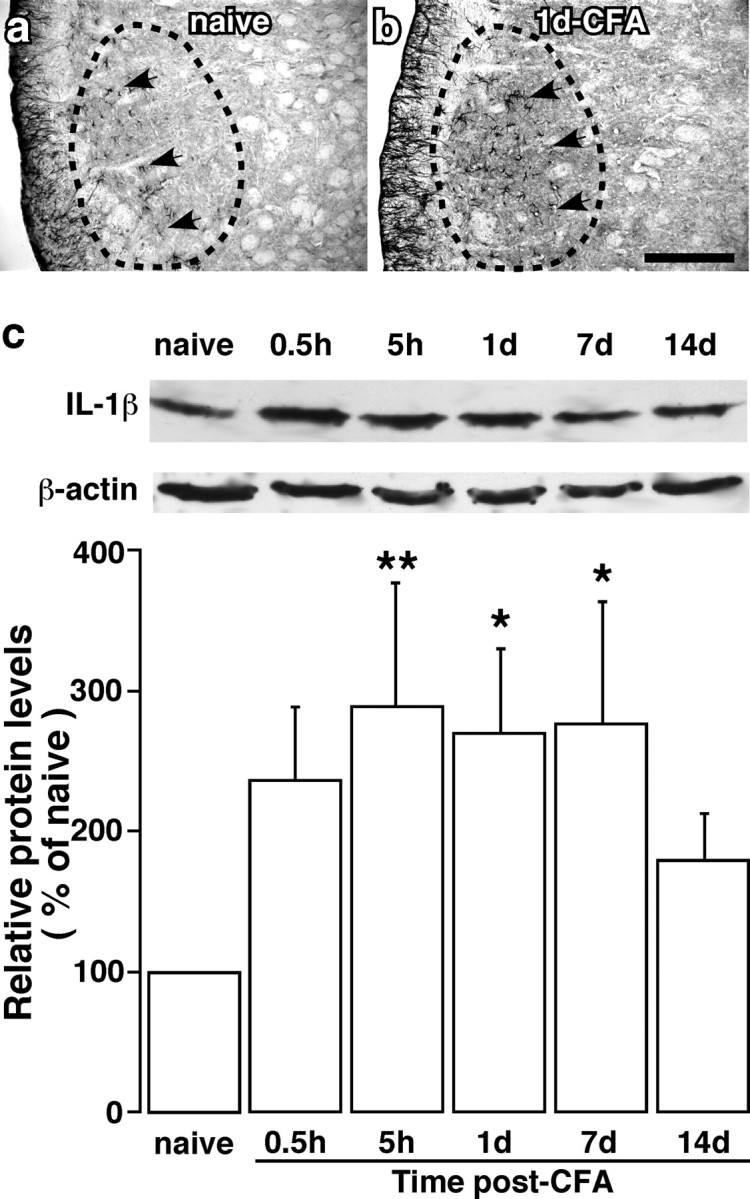
Masseter inflammation induced increases in IL-1β immunoreactivity in the Vi/Vc trigeminal transition zone. a, b, IL-1β-like immunostaining in the ventral Vi/Vc transition zone in noninflamed (a; naive) and CFA-inflamed (b; 1 d post-CFA) rats. The dashed circles indicate regions of the Vi/Vc transition zone. Arrows indicate examples of IL-1β-labeled profiles. Scale bar, 0.2 mm. c, Western immunoblot illustrating masseter inflammation-induced increase in IL-1β. An example of the blot is shown (top), and the relative protein levels are shown in the bottom histogram. *p < 0.05; **p < 0.01 vs naive rats. n = 4 for each time point. Error bars represent SEM.
Figure 4.

Colocalization of IL-1β with GFAP and the effect of a glial inhibitor on GFAP and IL-1β upregulation. a–i, Brainstem tissue sections were processed for IL-1β (a, d, g) and double labeled with GFAP (b), CD11b (e), or NeuN (h) 1 d after masseter inflammation. Photomicrograph images are from the ventral portion of the Vi/Vc transition zone. IL-1β immunoreactivity is shown as green fluorescence after reacting with anti-goat IgG conjugated with Cy2. GFAP-, CD11b-, and NeuN-LI are visualized with Cy3 conjugated with anti-rabbit IgG (red). Overlap of a and b reveals double fluorescence (c; yellow/orange), which was only identified for IL-1β and GFAP staining (arrows in a, b, c), indicating the selective induction of IL-1β in astroglia. d–f and g–i show lack of double labeling of IL-1β with CD11b, a marker of microglia, or NeuN, a marker of neurons. The single-labeled neurons in d–i are indicated by arrowheads. Scale bar, 0.1 mm. j, k, Western blots illustrating the effect of propentofylline (PPF), a glial inhibitor, on masseter inflammation-induced increase in GFAP (j) and IL-1β (k) in the ventral Vi/Vc transition zone. Propentofylline was injected intraperitoneally at 10 mg/kg first at 20 min before CFA, and the second PPF injection (10 mg/kg) was given 8 h after CFA. The Vi/Vc tissues were collected 1 d after CFA injection. Saline was injected as a vehicle control. Compared with naive rats, the GFAP and IL-1β immunoreactivity was increased at 1 d in rats receiving saline. The PPF treatment significantly attenuated CFA-induced increase in GFAP and IL-1β (p < 0.01; n = 3 per group.)
To demonstrate the association of CFA-induced cytokine with glial activation, we studied the effect of glial inhibitors. We injected propentofylline, a glial modulator, 20 min before CFA and 8 h after CFA injection (10 mg/kg per injection, i.p.), and the tissues from the ventral Vi/Vc transition zone were collected 1 d after CFA. Saline was injected as a vehicle control for propentofylline. Compared with naive rats, the GFAP and IL-1β immunoreactivity was increased at 1 d in saline-treated rats (Fig. 4j,k). The CFA-induced increases in GFAP levels were significantly reduced from 338 ± 13.6 to 138 ± 7.6% of naive rats (p < 0.01; n = 3), and the increased IL-1β levels were reduced from 341 ± 31.1 to 117 ± 6.5% of naive rats (p < 0.01; n = 3) after the propentofylline treatment. These results suggest that glial activation is upstream and critical to cytokine induction in the Vi/Vc after inflammation.
Glial activation depends on nerve input
An intriguing possibility for triggering glial activation and cytokine release is through neuronal signals. We hypothesize that this neuron-to-glia signaling plays a key role in glial activation, cytokine production, and neuronal hyperexcitability, and eventually leads to increased pain. We directly tested this hypothesis by producing local anesthetic block of the primary afferent input. A local anesthetic, lidocaine (2%, 0.05 ml), was infiltrated into tissues surrounding the masseter nerve at the level of the mandibular notch (Greene, 1935). The amount of lidocaine infiltration was small (0.05 ml), and the site of infiltration was ∼5 mm away from the site of inflammation, thus avoiding an effect of lidocaine on immune cells attracted to the site of CFA injection. Lidocaine was given 10 min before the injection of CFA into the masseter muscle. The Vi/Vc tissues were punched out 30 min after CFA injection. In rats pretreated with saline (0.9%, 0.05 ml), the GFAP level was significantly increased (p < 0.05; n = 4) after CFA injection (Fig. 5a). In lidocaine-treated rats, the CFA-induced increase in GFAP was abolished (n = 4) (Fig. 5a). The inflammation-induced increase in IL-1β was also blocked (Fig. 5b). The lidocaine treatment also blocked the early development of mechanical allodynia, confirming the effectiveness of local anesthesia. These findings indicate that primary afferent inputs associated with peripheral nociceptor activation after inflammation are necessary for the activation of astroglia and upregulation of IL-1β.
Figure 5.
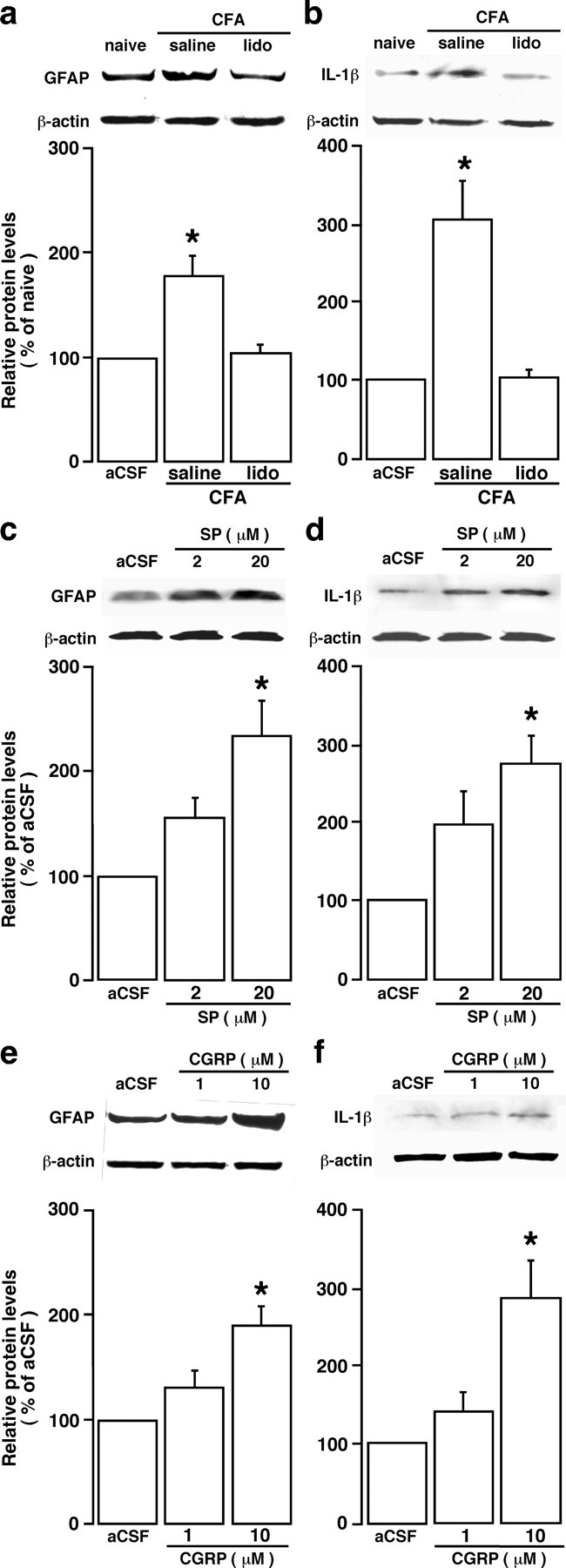
Neuronal signal contributes to inflammation-induced glial activation. a, b, The effect of local anesthesia on inflammation-induced astroglial activation. Lidocaine (lido; 0.05 ml, 2%) was infiltrated to the tissues surrounding the masseter nerve 10 min before CFA injection, and the Vi/Vc tissues were removed 30 min after CFA injection. Saline (0.05 ml, 0.9%) was injected as a control for lidocaine. Compared with saline-injected rats, the increase in GFAP (a) and IL-1β (b) was blocked by lidocaine treatment, suggesting that inflammation-induced astrocytic activation depended on the input from the injured site. c, d, Substance P (SP), a prototype neurotransmitter released from central terminals of primary afferent neurons, induced increases in GFAP (c) and IL-1β (d) levels in the Vi/Vc transition zone. Transverse medullary slices (0.5 mm thick) including the Vi/Vc were obtained from 8- to 10-week-old rats. The slice was incubated in artificial CSF (aCSF) with SP (2 and 20 μm) for 20 min and proteins in the ventral Vi/Vc extracted for Western blot analysis. e, f, CGRP induces GFAP and IL-1β in the Vi/Vc transition zone in vitro. Transverse medullary slices (0.5 mm thick) including the Vi/Vc were incubated in aCSF with CGRP (1 and 10 μm) for 20 min and proteins in the ventral Vi/Vc extracted for Western blot analysis. CGRP induced increases in GFAP (e) and IL-1β (f) levels in the Vi/Vc transition zone. For all panels, representative immunoblots against anti-GFAP and IL-1β antibodies are shown on top and the relative protein levels are shown in the bottom histogram. *p < 0.05 vs naive (a, b) rats or aCSF (c–f). n = 3–4 for each time point. Error bars represent SEM.
We next tested whether substance P, a prototype neurotransmitter in pain processing circuitry at spinal and trigeminal levels, could induce glial activation and cytokine increase. Medullary slices were obtained from naive adult Sprague Dawley rats. After a 20 min incubation of the slice with substance P (2 and 20 μm), the tissues of the Vi/Vc transition zone were punched out for Western blotting. We observed a significant increase in GFAP (Fig. 5c) and IL-1β (Fig. 5d) in the Vi/Vc transition zone. CGRP, the other neuropeptide released from primary afferent terminals (Henry et al., 1996), produced similar increases in GFAP and IL-1β (Fig. 5e,f). These results suggest that activation of either CGRP and substance P receptors can induce glial activation and cytokine induction.
Interestingly, we did not find colocalization of NK-1 (neurokinin-1) tachykinin receptors, the primary binding site for substance P in the nervous system, with GFAP in the Vi/Vc transition zone (Fig. 6a–d). We then tested whether nitric oxide, a gaseous membrane soluble molecule that is capable of regulating GFAP in astrocytes (Meller and Gebhart, 1993; Brahmachari et al., 2006), could act as a messenger of substance P between the neuron and glia (Meller and Gebhart, 1993; Holguin et al., 2004). At 10 min before the application of substance P (10 μm), the slice was incubated with a nitric oxide synthase inhibitor l-NAME (2 mm) or its inactive isomer, d-NAME (2 mm). Compared with vehicle and d-NAME-treated slices, l-NAME blocked the substance P-induced upregulation of GFAP and IL-1β (Fig. 6e). These results are consistent with a recent report that nitric oxide may act upstream to glial activation (Holguin et al., 2004). These convergent results suggest that neuronal input and related chemical mediators are essential for triggering trigeminal glial activation and cytokine induction after peripheral tissue injury.
Figure 6.
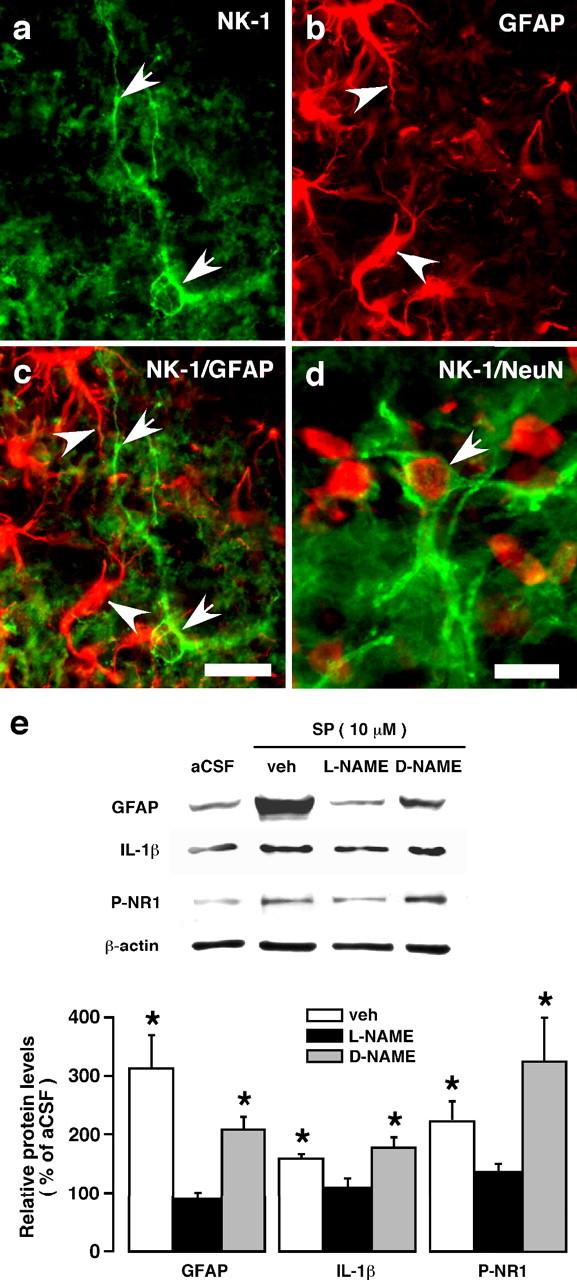
Lack of NK-1 (neurokinin-1) tachykinin receptor immunoreactivity in Vi/Vc astrocytes and the effect of a nitric oxide synthase inhibitor on substance P-induced upregulation of GFAP, IL-1β, and NMDAR phosphorylation (P-NR1) in vitro. a, NK-1-like immunoreactivity is shown by green immunofluorescence (Alexa Fluor 488, 1:500). Arrows indicate NK-1 staining profiles. b, The same field as panel a was stained with GFAP (red, Cy3; 1:500), a marker of astrocytes. Arrowheads indicate examples of GFAP staining profile. c, Overlap of panels a and b shows a lack of colocalization of GFAP (arrowheads) and NK-1 (arrows) immunoreactivity. d, Arrow indicates double label of NK-1 and NeuN, confirming neuronal localization of NK-1. Images represent a field of the trigeminal Vi/Vc transition zone from an adult rat at 24 h after masseter inflammation. Scale bars: a–c, 0.02 mm; d, 0.01 mm. e, Effect of a nitric oxide synthase inhibitor, l-NAME, on substance P-induced upregulation of GFAP, IL-1β, and NMDAR phosphorylation (P-NR1). Medullary slices were incubated with l-NAME (2 mm), d-NAME (2 mm, inactive isoform of l-NAME), or vehicle (saline) before incubation with substance P (10 μm). Representative immunoblots are shown on top, and the relative protein levels are shown in the bottom histogram. *p < 0.05 vs l-NAME. n = 3–4 per group. Error bars represent SEM.
Glial and IL-1 receptor antagonist inhibits behavioral hyperalgesia and NMDA receptor phosphorylation
To study the effect of glial activation on neuronal activity, we first examined NMDAR activation after masseter inflammation. Tissue injury induces NMDAR activation involving phosphorylation of the NR1 and NR2 subunits in the spinal dorsal horn (Guo et al., 2002, 2004; Brenner et al., 2004), which underlies the mechanisms of dorsal horn hyperexcitability and persistent pain. As shown in Figure 7a, masseter inflammation induced a time-dependent increase in phosphorylation at serine 896 of the NR1 subunit (P-ser896-NR1) in the Vi/Vc as early as 0.5 h after inflammation. There was also a time-dependent upregulation of the NR1 protein levels (Fig. 7a). The increase in the levels of NR1 proteins appeared late (1–7 d post-CFA; p < 0.05) and small in magnitude. Double immunostaining of anti-P-ser896-NR1 with NeuN confirmed that the increased NMDAR phosphorylation was in neurons 1 d after inflammation (Fig. 7b). Interestingly, the inflammation or SP-induced NMDAR phosphorylation was also blocked by propentofylline (p < 0.01) (Fig. 7c) or l-NAME (p < 0.05) (Fig. 6e).
Figure 7.
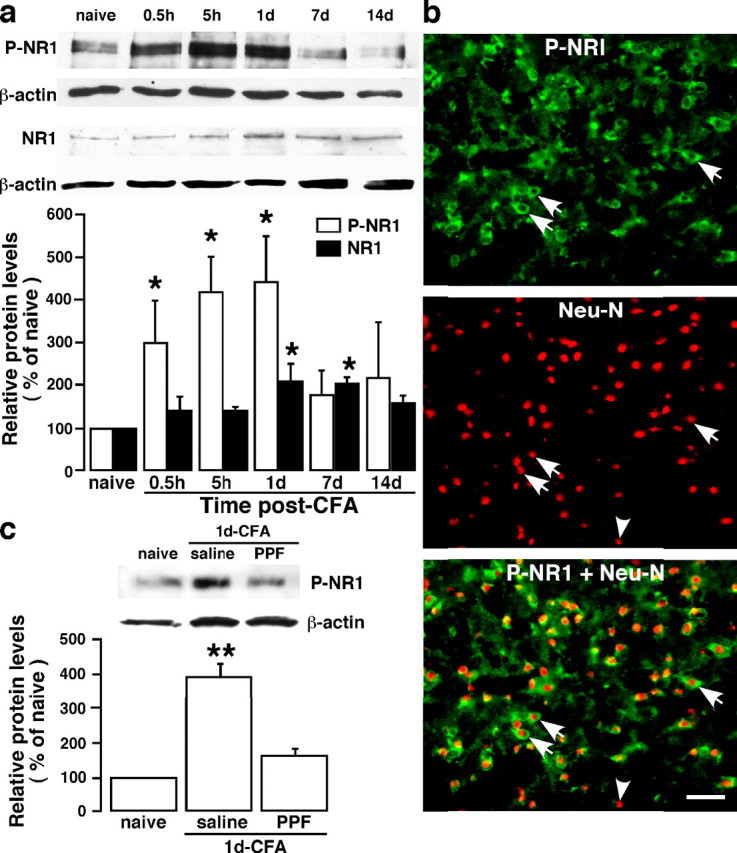
Inflammation-induced NMDA receptor phosphorylation and the effect of a glial inhibitor. a, Masseter inflammation induced a time-dependent increase in phosphoserine 896 NR1 (P-NR1) in the Vi/Vc. A late (1–7 d after CFA injection) and slight increase in the NR1 protein levels was also seen. Representative immunoblots against anti-P-NR1 and anti-NR1 antibodies are shown on top and the relative P-NR1 and NR1 levels are shown below. *p < 0.05 versus naive. n = 4 per time point. b, Localization of P-NR1 in Vi/Vc neurons 1 d after CFA injection. P-NR1 is shown as green fluorescence (Alexa Fluor 488; top). NeuN is visualized with Cy3 (red; middle). Overlap of left and middle panels reveals cells that exhibit double fluorescence of NeuN (nucleus) and P-NR1 (cytoplasm) in the same neurons (bottom). Examples of double- (arrows) and single- (arrowheads) labeled neurons are indicated. Scale bar, 0.05 mm. c, The effect of PPF on masseter inflammation-induced increase in P-NR1. Propentofylline (10 mg/kg, i.p.) was first injected 20 min before CFA, and the second PPF injection (10 mg/kg) was given 8 h after CFA injection. The Vi/Vc tissues were collected at 1 d after CFA. Saline was injected as a vehicle control for PPF. Compared with noninflamed rats (naive), the P-NR1 immunoreactivity was increased 1 d in rats receiving saline. The PPF treatment attenuated CFA-induced increase in P-NR1 (PPF). **p < 0.01 versus naive. n = 4 per group. Error bars represent SEM.
To test our hypothesis that trigeminal glial activation and inflammatory cytokine release affect or facilitate neuronal plasticity through interactions with neuronal glutamate receptors and play a critical role in the development of inflammatory hyperalgesia, we studied the effects of a glial inhibitor and an IL-1ra on masseter hyperalgesia and NMDAR phosphorylation. The mechanical allodynia/hyperalgesia developed after masseter inflammation was assessed by applying a series of von Frey microfilaments to the skin above the masseter muscle. The response frequencies to a range of von Frey filament forces were determined, a stimulus-response frequency relationship established, and EF50 values derived. After establishing the behavioral hyperalgesia at 1 d after CFA injection, fluorocitrate, a gliotoxin that is relatively selective against astroglia (Hosoi et al., 2004), was microinjected into the ventral Vi/Vc transition zone through a chronically implanted guide cannula (Fig. 8a). Fluorocitrate at 1.0 μg attenuated significantly mechanical hyperalgesia at 30–60 min time points tested, as indicated by an increase in EF50 values. Western blotting of Vi/Vc tissues from these rats dissected after behavioral test showed that the inflammation-induced P-ser896-NR1 levels were also reduced significantly (Fig. 8b).
Figure 8.
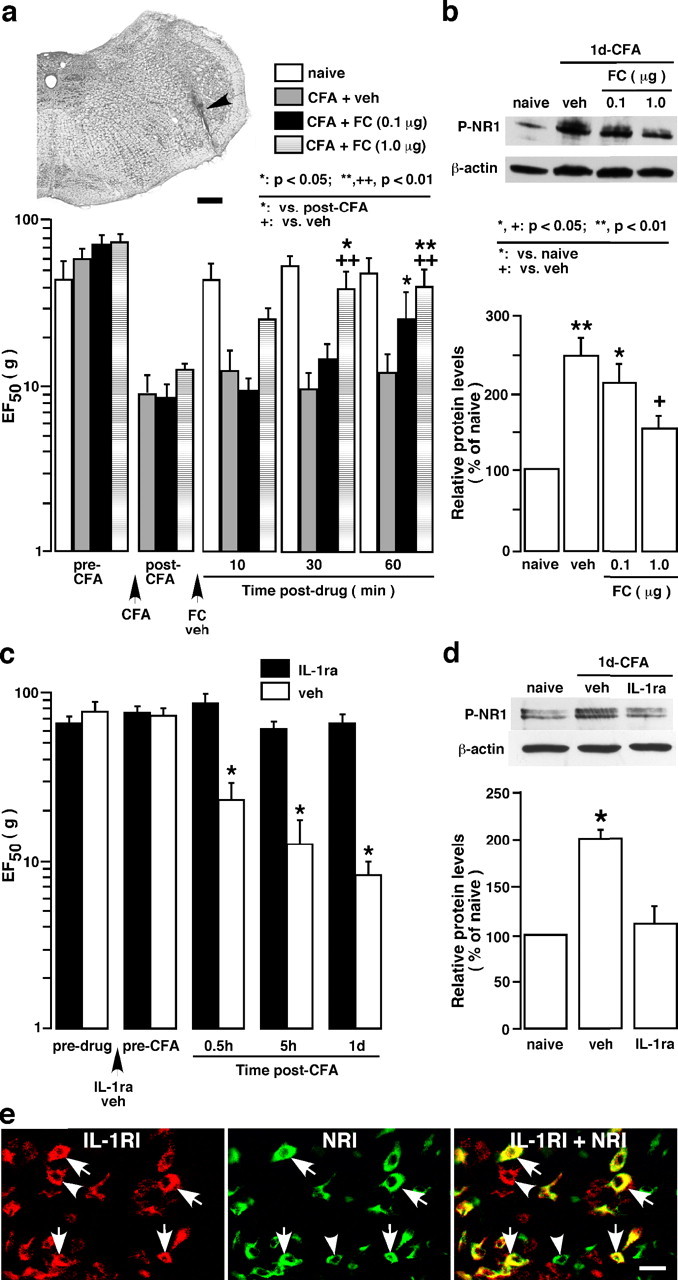
Effect of glial inhibitor fluorocitrate (FC) and IL-1ra on behavioral hyperalgesia and NMDAR phosphorylation after masseter inflammation. a, Effect of microinjection of FC into the Vi/Vc transition zone on hyperalgesia. The top image shows an example of the injection site (arrow). Scale bar, 0.4 mm. The bottom histogram shows EF50 values derived from stimulus–response curves in behavioral testing. Vertical bars indicate 95% confidence intervals of EF50. A significant decrease in EF50 indicates the presence of hyperalgesia and allodynia. Intra-Vi/Vc injection of fluorocitrate (0.1 and 1.0 μg; n = 6 per dose) attenuated the masseter hyperalgesia, as indicated by a significant increase in EF50 values. b, Rats in a were killed 70 min after drug injection, and the ventral Vi/Vc tissues were punched out for Western blotting. Fluorocitrate attenuated inflammation- induced increase in P-NR1 levels. c, Effect of IL-1ra on hyperalgesia. Compared with vehicle-injected rats, intrathecal infusion of IL-1ra (12.5 pg per 500 nl/h; n = 9) attenuated the masseter hyperalgesia. Asterisks denote significant differences from the vehicle saline- (veh; n = 6) treated rats (*p < 0.05; ANOVA with repeated-measures and post hoc tests). The infusion of the drug started 3 d before CFA injection. d, Rats in c were killed 1 d after inflammation and the ventral Vi/Vc tissues were punched out for the Western blot. Compared with vehicle-treated rats, IL-1ra attenuated inflammation-induced increase in P-NR1 levels. *p < 0.05 versus naive rats. e, Colocalization of IL-1R with NR1 in Vi/Vc neurons. IL-1R is shown as red fluorescence (Cy3, left). NR1 is visualized with Cy2 (green, middle). Overlap of left and middle panels reveals cells that exhibit double fluorescence (yellow/orange), suggesting colocalization of IL-1R and NR1. Examples of double- (arrows) and single- (arrowheads) labeled neurons are indicated. Scale bar, 0.02 mm. Error bars in a–d represent SEM.
IL-1ra (12.5 pg per 500 nl/h; n = 9) was administered intrathecally via osmotic pumps (model 2002; Alzet, Cupertino, CA) through a cannula implanted at the level of the obex. The infusion of the drug started 3 d before CFA injection. Saline was infused as the vehicle control (n = 6). The Vi/Vc tissues from these rats were dissected out 1 d after CFA injection to examine the level of P-ser896-NR1. The results showed that IL-1ra significantly attenuated behavioral hyperalgesia (Fig. 8c) and blocked the increase in NMDAR phosphorylation (Fig. 8d) after masseter inflammation. Our immunostaining experiments showed that IL-1 receptor (IL-R) and the NR1 subunit of the NMDAR colocalized in Vi/Vc neurons (Fig. 8e). These results are consistent with our hypothesis and suggest that glial activation and IL-1R signaling contributes directly to modulation of neuronal NMDAR function.
IL-1β induces NMDA receptor phosphorylation
We directly tested whether IL-1β induced NMDAR phosphorylation in our in vitro medullary slice preparation. The results showed that incubation of IL-1β (1.0 and 3.0 nm; n = 4 per dose) in the medullary slices induced a significant increase in P-ser896-NR1 levels (p < 0.05) in the Vi/Vc transition zone and the effect was apparently dose dependent (Fig. 9a). Because another prototype inflammatory cytokine, TNF-α, has also been shown to regulate neuronal activity by affecting AMPA receptor trafficking (Stellwagen et al., 2005), we tested the effect of TNF-α. Interestingly, the application of TNF-α (7.0 nm, n = 4) did not affect P-ser896-NR1 levels at the dose tested (Fig. 9b). We further show that IL-1β-induced NR1 phosphorylation was blocked by IL-1ra (0.01 mm, n = 4) (Fig. 9c) but not by fluorocitrate, a glial inhibitor (0.01 mm; n = 3) (Fig. 9d). Microinjection of IL-1β (1.5 pmol/500 nl) into the ventral Vi/Vc transition zone in vivo also produced an increase in P-ser896-NR1 levels that was blocked by IL-1ra (1 nmol) (Fig. 9e). Thus, IL-1R signaling may selectively regulate NMDAR function and increase synaptic strength via post-translational phosphorylation and contribute to inflammation-induced pain hypersensitivity.
Figure 9.

IL-1β induces NMDA receptor NR1 ser896-phosphorylation. Representative immunoblots are shown on top, and mean relative levels of P-NR1 proteins are shown in the bar graphs. a–d, The transverse medullary slices, including the Vi/Vc transition zone, were obtained from adult 8- to 10-week-old rats. The slices were incubated with IL-1β or TNF-α for 15 min, and the ventral Vi/Vc tissues were harvested by taking punches. Note that IL-1β induced a significant increase in P-NR1 levels (a), but TNF-α did not produce an increase in P-NR1 at the dose tested (b). c, d, Compared with vehicle (veh), the IL-1β-induced increase in P-NR1 was blocked by IL-1ra (0.01 mm; c), but not by fluorocitrate (FC; 0.01 mm), a glial inhibitor (d). e, IL-1β induced an increase in P-NR1 in vivo. IL-1β (1.5 pmol/500 nl) was microinjected into the ventral Vi/Vc transition zone and Vi/Vc tissues were punched out 30 min after IL-1β injection. Animals subjected to the injection procedure without receiving IL-1β were used as the SHAM control. Saline (500 nl) was injected as a control for IL-1β. Compared with naive rat, IL-1β induced an increase in P-ser896-NR1 that was blocked by IL-1ra (1 nmol). The SHAM procedure and saline did not have an effect on the P-NR1 levels. *p < 0.05; **p < 0.01 vs artificial CSF (aCSF)-treated slices (a–d) or naive rats (e; n = 3–4 per group). Error bars represent SEM.
We further examined the signal pathways leading to IL-1β-induced NMDAR phosphorylation. The results showed that IL-1β-induced NR1 phosphorylation was blocked by chelerythrine (0.01 mm; n = 3), a PKC inhibitor (Fig. 10a), confirming that PKC is involved in this effect. Furthermore, 2APB (0.072 μm, n = 4), a membrane-permeable IP3 receptor antagonist that does not affect Ca2+ release from the ryanodine-sensitive Ca2+ stores (Maruyama et al., 1997), also blocked IL-1β-induced NR1 phosphorylation (Fig. 10b). The NMDAR channel blocker MK-801 (0.03 mm; n = 3) did not affect the IL-1β-induced increase in P-ser896-NR1 (Fig. 10c).
Figure 10.
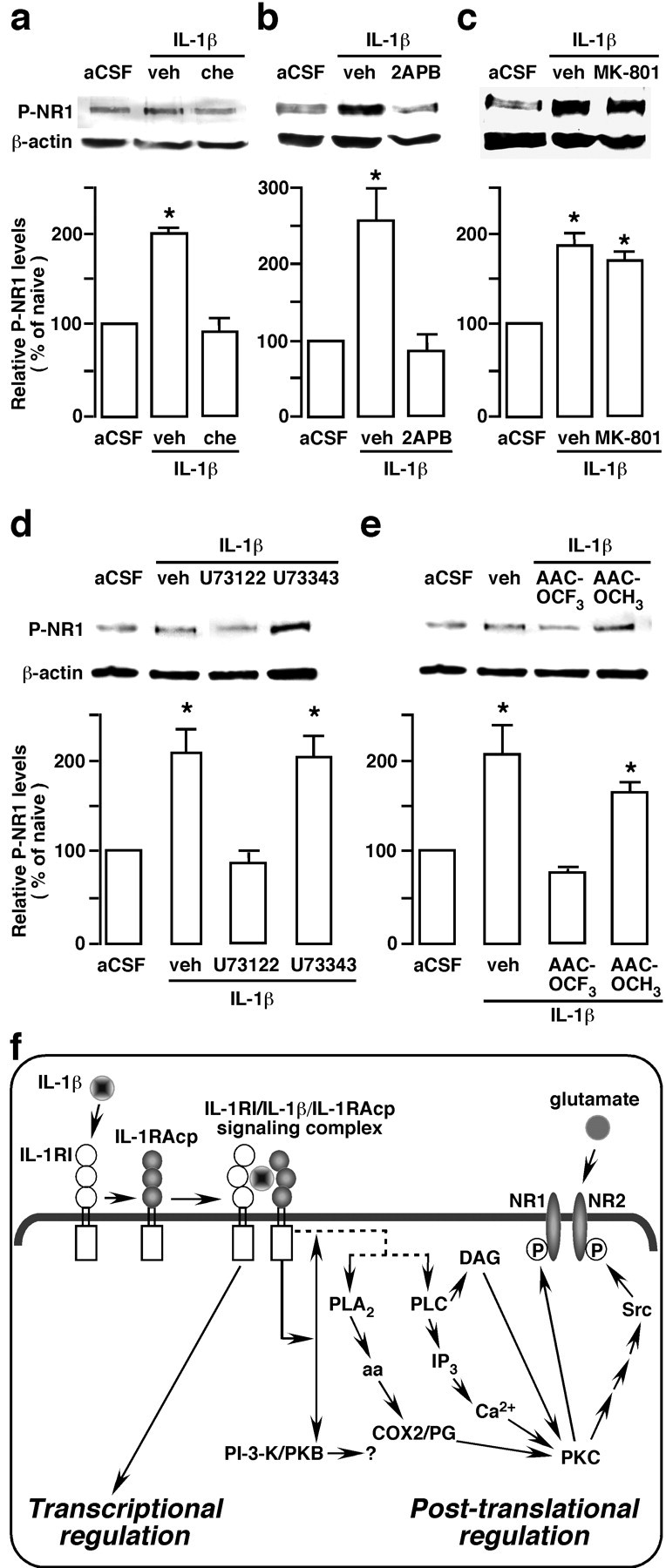
Signal pathways involved in the IL-1R NMDA receptor coupling. The transverse medullary slices, including the Vi/Vc transition zone, was obtained from adult 8- to 10-week-old rats. The slices were incubated with IL-1β for 15 min and the ventral Vi/Vc tissues were harvested by taking punches. a–c, Compared with vehicle (veh), the IL-1β-induced increase in P-NR1 was blocked by the following: chelerythrine (Che, 0.01 mm), a PKC inhibitor (a); 2APB (0.072 mm), an IP3 receptor antagonist (b); but not by an NMDAR channel blocker, MK-801 (0.03 mm; c). d, e, U73122 (0.01 mm; d), a phosphlipase C (PLC) inhibitor, and AACOCF3 (0.01 mm; e), a PLA2 inhibitor, also blocked the effect of IL-1β on P-NR1 levels. U73343 (0.01 mm) and AACOCH3 are inactive analogs of U73122 and AACOCF3, respectively, and were used as negative controls. All inhibitors and inactive analogs were applied 10 min before IL-1β. *p < 0.05 versus artificial CSF (aCSF)-treated slices (n = 3–4 per group). Error bars represent SEM. f, The proposed modulation of NMDAR phosphorylation involving IL-1R signaling. Binding of IL-1β to the IL-1R leads to the recruitment of the IL-1R accessory protein (IL-1RAcP) and the formation of a heterotrimeric complex. The signaling IL-1R complex may stimulate PLC or PLA2, or both, although the intermediate steps are unknown (dashed lines). The downstream effectors of PLA2 and PLC may include activation of PKC through the formation of arachidonic acid (aa), cyclooxygenase2 (COX2)/prostaglandin (PG), IP3 (inositol 1,4,5-trisphosphate), and diacylglycerol (DAG). PKC can directly phosphorylate serine 890 and 896 of the NR1 subunit of the NMDAR and indirectly lead to Src activation and NR2B tyrosine phosphorylation (P). The PI-3-K (phosphatidylinositol-3-kinase)/PKB (protein kinase B or Akt kinase) pathway is also downstream to IL-1R signaling. Whether it participates in regulation of NMDAR is unclear. It is well documented that IL-1R signaling leads to transcriptional regulation of cellular activity. Our results show that the IL-1R signaling is also involved in post-translational regulation of cellular activity through coupling to NMDARs, which may underlie mechanisms of persistent pain.
Because PKC is a downstream effector of arachidonic acid and diacylglycerol, the results of the PKC inhibitor experiment suggest that a key link between IL-1β and subsequent activation of NMDAR may involve phospholipase A2 (PLA2) or phospholipase C (PLC) (Chen et al., 2000; Molina-Holgado et al., 2000; Martin and Wesche, 2002). We directly tested whether PLC and PLA2 play a role in mediating IL-β-induced NMDAR phosphorylation. The results showed that the PLC inhibitor U73122 (0.01 mm; n = 3) (Fig. 10d) and PLA2 inhibitor AACOCF3 (0.01 mm; n = 3) (Fig. 10e) blocked the effect of IL-1β on NMDAR phosphorylation. U73343 and AACOCH3, the inactive analogs of U73122 and AACOCF3, respectively, did not block the effect of IL-1β on NMDAR phosphorylation (Fig. 10d,e). It is well known that IL-1R signaling leads to transcriptional regulation of cellular function (Martin and Wesche, 2002). Our results show that the effect of IL-1β on NMDAR phosphorylation is attributable to post-translational regulation by IL-1R signaling and that the intermediate pathways linking IL-1R and NMDAR activation involves PLC, PLA2, and subsequent PKC activation and intracellular Ca2+ release (Fig. 10f).
Discussion
In addition to supporting roles in normal neuronal function, CNS glial cells are increasingly known to be important regulators of synaptic activity and key functional units of the nervous system (Fields and Stevens-Graham, 2002). Glial cells are activated in response to stimuli related to trauma, ischemia, and invading pathogens. Peripheral tissue or nerve injury induces CNS glial activation, mainly involving astrocytes and microglia (Garrison et al., 1991; Meller et al., 1994; Tsuda et al., 2003; Tanga et al., 2005; Watkins and Maier, 2005; Hains and Waxman, 2006; Zhuang et al., 2006). It is not known how glial cells and associated cytokines contribute to CNS neuronal plasticity and the persistent pain state. Utilizing a trigeminal model of inflammation and persistent pain, the present study demonstrates interactions between astroglia and neurons through proinflammatory cytokines and NMDAR coupling in central hyperexcitability and inflammatory hyperalgesia.
In the trigeminal transition zone where masseteric primary afferents terminate (Wang et al., 2006), masseter inflammation activates glial cells and upregulates inflammatory cytokine IL-1β that was selectively localized to astroglial cells. Similarly, we have reported selective localization of IL-1β in astroglia in the spinal cord in a rat bone cancer pain model (Zhang et al., 2005). The activation of astroglia was further demonstrated by a selective upregulation of Cx43, an astrocytic gap junction protein, consistent with a role of gap junction proteins in astroglial function. The activation of astrocytes involves calcium waves spreading between astrocytes through the gap junction (Nedergaard, 1994). The gap junction may play a role in spinal nociceptive processing (Asghar et al., 2005) and pain facilitation (Spataro et al., 2004). Facial nerve transection results in a rapid increase in Cx43-LI in the facial nucleus of rats (Rohlmann et al., 1993, 1994). Peripheral nerve stimulation affects the phosphorylation state of Cx43 in rats (Li and Nagy, 2000).
Although earlier studies have attempted to correlate cytokines with trigeminal pain, the major effort was on action of cytokines in the periphery (Alstergren et al., 1999; Kacena et al., 2001). Cytokines including IL-1β, IL-6, and TNF-α are upregulated in the spinal cord and brain in the CFA model of inflammation (Raghavendra et al., 2004). An elevation of IL-6 level in the brainstem, including the spinal trigeminal complex, is found after trigeminal nerve injury in the rat (Anderson and Rao, 2001). Different from the response to direct brain injury (Yan et al., 1992), the induction of central cytokines after peripheral injury involves more complicated and still unknown pathways at both spinal and trigeminal levels. Our results suggest that the proinflammatory cytokine IL-1β was selectively induced in astroglia in the Vi/Vc transition zone after inflammation. These results strongly suggest that activated glial cells are a source of central IL-1β release after peripheral injury.
We further observed that blocking injury-related input blocked the increased GFAP, a marker of reactive astroglia and IL-1β, and a primary afferent neurotransmitter, substance P, induced an increase in GFAP and IL-1β in the Vi/Vc transition zone. The existing literature has suggested signaling between axons and glial cells (Chiu and Kriegler, 1994). Neuronal activity can generate glial calcium responses in the hippocampus (Dani et al., 1992). It is shown that substance P stimulates IL-1 production by astrocytes via intracellular calcium (Martin et al., 1992). Results from cultured astroglia implicate that glia may be activated by chemicals that are released from primary afferent terminals and involved in pain transmission (Takuma et al., 1996). However, no previous studies have directly shown that glial activation in the spinal cord and trigeminal system is dependent on peripheral nerve input, except that the increase in GFAP after nerve injury appears to depend on NMDAR activity (Garrison et al., 1994). Our results demonstrate that central glial activation depends on neuronal input and presumably concomitant transmitter release. We further showed that the effect of substance P on glial activation and cytokine induction was blocked by a nitric oxide synthase inhibitor. The nitric oxide synthase is distributed in the spinal trigeminal nucleus (Dohrn et al., 1994). Nitric oxide is engaged in neuronal signaling involving substance P in the brain (Vruwink et al., 2001) and spinal dorsal horn (Aimar et al., 1998) and has been shown to regulate expression of GFAP in astrocytes (Brahmachari et al., 2006). Holguin et al. (2004) show that nitric oxide mediates cytokine release and pain facilitation induced by gp120, an envelope protein of human immunodeficiency virus-1, which activate glia. Thus, our results are consistent with a role for nitric oxide in neuronal-glial signaling after masseter inflammation.
NMDARs are prominently involved in activity-dependent synaptic plasticity and persistent pain (Ren and Dubner, 1999; Woolf and Salter, 2000). Recent studies indicate that inflammation-induced NMDAR activation involves phosphorylation of the NR1 and NR2B subunits in the spinal dorsal horn (Guo et al., 2002, 2004; Brenner et al., 2004), which is correlated with behavioral hyperalgesia. Although studies have addressed the role of NMDAR in trigeminal plasticity (Bereiter and Bereiter, 1996; Chiang et al., 1998), there have been no reports on whether phosphorylation of the NMDAR in the trigeminal system is associated with the development of persistent orofacial pain. Our results indicate that NMDARs in the Vi/Vc transition zone were activated after masseter inflammation in a time-dependent manner and that the increased P-ser896-NR1 was localized to trigeminal neurons as opposed to glia. In contrast, GluR1 and GluR2/3 receptors were apparently downregulated in the spinal trigeminal nucleus after noxious stimulation (Florenzano and De Luca, 1999).
Most interestingly, the present study shows that a glial inhibitor blocked the upregulation of GFAP and IL-1β, as well as NMDAR activation after tissue injury. A nitric oxide synthase inhibitor similarly blocked the increased GFAP, IL-1β, and NMDAR phosphorylation after in vitro application of SP. Furthermore, fluorocitrate and IL-1ra attenuated behavioral hyperalgesia and NMDAR phosphorylation after inflammation. These findings indicate that glia and cytokines interact with neurons in response to peripheral injury. Previous studies suggest that glia are intimately involved in the control of neuronal activity (Araque et al., 1999; Frerking, 2004). Neurons sheathed by the reticulate network of astrocytes respond with large increases in their concentration of intracellular calcium (Nedergaard, 1994). CNS cytokines are released from glia to modulate neuronal function (Srinivasan et al., 2004). Glial TNF-α modulates synaptic strength in the brain (Beattie et al., 2002). The inflammatory cytokines are released by activated glia and play important roles in persistent pain states (DeLeo and Yezierski, 2001; Watkins et al., 2003).
Our findings that IL-1R colocalizes with NMDAR subunits in trigeminal neurons and that IL-1β directly induces NMDAR serine phosphorylation provide evidence supporting a close interaction of inflammatory cytokine signaling with neuronal NMDARs. The IL-1β signaling first involves binding to its receptor, IL-1R, a subfamily of the Toll/IL-1 receptor superfamily (Martin and Wesche, 2002). IL-1R contains at least two subtypes: the type I IL-1R (IL-1RI) is a transmembrane molecule and responsible for IL-1 signaling and the type II IL-1R lacks an intracellular domain and is incapable of signal transduction (Dayer, 2003). The IL-1R has been localized to the brain (Rothwell and Luheshi, 1994), including spinal neurons (Samad et al., 2001). Our results suggest a signal transduction cascade involving intracellular calcium release and PKC, by which IL-1β and NMDARs are linked and contribute to trigeminal sensory transmission in persistent pain (Fig. 10f). Interestingly, the events upstream to PKC activation appear to involve activation of both PLC and PLA2, consistent with previous reports that IL-1 signaling activates PLC (Chen et al., 2000) and PLA2 (Molina-Holgado et al., 2000) pathways (for review, see Martin and Wesche, 2002) and that NMDAR current is potentiated by arachidonic acid (Miller et al., 1992).
In the present study, MK-801, an NMDAR channel blocker, did not prevent IL-1β-induced NMDAR phosphorylation, although the doses used have been shown to attenuate hyperalgesia (Ren et al., 1992). This suggests that the NMDAR channel itself does not contribute to the initiation of the increased NR1 phosphorylation through a feedforward mechanism. Similarly, we have observed a lack of effect of MK-801 on the initiation of inflammation-induced NMDAR phosphorylation (Guo et al., 2004). These results suggest that (1) IL-1β-induced phosphorylation of the NMDAR does not depend on the opening status of the NMDAR channel and (2) MK-801 produces antihyperalgesic effects by acting on an event downstream to the initiation of NMDAR phosphorylation.
In summary, we have provided evidence to support a model of glial–cytokine–neuronal interactions underlying the mechanisms of inflammatory pain. We show that although neuronal input generated by injury induces neuronal hyperexcitability in the pain-processing circuitry, the input also activates glia and leads to the induction of cytokines and other chemical mediators in glial cells. Cytokines are then released and act back on neurons to facilitate central sensitization. The reciprocal interactions between glia and neurons through neurotransmitters and chemical mediators will amplify neuronal input and lead to increased pain. Advancing from previous studies, the model emphasizes activation of glia by tissue injury, concomitant cytokine release, and post-translational regulation of NMDAR through IL-1R signaling. These findings will help to identify novel targets and agents for clinical management of chronic pain.
Footnotes
This work was supported by National Institutes of Health–National Institute of Dental and Craniofacial Research Grants DE11964 and DE15374, National Institute on Drug Abuse Grant DA10275, and National Institute of Neurological Disorders and Stroke Grant NS41384. We thank E. Wade for technical assistance.
References
- Aimar P, Pasti L, Carmignoto G, Merighi A. Nitric oxide-producing islet cells modulate the release of sensory neuropeptides in the rat substantia gelatinosa. J Neurosci. 1998;18:10375–10388. doi: 10.1523/JNEUROSCI.18-24-10375.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alstergren P, Ernberg M, Kopp S, Lundeberg T, Theodorsson E. TMJ pain in relation to circulating neuropeptide Y, serotonin, and interleukin-1 beta in rheumatoid arthritis. J Orofac Pain. 1999;13:49–55. [PubMed] [Google Scholar]
- Anderson LC, Rao RD. Interleukin-6 and nerve growth factor levels in peripheral nerve and brainstem after trigeminal nerve injury in the rat. Arch Oral Biol. 2001;46:633–640. doi: 10.1016/s0003-9969(01)00024-3. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Asghar AU, Cilia La Corte PF, LeBeau FE, Al Dawoud M, Reilly SC, Buhl EH, Whittington MA, King AE. Oscillatory activity within rat substantia gelatinosa in vitro: a role for chemical and electrical neurotransmission. J Physiol (Lond) 2005;562:183–198. doi: 10.1113/jphysiol.2004.076398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bereiter DA, Bereiter DF. N-methyl-D-aspartate and non-N-methyl-D-aspartate receptor antagonism reduces Fos-like immunoreactivity in central trigeminal neurons after corneal stimulation in the rat. Neuroscience. 1996;73:249–258. doi: 10.1016/0306-4522(96)00038-3. [DOI] [PubMed] [Google Scholar]
- Bereiter DA, Hirata H, Hu JW. Trigeminal subnucleus caudalis: beyond homologies with the spinal dorsal horn. Pain. 2000;88:221–224. doi: 10.1016/S0304-3959(00)00434-6. [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26:4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner GJ, Ji RR, Shaffer S, Woolf CJ. Peripheral noxious stimulation induces phosphorylation of the NMDA receptor NR1 subunit at the PKC-dependent site, serine-896, in spinal cord dorsal horn neurons. Eur J Neurosci. 2004;20:375–384. doi: 10.1111/j.1460-9568.2004.03506.x. [DOI] [PubMed] [Google Scholar]
- Chen CC, Chen JJ, Chou CY. Protein kinase calpha but not p44/42 mitogen-activated protein kinase, p38, or c-Jun NH(2)-terminal kinase is required for intercellular adhesion molecule-1 expression mediated by interleukin-1beta: involvement of sequential activation of tyrosine kinase, nuclear factor-κB-inducing kinase, and IκB kinase 2. Mol Pharmacol. 2000;58:1479–1489. doi: 10.1124/mol.58.6.1479. [DOI] [PubMed] [Google Scholar]
- Chiang CY, Park SJ, Kwan CL, Hu JW, Sessle BJ. NMDA receptor mechanisms contribute to neuroplasticity induced in caudalis nociceptive neurons by tooth pulp stimulation. J Neurophysiol. 1998;80:2621–2631. doi: 10.1152/jn.1998.80.5.2621. [DOI] [PubMed] [Google Scholar]
- Chiu SY, Kriegler S. Neurotransmitter-mediated signaling between axons and glial cells. Glia. 1994;11:191–200. doi: 10.1002/glia.440110213. [DOI] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford) 2003;42:ii3–i10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Dohrn CS, Mullett MA, Price RH, Beitz AJ. Distribution of nitric oxide synthase-immunoreactive interneurons in the spinal trigeminal nucleus. J Comp Neurol. 1994;346:449–460. doi: 10.1002/cne.903460308. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ren K. Brainstem mechanisms of persistent pain following injury. J Orofac Pain. 2004;18:299–305. [PubMed] [Google Scholar]
- Dworkin SF, LeResche L. Res diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandib Disord. 1992;6:301–355. [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florenzano F, De Luca B. Nociceptive stimulation induces glutamate receptor down-regulation in the trigeminal nucleus. Neuroscience. 1999;90:201–207. doi: 10.1016/s0306-4522(98)00388-1. [DOI] [PubMed] [Google Scholar]
- Frerking M. When astrocytes signal, kainate receptors respond. Proc Natl Acad Sci USA. 2004;101:2649–2650. doi: 10.1073/pnas.0400474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol. 1994;129:237–243. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- Greene EC. The anatomy of the rat. Philadelphia: The American Philosophical Society; 1935. [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–6217. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wei F, Zou S, Robbins MT, Sugiyo S, Ikeda T, Tu JC, Worley PF, Dubner R, Ren K. Group I metabotropic glutamate receptor NMDA receptor coupling and signaling cascade mediate spinal dorsal horn NMDA receptor 2B tyrosine phosphorylation associated with inflammatory hyperalgesia. J Neurosci. 2004;24:9161–9173. doi: 10.1523/JNEUROSCI.3422-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathaway CB, Hu JW, Bereiter DA. Distribution of Fos-like immunoreactivity in the caudal brainstem of the rat following noxious chemical stimulation of the temporomandibular joint. J Comp Neurol. 1995;356:444–456. doi: 10.1002/cne.903560311. [DOI] [PubMed] [Google Scholar]
- Henry MA, Johnson LR, Nousek-Goebl N, Westrum LE. Light microscopic localization of calcitonin gene-related peptide in the normal feline trigeminal system and following retrogasserian rhizotomy. J Comp Neurol. 1996;365:526–540. doi: 10.1002/(SICI)1096-9861(19960219)365:4<526::AID-CNE2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Holguin A, O'Connor KA, Biedenkapp J, Campisi J, Wieseler-Frank J, Milligan ED, Hansen MK, Spataro L, Maksimova E, Bravmann C, Martin D, Fleshner M, Maier SF, Watkins LR. HIV-1 gp120 stimulates proinflammatory cytokine-mediated pain facilitation via activation of nitric oxide synthase-I (nNOS) Pain. 2004;110:517–530. doi: 10.1016/j.pain.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Hosoi R, Okada M, Hatazawa J, Gee A, Inoue O. Effect of astrocytic energy metabolism depressant on 14C-acetate uptake in intact rat brain. J Cereb Blood Flow Metab. 2004;24:188–190. doi: 10.1097/01.WCB.0000098606.42140.02. [DOI] [PubMed] [Google Scholar]
- Kacena MA, Merrel GA, Konda SR, Wilson KM, Xi Y, Horowitz MC. Inflammation and bony changes at the temporomandibular joint. Cells Tissues Organs. 2001;169:257–264. doi: 10.1159/000047889. [DOI] [PubMed] [Google Scholar]
- Lee HL, Lee KM, Son SJ, Hwang SH, Cho HJ. Temporal expression of cytokines and their receptors mRNAs in a neuropathic pain model. NeuroReport. 2004;15:2807–2811. [PubMed] [Google Scholar]
- Li WE, Nagy JI. Activation of fibres in rat sciatic nerve alters phosphorylation state of connexin-43 at astrocytic gap junctions in spinal cord: evidence for junction regulation by neuronal-glial interactions. Neuroscience. 2000;97:113–123. doi: 10.1016/s0306-4522(00)00032-4. [DOI] [PubMed] [Google Scholar]
- Martin FC, Charles AC, Sanderson MJ, Merrill JE. Substance P stimulates IL-1 production by astrocytes via intracellular calcium. Brain Res. 1992;599:13–18. doi: 10.1016/0006-8993(92)90846-2. [DOI] [PubMed] [Google Scholar]
- Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592:265–280. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain. 1993;52:127–136. doi: 10.1016/0304-3959(93)90124-8. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131:152–159. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier; 2005. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ren K. An improved method for assessing mechanical allodynia in the rat. Physiol Behav. 1999;67:711–716. doi: 10.1016/s0031-9384(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Central nervous system plasticity and persistent pain. J Orofac Pain. 1999;13:155–163. 164–171. discussion. [PubMed] [Google Scholar]
- Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50:331–344. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- Ro JY, Harriott A, Crouse U, Capra NF. Innocuous jaw movements increase c-fos expression in trigeminal sensory nuclei produced by masseter muscle inflammation. Pain. 2003;104:539–548. doi: 10.1016/S0304-3959(03)00093-9. [DOI] [PubMed] [Google Scholar]
- Rohlmann A, Laskawi R, Hofer A, Dobo E, Dermietzel R, Wolff JR. Facial nerve lesions lead to increased immunostaining of the astrocytic gap junction protein (connexin 43) in the corresponding facial nucleus of rats. Neurosci Lett. 1993;154:206–208. doi: 10.1016/0304-3940(93)90208-3. [DOI] [PubMed] [Google Scholar]
- Rohlmann A, Laskawi R, Hofer A, Dermietzel R, Wolff JR. Astrocytes as rapid sensors of peripheral axotomy in the facial nucleus of rats. NeuroReport. 1994;5:409–412. doi: 10.1097/00001756-199401120-00009. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi G. Pharmacology of interleukin-1 actions in the brain. Adv Pharmacol. 1994;25:1–20. doi: 10.1016/s1054-3589(08)60428-7. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Spataro LE, Sloane EM, Milligan ED, Wieseler-Frank J, Schoeniger D, Jekich BM, Barrientos RM, Maier SF, Watkins LR. Spinal gap junctions: potential involvement in pain facilitation. J Pain. 2004;5:392–405. doi: 10.1016/j.jpain.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type-specific interleukin-1β signaling in the CNS. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassman AM, Vos BP. Somatotopic and laminar organization of fos-like immunoreactivity in the medullary and upper cervical dorsal horn induced by noxious facial stimulation in the rat. J Comp Neurol. 1993;331:495–516. doi: 10.1002/cne.903310406. [DOI] [PubMed] [Google Scholar]
- Sugiyo S, Takemura M, Dubner R, Ren K. Trigeminal transition zone/rostral ventromedial medulla connections and facilitation of orofacial hyperalgesia after masseter inflammation in rats. J Comp Neurol. 2005;493:510–523. doi: 10.1002/cne.20797. [DOI] [PubMed] [Google Scholar]
- Takuma K, Matsuda T, Hashimoto H, Kitanaka J, Asano S, Kishida Y, Baba A. Role of Na(+)-Ca2+ exchanger in agonist-induced Ca2+ signaling in cultured rat astrocytes. J Neurochem. 1996;67:1840–1845. doi: 10.1046/j.1471-4159.1996.67051840.x. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vruwink M, Schmidt HH, Weinberg RJ, Burette A. Substance P and nitric oxide signaling in cerebral cortex: anatomical evidence for reciprocal signaling between two classes of interneurons. J Comp Neurol. 2001;441:288–301. doi: 10.1002/cne.1413. [DOI] [PubMed] [Google Scholar]
- Wang H, Wei F, Dubner R, Ren K. Selective distribution and function of primary afferent nociceptive inputs from deep muscle tissue to the brainstem trigeminal transition zone. J Comp Neurol. 2006;498:390–402. doi: 10.1002/cne.21062. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Guo W, Zou S, Sugiyo S, Dubner R, Ren K. Antibody array analysis of peripheral and blood cytokine levels in rats after masseter inflammation. Neurosci Lett. 2005;382:128–133. doi: 10.1016/j.neulet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Immune regulation of central nervous system functions: from sickness responses to pathological pain. J Intern Med. 2005;257:139–155. doi: 10.1111/j.1365-2796.2004.01443.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1–21. [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yan HQ, Banos MA, Herregodts P, Hooghe R, Hooghe-Peters EL. Expression of interleukin (IL)-1 beta, IL-6 and their respective receptors in the normal rat brain and after injury. Eur J Immunol. 1992;22:2963–2971. doi: 10.1002/eji.1830221131. [DOI] [PubMed] [Google Scholar]
- Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034:172–179. doi: 10.1016/j.brainres.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, Lao L. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118:125–136. doi: 10.1016/j.pain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]