

Abstract
p53 is a powerful tumor suppressor and is an attractive cancer therapeutic target because it can be functionally activated to eradicate tumors. The gene encoding p53 protein is mutated or deleted in half of human cancers, which inactivates its tumor suppressor activity. In the remaining cancers with wild-type p53 status, its function is effectively inhibited through direct interaction with the human murine double minute 2 (MDM2) oncoprotein. Blocking the MDM2-p53 interaction to reactivate the p53 function is a promising cancer therapeutic strategy. This review will highlight the advances in the design and development of small-molecule inhibitors of the MDM2-p53 interaction as a cancer therapeutic approach.
Background
Tumor suppressor p53 is a powerful transcription factor and plays a central role in the regulation of cell cycle, apoptosis, DNA repair, senescence, and angiogenesis (1–3). The p53 protein was identified in 1979 (4–6), and its gene, called TP53, was cloned in 1983 (7). In 1989, p53 was shown to block the transformation of rat embryo fibroblasts (8) and in 1990, the p53 gene was implicated in most cases of Li-Fraumeni syndrome, a rare inherited condition which is associated with frequent occurrence of several types of cancer in affected families. Subsequently, the p53 gene was found to be altered in a wide variety of cancers. Due to the “exhilarating possibilities for prevention and cure of cancer,” p53 was crowned as the “Molecule of the Year” in 1993 (9).
Because of its prominent role as a tumor suppressor, p53 is functionally impaired by mutation or deletion in nearly 50% of human cancers (10). In the remaining human cancers, p53 retains wild-type status but its function is inhibited by its primary cellular inhibitor, the murine double minute 2 (MDM2; HDM2 in humans). MDM2 was initially discovered as the product of an oncogene found overexpressed by amplification in a spontaneously transformed mouse cell line (11). MDM2 is an essential regulator of p53 in normal cells, but its deregulated expression provides growth advantage to cells. Overexpression of MDM2 due to the amplification of the MDM2 gene was first found in sarcomas retaining wild-type p53 (12), and this amplification was later observed in several other human cancers (13).
Regulation of p53 and MDM2
Soon after its discovery, MDM2 was shown as a negative regulator of p53-mediated transactivation (14). MDM2 and p53 regulate each other through an autoregulatory feedback loop (Fig. 1; ref. 15). Upon activation, p53 transcribes the MDM2 gene and, in turn, the MDM2 protein inhibits p53 activity: MDM2 (a) binds to the p53 transactivation domain and inhibits its transcriptional activity (14, 16); (b) exports p53 out of the nucleus, promoting its degradation and rendering it inaccessible to the target genes (17); and (c) promotes proteasome-mediated degradation of p53 by functioning as an E3 ubiquitin ligase (18). In this manner, MDM2 functions as an effective inhibitor of p53 activity. The convincing physiologic relevance of MDM2 as a critical inhibitor of p53 was provided by genetic studies which showed that the embryonic lethality of MDM2-null mice can only be rescued by the simultaneous deletion of the p53 gene (19, 20).
Fig. 1.

Regulation of p53 and MDM2 and the outcomes of p53 activation. A, MDM2 inhibits p53 through an autoregulatory loop. MDM2 directly binds to the transactivation domain of p53 and inhibits its transcriptional activity, causes the ubiquitination and proteasomal degradation of p53, and exports p53 out of the nucleus which promotes p53 degradation and inhibits its activity. MDMX, a homologue of MDM2, also directly binds to the transactivation domain of p53 and inhibits p53 activity, but does not induce p53 degradation. ARF binds to MDM2 and sequesters MDM2 into the nucleolus, leading to the stabilization of p53. B, activation of p53 can also lead to induction of apoptosis via intrinsic (mitochondrial) and extrinsic (death receptor) apoptosis pathways. Apoptosis can be transcriptional-dependent or -independent because p53 itself can participate in mitochondrial mediated apoptosis through interaction with proapoptotic and antiapoptotic members of the Bcl-2 family. C, activation of p53 can halt cell cycle progression in the G1-S and G2-M boundaries of cell cycle through the up-regulation of the p21, Gadd45, and 14-3-3-σ proteins. Transition into the S-phase requires cyclin-dependent kinases (CDK), such as CDK2, which phosphorylates and inactivates Rb, rendering E2F free and transcriptionally active, leading to cell cycle progression. However, p53 activation induces the CDK inhibitor p21, which leads to cell cycle arrest. Furthermore, Cdc2/cyclinE activity is essential for entry into mitosis, and this activity can be inhibited by p21, Gadd45, and 14-3-3-σ, resulting in G2-M phase arrest. D, senescence is a potent tumor suppressor mechanism of p53. Telomere erosion, DNA damage, and oxidative stress or oncogenic stress can signal p53 activation, triggering senescence response via the p21-Rb-E2F signaling pathway. Oncogenic Ras can activate MAPkinase pathway that phosphorylates and activates p53 and also induces the expression of ARF, which in turn binds to and inhibits MDM2, leading to the up-regulation of p53 and the induction of senescence. E, p53 can suppress angiogenesis through the down-regulation of proangiogenic proteins and up-regulation of antiangiogenic proteins. In addition, p53 can bind to HIF-1α, a promoter of angiogenesis during hypoxia, and target it for degradation by MDM2. In a p53-independent manner, HIF-1α interacts with the p53-binding domain of MDM2, and transcriptionally up-regulates the vascular endothelial growth factor (VEGF), promoting angiogenesis. F, p53 plays a critical role in DNA damage repair. DNA damage and replication errors can activate ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad-related (ATR) kinases, which trigger several cellular responses, including DNA repair. ATM and ATR can phosphorylate DNA repair protein 53BP1 as well as induce the accumulation of p53 through phosphorylation directly or via CHK1and CHK2 kinases. p53 participates in DNA repair in a transactivation-dependent manner through the up-regulation of proteins such as p53R2 (p53-inducible small subunit of ribonucleotide reductase), p48 (gene product of DDB2 gene), and XPC (xeroderma pigmentosum group C protein), and in an independent manner through interaction with other DNA repair proteins, such as 53BP1.
Structural basis of the MDM2-p53 interaction
The MDM2-p53 interaction was mapped to the first ~120 NH2-terminal amino acids in MDM2 and the NH2 terminus of the transactivation domain of p53 (16, 21). The high-resolution crystal structures of the NH2-terminal domains of human and Xenopus laevis MDM2 complexed with short p53 peptides (residues 15–29; ref. 22) have provided atomic details of the interaction. These structures show that the MDM2-p53 interaction is mediated by a well-defined hydrophobic surface pocket in MDM2 and four key hydrophobic residues in p53, namely Phe19, Leu22, Trp23, and Leu26 (Fig. 2A). This well-defined interaction has provided the basis for the design of nonpeptide, drug-like small-molecule inhibitors of the MDM2-p53 interaction to reactivate p53.
Fig. 2.

Binding mode of (A) p53 peptide (PDB:1YCR) and (B) a Nutlin analogue (PDB:1RV1), and (C) predicted binding model of MI-219 to MDM2. Side chains of p53 residues involved in the MDM2-p53 interaction are shown in stick representation, whereas Nutlin-2, an analogue of Nutlin-3, and MI-219 are shown in ball-and-stick representation. Nutlin-2 is shown with carbons in cyan, nitrogen in blue, oxygen in red and bromine in brown. MI-219 is shown with carbons in cyan, nitrogen in blue, oxygen in red, fluorine in light blue, and chlorine in green. The surface representation of MDM2 in each case is shown with carbons in gray, nitrogen in blue, oxygen in red, and sulfur in yellow. Hydrogen bonds are depicted with yellow lines and hydrogen atoms are excluded for clarity. The figures were generated by Dr. Denzil Bernard using the program Pymol.
Reactivation of p53 as a therapeutic strategy
Recent genetic studies (23) have shown that the loss of p53 induces tumor formation in mice, whereas its restoration leads universally to a rapid regression of established in situ tumors, further showing the cancer-therapeutic potential of p53 restoration. Over the last two decades, a number of distinct therapeutic strategies have been pursued to restore p53 function for cancer treatment (24, 25). Because the interaction between MDM2 and p53 is a primary mechanism for inhibition of the p53 function in cancers retaining wild-type p53, targeting the MDM2-p53 interaction by small molecules to reactivate p53 has emerged as a promising new cancer therapeutic strategy, and is the focus of this review.
Targeting the MDM2-p53 Interaction
Design of nonpeptidic small-molecule MDM2 inhibitors of the MDM2-p53 interaction
The progress in the design of non-peptidic, small-molecule inhibitors of the MDM2-p53 interaction (mentioned herein as MDM2 inhibitors; sometimes also called as HDM2 inhibitors) proceeded very slowly for almost a decade after the publication of the crystal structures. The very first class of bona fide, potent, nonpeptidic, small-molecule MDM2 inhibitors, known as Nutlins, was reported in 2004 (Table 1; Fig. 2B; ref. 26). The Nutlins contain a cis-imidazoline core structure (Table 1) and one analogue, Nutlin-3, has potent in vivo antitumor activity in xenograft models of human cancer-retaining wild-type p53 (26–28). The discovery of the Nutlins provided the important proof-of-concept and fueled enthusiasm for the design and development of small-molecule MDM2 inhibitors. In the last 4 years, several new classes of small-molecule MDM2 inhibitors have been discovered using different approaches (24, 29, 30). Using a computational structure-based de novo design strategy, our laboratory has reported the design of spiro-oxindoles as a new class of potent small-molecule MDM2 inhibitors (31), as exemplified by MI-63 (32) and MI-219 (Table 1; Fig. 2C; ref. 33).
Table 1.
Nonpeptidic small-molecule inhibitors of the MDM2-p53 interaction
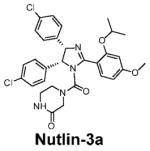
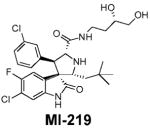
Molecular mechanism of p53 activation by MDM2 inhibitors
The availability of potent and specific MDM2 inhibitors, such as Nutlin-3 and MI-219, has provided the opportunity to examine in detail the molecular mechanism of p53 activation. In competition with a p53-based peptide, Nutlin-3 and MI-219 bind with high affinity to MDM2 (Ki = 36 and 5 nmol/L, respectively; ref. 33). They block the intracellular MDM2-p53 interaction and induce the accumulation of p53 and the activation of the p53 pathway in tumor and normal cells (26, 33–37). Conventional genotoxic anticancer agents and radiation also induce the accumulation and activation of p53, but they do so by posttranslational modifications of p53, such as phosphorylation. In contrast, Nutlin-3 induces neither DNA damage nor p53 phosphorylation in cells (38). Hence, small-molecule MDM2 inhibitors represent a new class of nongenotoxic agents that can reactivate the p53 function.
Antitumor activity of small-molecule MDM2 inhibitors
MDM2 inhibitors, through the activation of p53, could elicit a wide variety of cellular responses in normal and tumor cells (Fig. 1B–F). Studies using Nutlin-3 and MI-219 show that MDM2 inhibitors yield both common and different cellular responses in normal and tumor cells (26, 33). In normal cells, the activation of p53 by MDM2 inhibitors induces cell cycle arrest but not cell death (26, 33). In tumor cells, the activation of p53 by the inhibitors induces not only cell cycle arrest but also cell death (26, 27, 33). These cellular effects depend upon p53 activation because the knock-down or knockout of p53 abrogates the cell cycle arrest and the cell death induced by Nutlin-3 and MI-219 (33, 35). Furthermore, cellular activity of MDM2 inhibitors is dependent on their binding to MDM2 because inactive analogues have no effect on both cancer and normal cells.
In mouse models of human cancer with wild-type p53, oral administration of Nutlin-3 or MI-219 activates the p53 pathway in xenograft tumor tissues, as indicated by the accumulation of p53 and the up-regulation of p21, a p53-targeted gene product (27, 33). Nutlin-3 (26–28) and MI-219 (33) show strong antitumor activity in several xenograft models of human cancer with wild-type p53, including osteosarcoma and prostate cancer, but lack activity against tumors deficient in wild-type p53. Importantly, the antitumor activity of both small molecules is achieved without causing visible signs of toxicity in the animals, as assessed by necropsy studies and body weight loss (26–28, 33).
The induction of senescence, which is a permanent form of cell growth arrest, and apoptosis are two major tumor suppressor mechanisms of p53 (Fig. 1B and D; refs. 3, 39). Genetic mouse models have shown that the restoration of p53 function leads to tumor regression through different mechanisms depending upon the tumor type (23). Regression of liver tumors and sarcomas is through induction of senescence and activation of the innate immune system, and of lymphomas by apoptosis. MDM2 inhibitors also induce senescence in vitro (40), and apoptosis both in vitro and in vivo (26, 27, 33), but the primary mechanisms of the in vivo antitumor response to MDM2 inhibitors have not been elucidated.
Lack of toxicity to normal tissues
The effect of p53 activation by an MDM2 inhibitor in normal tissues is of immense interest from a therapeutic perspective. Radio-sensitive tissues, such as small-intestine crypts and thymus are extremely susceptible to p53-induced apoptosis (41, 42). Restoration of p53 by a genetic approach in the absence of MDM2 results in severe pathologic damage to radio-sensitive mouse tissues and the death of all animals within five days (43). In contrast, both Nutlin-3 (26) and MI-219 (33) show little toxicity to animals at therapeutically efficacious dose-schedules. Whereas both γ-radiation and irinotecan chemotherapy induce profound apoptosis in small-intestine crypts and thymus, MI-219, in either single or repeated doses, does not cause apoptosis or damage in either radio-sensitive or radio-resistant normal mouse tissues, indicating that MDM2 inhibitors display a therapeutic window (33). The precise mechanism for the lack of toxicity of MDM2 inhibitors to normal tissues remains to be elucidated.
MDMX is a modulator of the activity of MDM2 inhibitors
MDMX has been shown to be a key modulator of the activity of MDM2 inhibitors. MDMX is a homologue of MDM2. MDMX also binds directly to p53 and inhibits its transcriptional activity, but does not induce p53 degradation (Fig. 1A; ref. 44).
Nutlin-3 and MI-219 bind to MDM2 with a much higher affinity than to MDMX (33–37). In the presence of MDMX, MDM2 inhibitors may not be able to fully activate p53, thus attenuating the activity of MDM2 inhibitors. Indeed, studies using ectopic expression of MDMX and/or its down-regulation by RNAi show that MDMX attenuates the p53 activation by MDM2 inhibitors and inhibits the cellular activity of MDM2 inhibitors (33–36). Intriguingly, in some cancer cell lines, MDM2 inhibitors can induce MDMX degradation, presumably mediated through p53-dependent up-regulation of MDM2 (33, 35, 36), which is known to ubiquitinate and degrade MDMX during DNA damage (45). Such cancer cell lines are more susceptible to Nutlin-3 than those in which Nutlin-3 fails to induce MDMX degradation. Thus, the antitumor activity of the MDM2 inhibitors could be compromised in certain human tumors which overexpress MDMX. In this regard, small molecule inhibitors targeting both MDM2 and MDMX could be more efficacious than those that are specific for either MDM2 or MDMX.
p53-independent effects of MDM2 inhibitors
Studies using in vitro and in vivo models of human cancer have shown that the antitumor activity of MDM2 inhibitor is dependent on wild-type p53 status. However, in addition to p53, MDM2 interacts with a number of other proteins, such as p53 homologues p73 and p63, and also with E2F-1, hypoxia-inducible factor-1α (HIF-1α), and Numb (46–49). A small-molecule MDM2 inhibitor, which interferes with the binding of MDM2 with these proteins, especially those which have homology to p53 and/or interact with the p53-binding site of MDM2, may have a p53-independent effect. Using in vitro cell line models, it has been shown that Nutlin-3 disrupts the interaction of MDM2 with p73 (46), E2F-1 (47), and HIF-1α (48), thereby producing p53-independent effects. However, higher concentrations of Nutlin-3 (for a p73-dependent effect) or a DNA damage signal (for an E2F-1-dependent effect) was required for p53-independent effects.
Inhibition of angiogenesis by MDM2 inhibitors
There is accumulating evidence that activation of p53 may effectively inhibit angiogenesis (Fig. 1E; ref. 1). Therefore, in addition to the direct effect of targeting tumor cells, MDM2 inhibitors may inhibit angiogenesis (50, 51). Activation of p53 can up-regulate several antiangiogenic factors, including thrombospondin-1 (TSP1), and brain-specific angiogenesis inhibitor 1 (BAI1), and down-regulate several proangiogenic factors, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), basic fibroblast growth factor binding protein, and cyclooxygenase-2 (COX-2; ref. 1). Thus, through the activation of p53, MDM2 inhibitors can inhibit angiogenesis; in fact, a recent study (50) showed the in vitro and in vivo antiangiogenesis activity of Nutlin-3. Because this activity is through targeting endothelial cells with wild-type p53, MDM2 inhibitors could also be useful in the treatment of tumors lacking functional p53 through the inhibition of angiogenesis.
Under hypoxia, HIF-1α transcriptionally up-regulates proangiogenic VEGF and promotes neovascularization (1). Hypoxic stress also stabilizes p53, which in turn up-regulates E3 ligase MDM2, promoting the ubiquitination and degradation of HIF-1α (1, 52), causing diminished VEGF production, and suppressing angiogenesis (Fig. 1E). Interestingly, HIF-1α also binds to the p53-binding site in MDM2, and Nutlin-3 was shown to block the MDM2-HIF-1α interaction, leading to the decreased transcriptional activity of HIF-1α and the down-regulation of its target gene VEGF under conditions of normoxia or hypoxia (48). Thus, MDM2 inhibitors can inhibit angiogenesis in both p53-dependent and -independent manner.
Clinical-Translational Potential of MDM2 Inhibitors
Three small-molecule MDM2 inhibitors, each from a different chemical class, have reached the preclinical stage of development (Table 1).
Predictors of response to MDM2 inhibitors
Studies using Nutlin-3, MI-219, and MI-63 in cancer cell lines in vitro (26, 33, 35, 53), xenograft models in vivo (27, 33), and B-cell chronic lymphocytic leukemia (B-CLL; refs54, 55) and acute myeloid leukemia (56) patient samples ex vivo have shown that the wild-type status of p53 is the major determinant of the antitumor activity of MDM2 inhibitors. Furthermore, because both Nutlin-3 and MI-219 target the MDM2-p53 interaction with a high degree of selectivity over the MDMX-p53 interaction, they are highly effective against cancer cell lines in which MDM2 is overexpressed and is the dominant inhibitor of p53 (27, 33, 57). Tumors use several mechanisms to overexpress the MDM2 protein, such as increased transcription, increased translation, amplification, and single-nucleotide polymorphism (SNP) of the MDM2 gene (58, 59). MDM2 gene amplification and homozygous or heterozygous SNP from T to G at nucleotide 309 of the MDM2 promoter (SNP309) have been associated with the early onset of cancer, drug resistance, and poor overall survival in a variety of human tumors, including sarcomas, lung, breast, liver, colon, neuroblastoma, and B-CLL (58–61). It is predicted that patients carrying such tumors will benefit from therapy based on MDM2 inhibitors. Thus, wild-type p53 status, MDM2 overexpression, and low MDMX expression may serve as diagnostic markers in the treatment with MDM2 inhibitors.
In addition, it can also be predicted that defects in upstream signaling to p53 will not adversely affect the activity of MDM2 inhibitors. For example, a low level of the ataxia telangiectasia mutated protein in CLL renders fludarabine ineffective, but does not have a significant influence on the activity of MDM2 inhibitors (54, 55).
Use of MDM2 inhibitors in combination setting
In the clinic, anticancer drugs are more often than not used in combination. Two desired outcomes of the combination regimens are enhanced antitumor activity and the protection of normal healthy tissues. Many traditional genotoxic anticancer drugs that induce p53 also cause collateral damage to normal cells. A rationale strategy to minimize the toxic effects of these drugs is to combine them with nongenotoxic agents of p53 activation, such as MDM2 inhibitors, which may also yield better antitumor efficacy. In fact, ex vivo experiments in patient samples retaining wild-type p53 show that Nutlin-3 synergizes with doxorubicin, chlorambucil, and fludarabine in B-CLL (54, 62, 63); with doxorubicin and 1-β-D-arabinofuranosylcytosine (Ara-C) in acute myeloid leukemia (56, 64); and with doxorubicin in Hodgkin and Reed-Sternberg cells (65) in inducing apoptosis. Significantly, in these experiments Nutlin-3 as a single agent or in combination was nontoxic toward normal hematopoietic cells. Additionally, targeting extrinsic and intrinsic pathways of apoptosis by tumor necrosis factor-α-related apoptosis-inducing ligand (TRAIL) and an MDM2 inhibitor, respectively, is another rational strategy to enhance apoptosis induction. TRAIL is known to bind to TRAIL-R2, a cell surface receptor, and transduces an apoptosis signal. It was shown that in acute myeloid leukemia cells expressing wild-type p53, Nutlin-3 induces p53-dependent production of TRAIL-R2, leading to synergistic cell death when used in combination with TRAIL (64).
Studies using Nutlin-3 in cell lines show that the cell cycle arrest function of MDM2 inhibitors can be exploited to protect normal cells from the toxic effects of chemotherapy. MDM2 inhibitors halt cell cycle progression at the G1-S and G2-M phases, and can thus abolish the activity of S-phase–and M-phase –specific drugs. Indeed, pretreatment with Nutlin-3 protects the normal proliferating fibroblasts from taxanes, which kill cells in the M-phase (53), and gemcitabine and Ara-C, which kill cells in the S-phase (66). Importantly, Nutlin-3 does not abolish the activity of these cell phase–specific drugs in cells lacking wild-type p53, indicating the utility of MDM2 inhibitors as chemoprotective agents in tumors lacking wild-type p53. These in vitro studies await further confirmation in vivo.
Biomarkers for p53 activation by MDM2 inhibitors
Because the antitumor activity of MDM2 inhibitors is largely p53-dependent, it is important to develop a robust and reliable biomarker of p53 activation for their optimal clinical use. Such a biomarker should be a specific indicator of p53 activation by MDM2 inhibitors, be minimally invasive, and show significant selectivity for tumors over normal tissues. Although p53 has many target genes, their gene products are localized in cells and their analysis requires invasive tissue biopsy. One attractive candidate biomarker of p53 activation is macrophage inhibitory cytokine-1 (MIC-1), a p53-target gene and a member of the transforming growth factor-β superfamily. The activation of p53 by treatment with doxorubicin induces the secretion of human MIC-1 in mouse serum and the up-regulation of p21 in an HCT-116 xenograft model (67), and both these markers of p53 correlate with each other. Activation of p53 by Nutlin-3 in CLL patient samples transcriptionally up-regulates MIC-1 (reported as GDF15) which correlates with several other p53 target genes (62). In endothelial cells, activation of p53 by Nutlin-3 induces MIC-1 secretion which can be quantified by ELISA (50). Thus MIC-1 represents a promising candidate biomarker of p53 activation.
Resistance to MDM2 inhibitors
Because p53 activation is critical for the antitumor activity of MDM2 inhibitors, persistent exposure to MDM2 inhibitors may select for tumors that are defective in p53 function. Defects in p53 can arise due to deletion or mutations of the p53 gene or other impairments in the p53 pathway. For example, in cells with wild-type p53, tumor suppressor p14ARF (p19ARF in mice) binds to MDM2, inactivates the E3 ubiquitin ligase activity of MDM2 protein, and stabilizes p53 (Fig. 1A and D). A genetic study using a mouse model, which recapitulates human Burkitt’s lymphoma/leukemia, has shown that although the restoration of p53 is therapeutically effective, it selects for secondary resistant tumors, due to either loss of p53 or p19ARF (23, 68). Therefore, the use of the MDM2 inhibitor as a single agent in the clinic may also result in similar tumor resistance. For this reason, combination strategies may be highly desirable.
Concluding Remarks
MDM2 is the primary cellular inhibitor of p53 in cancers retaining wild-type p53 and targeting the MDM2-p53 protein-protein interaction is an attractive cancer therapeutic strategy. Highly potent and specific small-molecule inhibitors with desirable pharmaceutical properties such as Nutlin-3 and MI-219 are now available. Studies using these inhibitors in preclinical models have already provided strong evidence that targeting the MDM2-p53 interaction using small-molecule inhibitors is a promising cancer therapeutic approach. Clinical testing of these new agents should provide the ultimate proof for this therapeutic strategy. A number of these inhibitors are now in advanced preclinical development and expected to progress into human clinical trials in the near future.
Acknowledgments
Grant support: National Cancer Institute/NIH, the Prostate Cancer Foundation, the Leukemia and Lymphoma Society, and AscentaTherapeutics.
We thank Dr. Denzil Bernard for the preparation of Fig. 2.
Footnotes
Disclosure of Potential Conflicts of Interest
S. Wang is a consultant for Ascenta, has received a commercial research grant from Ascenta, and has an ownership interest in Ascenta.
References
- 1.Teodoro JG, Evans SK, Green MR. Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. JMol Med. 2007;85:1175–86. doi: 10.1007/s00109-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 2.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 4.Lane DP, Crawford LV. Tantigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 5.DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A. 1979;76:2420–4. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 7.Oren M, Levine AJ. Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen. Proc Natl Acad Sci U S A. 1983;80:56–9. doi: 10.1073/pnas.80.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–93. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 9.Koshland DE., Jr Molecule of the year. Science. 1993;262:1953. doi: 10.1126/science.8266084. [DOI] [PubMed] [Google Scholar]
- 10.Feki A, Irminger-Finger I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit Rev Oncol Hematol. 2004;52:103–16. doi: 10.1016/j.critrevonc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565–9. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 13.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–9. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–14. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–93. doi: 10.1128/mcb.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 19.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 20.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 21.Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;-fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene. 1994;9:2523–9. [PubMed] [Google Scholar]
- 22.Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 23.Kastan MB. Wild-type p53: tumors can’t stand it. Cell. 2007;128:837–40. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Wiman KG. Strategies for therapeutic targeting of the p53 pathway in cancer. Cell Death Differ. 2006;13:921–6. doi: 10.1038/sj.cdd.4401921. [DOI] [PubMed] [Google Scholar]
- 26.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 27.Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarek G, Kurki S, Enback J, et al. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. JClin Invest. 2007;117:1019–28. doi: 10.1172/JCI30945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koblish HK, Zhao S, Franks CF, et al. Benzodiazepinedione inhibitors of the Hdm2:p53 complex suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo. Mol Cancer Ther. 2006;5:160–9. doi: 10.1158/1535-7163.MCT-05-0199. [DOI] [PubMed] [Google Scholar]
- 30.Grasberger BL, Lu T, Schubert C, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48:909–12. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 31.Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–1. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 32.Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2 – 53 interaction. J Med Chem. 2006;49:3432–5. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 33.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105:3933–8. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–5. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 35.Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 2006;66:3169–76. doi: 10.1158/0008-5472.CAN-05-3832. [DOI] [PubMed] [Google Scholar]
- 36.Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. Hdmx modulates the outcome of p53 activation in human tumor cells. J Biol Chem. 2006;281:33036–44. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- 37.Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 38.Thompson T, Tovar C, Yang H, et al. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–22. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- 39.Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem. 2001;268:2784–91. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 40.Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-Dependent Senescence by the MDM2 Antagonist Nutlin-3a in Mouse Cells of Fibroblast Origin. Cancer Res. 2007;67:7350–7. doi: 10.1158/0008-5472.CAN-07-0200. [DOI] [PubMed] [Google Scholar]
- 41.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–9. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 42.Potten CS, Wilson JW, Booth C. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells. 1997;15:82–93. doi: 10.1002/stem.150082. [DOI] [PubMed] [Google Scholar]
- 43.Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501–14. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Bottger V, Bottger A, Garcia-Echeverria C, et al. Comparative study of the p53 – mdm2 and p53-MDMX interfaces. Oncogene. 1999;18:189–99. doi: 10.1038/sj.onc.1202281. [DOI] [PubMed] [Google Scholar]
- 45.Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278:45946–53. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- 46.Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73–2 binding and enhances p73 function. Oncogene. 2008;27:997–1003. doi: 10.1038/sj.onc.1210707. [DOI] [PubMed] [Google Scholar]
- 47.Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. 2007;26:3473–81. doi: 10.1038/sj.onc.1210136. [DOI] [PubMed] [Google Scholar]
- 48.LaRusch GA, Jackson MW, Dunbar JD, Warren RS, Donner DB, Mayo LD. Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1α and Hdm2. Cancer Res. 2007;67:450–4. doi: 10.1158/0008-5472.CAN-06-2710. [DOI] [PubMed] [Google Scholar]
- 49.Colaluca IN, Tosoni D, Nuciforo P, et al. NUMB controls p53 tumour suppressor activity. Nature. 2008;451:76–80. doi: 10.1038/nature06412. [DOI] [PubMed] [Google Scholar]
- 50.Secchiero P, Corallini F, Gonelli A, et al. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res. 2007;100:61–9. doi: 10.1161/01.RES.0000253975.76198.ff. [DOI] [PubMed] [Google Scholar]
- 51.Binder BR. A novel application for murine double minute 2 antagonists: the p53 tumor suppressor network also controls angiogenesis. Circ Res. 2007;100:13–4. doi: 10.1161/01.RES.0000255897.84337.38. [DOI] [PubMed] [Google Scholar]
- 52.Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 53.Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005;65:1918–24. doi: 10.1158/0008-5472.CAN-04-3576. [DOI] [PubMed] [Google Scholar]
- 54.Kojima K, Konopleva M, McQueen T, O’Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108:993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saddler C, Ouillette P, Kujawski L, et al. Comprehensive biomarker and genomic analysis identifies P53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood. 2007;111:1584–93. doi: 10.1182/blood-2007-09-112698. [DOI] [PubMed] [Google Scholar]
- 56.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106:3150–9. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia M, Knezevic D, Tovar C, Huang B, Heimbrook DC, Vassilev LT. Elevated MDM2 boosts the apoptotic activity of p53 – 2 binding inhibitors by facilitating MDMX degradation. Cell Cycle. 2008;7:1604–12. doi: 10.4161/cc.7.11.5929. [DOI] [PubMed] [Google Scholar]
- 58.Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 59.Bond GL, Levine AJ. A single nucleotide polymorphism in the p53 pathway interacts with gender, environmental stresses and tumor genetics to influence cancer in humans. Oncogene. 2007;26:1317–23. doi: 10.1038/sj.onc.1210199. [DOI] [PubMed] [Google Scholar]
- 60.Cattelani S, Defferrari R, Marsilio S, et al. Impact of a Single Nucleotide Polymorphism in the MDM2 Gene on Neuroblastoma Development and Aggressiveness: Results of a Pilot Study on 239 Patients. Clin Cancer Res. 2008;14:3248–53. doi: 10.1158/1078-0432.CCR-07-4725. [DOI] [PubMed] [Google Scholar]
- 61.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer DrugTargets. 2005;5:27–41. doi: 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- 62.Secchiero P, Barbarotto E, Tiribelli M, et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL) Blood. 2006;107:4122–9. doi: 10.1182/blood-2005-11-4465. [DOI] [PubMed] [Google Scholar]
- 63.Coll-Mulet L, Iglesias-Serret D, Santidrian AF, et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood. 2006;107:4109–14. doi: 10.1182/blood-2005-08-3273. [DOI] [PubMed] [Google Scholar]
- 64.Secchiero P, Zerbinati C, di Iasio MG, et al. Synergistic cytotoxic activity of recombinant TRAIL plus the non-genotoxic activator of the p53 pathway nutlin-3 in acute myeloid leukemia cells. Curr Drug Metab. 2007;8:395–403. doi: 10.2174/138920007780655432. [DOI] [PubMed] [Google Scholar]
- 65.Drakos E, Thomaides A, Medeiros LJ, et al. Inhibition of p53-murine double minute 2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res. 2007;13:3380–7. doi: 10.1158/1078-0432.CCR-06-2581. [DOI] [PubMed] [Google Scholar]
- 66.Kranz D, Dobbelstein M. Nongenotoxic p53 activation protects cells against S-phase-specific chemotherapy. Cancer Res. 2006;66:10274–80. doi: 10.1158/0008-5472.CAN-06-1527. [DOI] [PubMed] [Google Scholar]
- 67.Yang H, Filipovic Z, Brown D, Breit SN, Vassilev LT. Macrophage inhibitory cytokine-1: a novel biomarker for p53 pathway activation. Mol CancerTher. 2003;2:1023–9. [PubMed] [Google Scholar]
- 68.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]