

Abstract
Multiple cellular stressors, including activation of the tumour suppressor p53, can stimulate autophagy. Here we show that knockout, knockdown or pharmacological inhibition of p53 can induce autophagy in human, mouse and nematode cells. Enhanced autophagy improved the survival of p53-deficient cancer cells under conditions of hypoxia and nutrient depletion, allowing them to maintain high ATP levels. Inhibition of p53 led to autophagy in enucleated cells, and cytoplasmic, not nuclear, p53 was able to repress the enhanced autophagy of p53-/- cells. Many different inducers of autophagy (for example, starvation, rapamycin and toxins affecting the endoplasmic reticulum) stimulated proteasome-mediated degradation of p53 through a pathway relying on the E3 ubiquitin ligase HDM2. Inhibition of p53 degradation prevented the activation of autophagy in several cell lines, in response to several distinct stimuli. These results provide evidence of a key signalling pathway that links autophagy to the cancer-associated dysregulation of p53.
Autophagy (`self-eating') is an important eukaryotic response to cellular stress. During autophagy, portions of the cytosol and cytoplasmic organelles are sequestered within characteristic double-or multi-membraned autophagosomes and delivered to lysosomes for bulk degradation. By promoting catabolic reactions, autophagy generates new metabolic substrates that meet the bioenergetic needs of cells and allows for adaptive protein synthesis. Autophagy also constitutes a homeostatic `clean-up' process to rid cells of intracellular parasites, damaged organelles and potentially toxic, aggregate-prone proteins. Finally, autophagy has been viewed as a self-destructive process in which stressed cells succumb to the so-called autophagic cell death1.
Autophagy is essential for the long-term survival of mammalian cells and a partial reduction in the autophagic capacity may constitute an oncogenic event. At least one of the phylogenetically conserved autophagy genes, atg6/beclin 1, is frequently inactivated at one locus in human cancers, and mouse studies have confirmed that beclin 1 is a haploinsufficient tumour suppressor2. There are two non-exclusive hypotheses to explain how inhibition of autophagy may stimulate oncogenesis and tumour progression. First, failure to undergo autophagy may favour necrotic cell death3 or compromise the clearance of dying cells4. This may exacerbate local inflammation and hence favour tumour growth. A second possibility is that inhibition of autophagy may lead to genomic instability5. A partial defect in autophagy can contribute to oncogenesis, at least in some cancers. Indeed, many human tumours manifest an aberrant level of autophagy6, and it can be stimulated by exogenous and endogenous stress, including chemotherapy, radiotherapy and hypoxia. Inhibition of this autophagic response reduces cell survival in most instances7,8, underscoring the importance of autophagy as a cellular defence mechanism.
The most extensively characterized tumour suppressor protein is p53, a master regulator with pleiotropic effects on metabolism, anti-oxidant defence, genomic stability, proliferation, senescence and cell death9. DNA damage induces autophagy in a p53-dependent fashion10. Moreover, re-expression of p53 in p53-deficient cancer cells has been shown to cause tumour regression through the re-activation of cell-intrinsic barriers against oncogenesis, such as senescence and apoptosis11-13. In the course of p53 re-activation, autophagy is induced13, presumably as a result of the p53-mediated transactivation of autophagy inducers14. This autophagic response to p53 may either contribute to cell death14 or constitute a cellular defence response, and its inhibition may improve the therapeutic effects of p53 re-activation on B cell lymphomas13.
Although the activation of the p53 system can induce autophagy, we made the surprising observation that removal of p53 can stimulate autophagy as well. Here, we provide evidence that p53 functions as an endogenous repressor of autophagy. Our results add to an increasingly complex homeostatic regulation in which p53 and autophagy are interconnected in a hitherto unexpected fashion.
RESULTS
Autophagic vacuolization after deletion, depletion or inhibition of p53
HCT116 colon cancer cells showed cytoplasmic accumulation of autophagosomes and/or autolysosomes, a morphological correlate of autophagy, as determined by transmission electron microscopy (TEM), when p53 was knocked down with a specific short interfering (si) RNA (Fig. 1a), knocked out by homologous recombination15 or inhibited with cyclic pifithrin-α (PFT-α), a pharmacological antagonist of p53 (ref. 16; Fig. 1b). Depletion or inhibition of p53 also induced TEM-detectable autophagy in non-transformed HFFF2 human fibroblasts, SH-SY5Y neuroblastoma and HeLa cervical cancer cells (Supplementary Information, Fig. S1a, b). Inhibition, depletion or deletion of p53 increased two biochemical signs of autophagy17, namely the conversion of LC3-I into LC3-II and reduced expression of p62/SQSTM1 (Fig. 1c). PFT-α, p53 knockout or p53 knockdown stimulated the redistribution of GFP-LC3 fusion protein from a ubiquitous, diffuse pattern towards autophagosomes, which became visible as cytoplasmic puncta, in HCT116 cells (Fig. 1d; Supplementary Information, Fig. S1c), mouse embryonic fibroblasts (MEFs; Fig. 1e; Supplementary Information, Fig. S1d) and human HFFF2, SH-SY5Y and HeLa cells (Supplementary Information, Fig. S1e-g).
Figure 1.

Induction of autophagic vacuolization by deletion, depletion or inhibition of p53. (a) Ultrastructural evidence of autophagy induced by depletion of p53 with a specific siRNA or pharmacological inhibition of p53 with PFT-α in HCT116 cells. (b) Autophagy induced by PFT-α, p53 knockdown, or p53 knockout in HCT116 cells. The number of autophagosomes and autolysosomes was determined for at least 50 cells in 3 independent experiments (mean ± s.e.m.; *P < 0.05). Culture in nutrient-free (NF) conditions was used as a positive control. (c) Effect of p53 on the maturation of LC3. Immunoblots are shown for wild-type (WT) or p53-/- HCT116 (n = 3). (d) GFP-LC3 puncta induced by PFT-α, p53 knockdown or knockout. WT and p53-/- HCT116 cells were transfected with control or p53-specific siRNAs, re-transfected with a GFP-LC3 plasmid, cultured in complete medium for 24 h and kept for 6 h in the presence or absence of 30 µM PFT-α. The percentage of cells showing accumulation of GFP-LC3 in puncta (GFP-LC3vac) is reported (mean ± s.d., n = 3; *P < 0.05). (e) GFP-LC3 puncta in p53-/- MEF, compared with WT MEF transfected with GFP-LC3 (mean ± s.d., n = 3; *P < 0.05). (f) GFP-LC3 dots in tissues from p53+/+ or p53-/- mice expressing a GFP-LC3 transgene (mean ± s.d.; *P < 0.05), without or after 24 h of starvation. The number of GFP-LC3 dots per area was determined for a minimum of 4 fields (at ×400 magnification for 4 slides per animal, 3 animals per condition). (g) TEM pictures of p53-/- and p53+/- mouse livers. p53-/- hepatocytes carry approximately 3 autophagosomes, p53+/- controls approximately 1 per cell. (h) Modulation of DsRed::LGG-1 levels by the p53 orthologue CEP-1 in C. elegans. Representative pictures are shown for WT animals and for nematodes carrying a cep-1 deletion allele (gk138). The count of puncta per cell was 6.9 ± 3.3 for WT, 52.2 ± 9.4 for cep-1 (gk138) embryos (mean ± s.e.m. for 20 individuals, 5 cells per embryo). The quantification of DsRed::LGG-1 puncta is shown for the indicated genetic backgrounds and conditions (dots represent pixel intensity for individual embryos).
Various tissues (pancreas, liver and kidney, but not muscle) from adult mice expressing a GFP-LC3 transgene18 on a p53-/- background showed a higher level of GFP-LC3 puncta than p53+/+ littermate controls (Fig. 1f; Supplementary Information, Fig. S2). Starvation for 24 h induced autophagy in GFP-LC3-transgenic p53+/+ mice18, whereas no further increase in GFP-LC3 puncta was observed in p53-/- mice (Fig. 1f; Supplementary Information, Fig. S2d), suggesting either that autophagy was occurring at a maximal level in the pancreas, liver and kidney, or that p53 is required for the induction of autophagy in response to starvation. TEM confirmed that p53-/- hepatocytes (Fig. 1g) and pancreatic epithelial cells (Supplementary Information, Fig. S2e) showed more autophagy than heterozygous p53+/- littermates. Systemic administration of PFT-α into mice also induced autophagy in the cerebellum and heart (Supplementary Information, Fig. S3a-f). Finally, knockout or RNAi-mediated depletion of the p53 orthologue cep-1 in Caenorhabditis elegans stimulated autophagy, as demonstrated by the increased expression and cytoplasmic aggregation of the DsRed::LGG-1 reporter gene product (LGG-1 is the orthologue of LC3) in embryos (Fig. 1h) and in adult pharyngeal cells (Supplementary Information, Fig. S3g, h). These data indicate that regulation of autophagy by p53 is phylogenetically conserved.
Mechanisms of enhanced autophagic vacuolization after p53 inhibition
GFP-LC3 puncta induced by p53 neutralization were suppressed by the depletion of Atg5, Beclin 1/Atg6, Atg10, Atg12 or hVps34 with previously validated siRNAs7,19, indicating that this phenomenon follows a canonical autophagic pathway (Fig. 2a, b). Similarly, MCF7 breast cancer cells, which are normally autophagy-incompetent due to deficient Beclin 1 expression20, failed to activate autophagy after p53 knockdown; however, the autophagic programme was restored when Beclin 1 expression was induced (Fig. 2c). The accumulation of autophagosomes and autolysomes by p53 depletion could involve an enhanced autophagic sequestration (on-rate) or a reduced degradation of autophagic material (off-rate). To distinguish between these possibilities, we assessed PFT-α-induced autophagic vacuolization by monitoring colocalization of GFP-LC3 with the lysosomal marker Lamp-2, either in the absence or in the presence of bafilomycin A1, which inhibits fusion beween autophagosomes and lysosomes7 (Fig. 2d, e). Bafilomycin A1 further enhanced the PFT-α-triggered accumulation of GFP-LC3 puncta (Fig. 2f). Similarly, the lysosomal protease inhibitor leupeptin21 further increased the PFT-α-triggered induction of LC3-II (Fig. 2g). These data suggest that p53 inhibition increases the on-rate of autophagy, at least in vitro, in cultured cells.
Figure 2.

PFT-α triggers autophagic vacuolization and has no effect on vacuole-lysosome fusion. (a) PFT-α or p53 siRNA-induced GFP-LC3 puncta in WT HCT116 cells transfected with siRNAs to deplete the essential autophagy gene products Atg5, Atg10, Atg12, Beclin 1 or hVps34. Culture in nutrient-free (NF) conditions was used as a control of autophagy induction. (b) PFT-α or p53 siRNA-mediated GFP-LC3 puncta in MEFs transfected with siRNAs specific for mouse Atg5 or Beclin 1. (c) Requirement of Beclin 1 expression for autophagy induction by p53 depletion in MCF7 cells. MCF7 cells stably transfected with a tetracyclinerepressible Beclin 1 construct were maintained in conditions that prevent or allow Beclin 1 expression, transfected with GFP-LC3 and subjected to p53 knockdown before quantification of GFP-LC3 puncta. Data in a-c are mean ± s.d. of 3 independent experiments. (d-f) Effect of bafilomycin A1 (BafA1) on PFT-α-induced GFP-LC3 puncta. GFP-LC3-expressing HeLa cells were treated with PFT-α and/or BafA1 and then immunostained for Lamp-2 to observe the colocalization between GFP-LC3 and Lamp-2. Representative confocal microphotographs are shown together with the profiles of colocalization (d) within the area of interest, indicated by the orientation of the arrow. Percentage overlap was plotted for control cells and cells cultured in the presence of PFT-α and/or BafA1 after 6 h (d, e). Columns represent the percentage of colocalization of GFP-LC3 and Lamp-2 (mean ± s.d.; *P < 0.05), quantified for at least 50 cells for each condition. The kinetics of GFP-LC3 redistribution was quantified by conventional fluorescence microscopy (mean ± s.d., 3 experiments, f). (g) Immunoblot detection of LC3-I/II in cells treated with PFT-α and/or leupeptin in WT HCT116 cells (n = 3).
Autophagy is controlled by several kinases including AMP kinase (AMPK), which induces autophagy22, and the mammalian target of rapamycin (mTOR), which suppresses autophagy23. In p53-/- cells, AMPK and the AMPK substrates tuberous sclerosis complex 2 (TSC2) and acetyl CoA carboxylase (ACCα) were hyperphosphorylated (which indicates AMPK activation), whereas p70S6K, an mTOR substrate, was hypophosphorylated (which reflects mTOR inhibition; Fig. 3a). siRNA-mediated depletion of AMPKα1 and AMPKα2, which inhibited the hyperphosphorylation of its substrate ACCα in p53-/- cells (Fig. 3c), or inhibition of mTOR with rapamycin (Fig. 3d) eliminated the difference in autophagy between p53-inhibited and control HCT116 (Fig. 3b-d) or SH-SY5Y cells (not shown). These results are consistent with the possibility that p53 controls autophagy through the AMPK/mTOR-dependent pathway.
Figure 3.

Role of AMPK, p70S6K and mTOR in the p53-mediated modulation of autophagy. (a) Immunoblot detection of the phosphorylation status of AMPKα, ACCα, TSC2 and p70S6K in HCT116 cells. Hypophosphorylation of p70S6K and hyperphosphorylation of AMPKα, ACCα and TSC2 was detected in p53-/- HCT116, compared with WT HCT116 cells after culture in complete medium (n = 5). (b) Quantification of GFP-LC3vac cells in WT and p53-/- HCT116 cells transfected with siRNAs specific for the catalytic α1 and α2 subunits of AMPK (mean ± s.d. of 3 independent experiments; *P < 0.05). The inset in b demonstrates the efficiency of siRNA-mediated downregulation of AMPKα1 and AMPKα2, as assessed by immunoblot analysis (n = 3). Note that the antibody recognizes both AMPKα1 and AMPKα2. (c) Immunoblot detection of ACCα phosphorylation in WT and p53-/- HCT116 cells subjected to knockdown of AMPKα1 and AMPKα2 (n = 5); Co, control. (d) Quantification of GFP-LC3vac cells (means ± s.d., n = 3 separate experiments) in WT HCT116 and p53-/- HCT116 treated with PFT-α and/or rapamycin.
Inhibition or deletion of p53 leads to stress resistance by induction of autophagy
Autophagy allows cells to maintain high ATP levels under conditions of starvation24. When wild-type HCT116 cells were transferred from a glucose-containing to a glucose-free medium, the cytosolic ATP levels were reduced within minutes (Fig. 4a; Supplementary Information, Fig. S4a). In wild-type cells, this reduction in ATP was blunted by supply of the glycolytic product pyruvate (provided as membrane-permeable methylpyruvate; Fig. 4b; Supplementary Information, Fig. S4b) and by the autophagy inducer rapamycin (Fig. 4c; Supplementary Information, Fig. S4c). In contrast, p53-/- cells maintained high ATP levels even in the absence of glucose (Fig. 4a). However, the capacity of p53-/- cells to maintain high ATP levels under glucose-free conditions was compromised when autophagy was inhibited by depletion of AMPKα, Beclin 1 (Fig. 4c), Atg5 or Atg10 (data not shown). This affects cell survival under conditions of metabolic stress (nutrient deprivation and hypoxia). After 48 h of metabolic stress, the mitochondrial transmembrane potential (ΔΨm measured with the fluorochrome DiOC6(3)) was reduced in a significant number of wild-type cells, a sign of imminent cell death and/or a loss of viability (measured with the vital dye propidium iodide or clonogenic assays). p53-/- cells were relatively resistant to metabolic stress; however, this resistance was reversed by knockdown of AMPKα or the essential autophagy gene products Atg5, Atg10 (Fig. 4d, e) or Beclin 1 (Supplementary Information, Fig. S4f). Methylpyruvate partially reversed the lethal effect of metabolic stress (Supplementary Information, Fig. S4d, e). Similar results were obtained when p53 and Beclin 1 levels were manipulated in MCF7 cells (autophagy-incompetent unless Beclin 1 expression is induced20; Fig. 4f), as well as in MEFs (autophagy-competent, except when Beclin 1 is depleted; Fig. 1e). Irrespective of the cell type, p53 depletion conferred an enhanced resistance to metabolic stress, but only when the cells expressed Beclin 1 and hence were capable of autophagy (Fig. 4f, g). These results indicate that the resistance of p53-/- cells to metabolic stress depends on increased autophagy.
Figure 4.
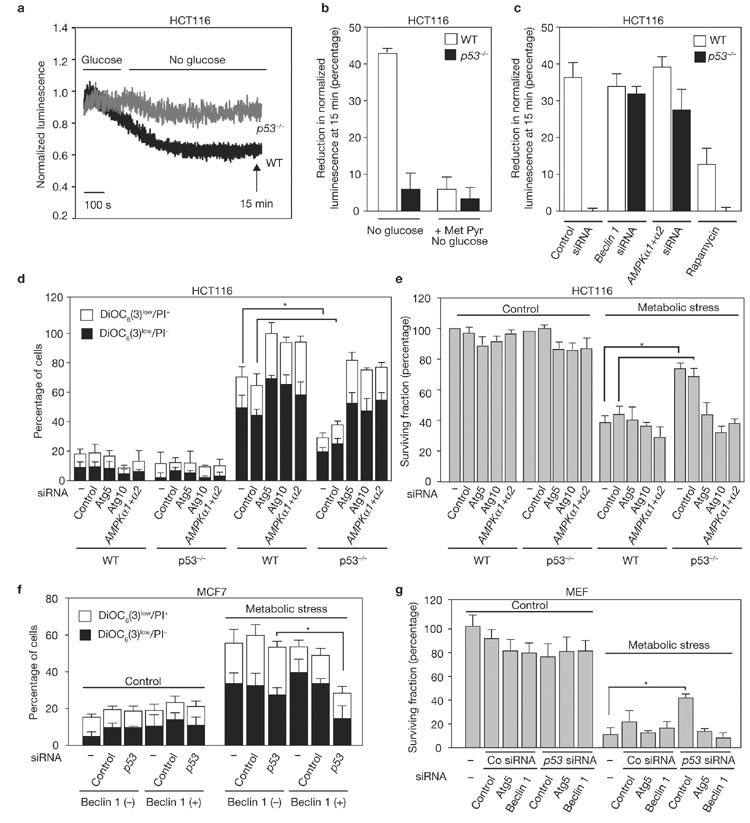
p53 deletion attenuates ATP depletion during glucose deprivation and favours survival under metabolic stress. (a) Cytosolic ATP levels in WT and p53-/- HCT116 cells in the absence of glucose. Measurements were performed on luciferase-expressing HCT116, after perfusion of the cells with medium first containing then missing glucose. (b) Effect of methylpyruvate (MetPyr) on ATP levels in WT p53-/- HCT116 cells depleted of glucose. ATP levels were measured as in a, in the presence of glucose and 15 min after its withdrawal or replacement by MetPyr. (c) Effect of autophagy on the ATP levels of WT and p53-/- HCT116 cells depleted of glucose. Cells were transfected with siRNAs specific for Beclin 1 and AMPKα, or they were treated with rapamycin for 6 h, followed by measurement of ATP levels as in a, before or after glucose withdrawal (means ± s.d. of triplicates, 3 independent experiments). (d) Metabolic stress-induced cell death is attenuated in the absence of p53. HCT116 cells were transfected with control, Atg5-, Atg10-, or AMPKα-specific siRNAs and were subjected 48 h later to metabolic stress (cultured for 48 h in nutrient-free, hypoxic conditions) and stained with DiOC6(3) and PI. The black and white portions of the columns refer to the DiOC6(3)low PI- (dying) and DiOC6(3)low PI+(dead) population, respectively. (e) Metabolic stress-induced decrease of clonogenic survival was attenuated in the absence of p53. HCT116 cells subjected to metabolic stress as in d were monitored for clonogenic survival. (f) Expression of Beclin 1 in MCF7 cells restores the survival advantage conferred by p53 depletion. MCF7 cells that carry a tetracycline-repressible Beclin 1 expression construct were cultured to avoid Beclin 1 expression or to induce it, transfected with a control siRNA or a p53-specific siRNA, subjected to metabolic stress, and finally stained with DiOC6(3)/PI. (g) Effect of p53 and autophagy on the survival of metabolically stressed MEFs. WT MEFs were transfected with the indicated siRNAs and then subjected to metabolic stress before performing clonogenic assays. Results in d-g are mean ± s.d. of 3 separate experiments (*P < 0.05); Co, control.
Cytoplasmic, not nuclear, p53, is required for inhibition of autophagy
To understand the mechanism by which p53 inhibits autophagy, we determined the transcriptome of wild-type and p53-/- HCT116 cells, but failed to identify changes in autophagy-related transcripts listed in a gene ontology program (www.ingenuity.com). Therefore, we investigated whether p53 suppression of autophagy is transcription-independent using cytoplasts (anucleate cells). In response to PFT-α, HeLa cytoplasts (arrows in Fig. 5a) were still able to accumulate GFP-LC3 puncta (Fig. 5a, b; Supplementary Information, Fig. S5a), indicating that nuclei (and by extension transcription) were not required for PFT-α-stimulated autophagy.
Figure 5.

Inhibition of autophagy by cytoplasmic p53. (a, b) PFT-α-mediated stimulation of autophagy in cytoplasts. GFP-LC3-transfected HeLa cells were enucleated by density-gradient centrifugation after cytochalasin B treatment and cultured on polylysine-coated coverslips. Mixtures of cells and cytoplasts (arrows, identified by the lack of Hoechst 33342 staining) were then exposed to three different autophagy inducers: PFT-α, nutrient starvation and rapamycin. Results are means ± s.d. n = 3 experiments). (c-e) Effect of p53 mutants on autophagy in p53-/-HCT116 cells. The p53 domains and mutants used in this study are schematically represented in c. Representative micrographs of cells transfected with plasmids expressing WT, nuclear and cytoplasmic p53 are shown in d. Quantification of GFP-LC3 puncta of p53-/- HCT116 cells transiently transfected with p53 mutants is shown in e (mean ± s.d., n = 3 experiments, *P < 0.05). (f) Detection of the phosphorylation status of AMPKα, AACα, TSC2 and p70S6K in p53-/- HCT116 cells transiently expressing WT or mutant p53 (n = 5).
Next, we re-transfected p53-/- HCT116 cells with p53 targeted to nuclear or extranuclear locations (Fig. 5c). Both wild-type and endothelium reticulum (ER)-targeted p53 (p53ER)25 inhibited autophagy, whereas a p53 form locked into the nucleus (using a disrupted nuclear export signal, NES) failed to inhibit autophagy (Fig. 5d, e; Supplementary Information, Fig. S5b). The mutation R175H, which is known to inhibit the nuclear and cytoplasmic effects of p53 (refs 26, 27), prevented inhibition of autophagy by p53 and p53ER. A p53 mutant with a mutation in the nuclear localization sequence (NLS), which causes cytoplasmic retention of p53, efficiently inhibited autophagy. The p53ERTAM fusion construct (the hormone-binding region of the oestrogen receptor fused to p53) is inactive unless 4-hydroxytamoxifen (4-OHT) is added28. Transfection of p53-/- cells with p53ERTAM only inhibited autophagy in the presence of 4-OHT. Removal of the Pro-rich domain (ΔPP) curtailed the inhibitory effect of p53ERTAM on autophagy, arguing against non-specific effects (Fig. 5e). Inhibition of autophagy by wild-type or cytoplasm-targeted p53 was accompanied by reduced phosphorylation of AMPKα, ACCα and TSC2, as well as increased phosphorylation of p70S6K, underscoring the impact of p53 on the AMPK/mTOR axis (Fig. 5f). These results indicate that p53 inhibits autophagy through a transcription-independent effect exerted from a cytoplasmic localization.
Implication of p53 in the physiological regulation of autophagy
Inhibition of p53 by PFT-α induced autophagy rapidly, with similar kinetics as those for rapamycin (see below). Shortly after addition of PFT-α (2 h), newly formed GFP-LC3+ puncta colocalized with the ER markers ERp57 (Fig. 6a) or calreticulin (data not shown), whereas colocalization with mitochondrial markers, such as the core 2 subunit of the OXPHOS complex, occurred at later time points (Fig. 6b). The level of ER markers in autophagic cells decreased 4-6 h after addition of PFT-α (Fig. 6a). Hence, inhibition of p53 initially induces efficient reticulophagy (autophagy of the ER), which is followed by mitophagy (autophagy of mitochondria). Reticulophagy is also induced by ER stressors such as thapsigargin, tunicamycin and brefeldin A, through a pathway that relies on IRE-1α-dependent phosphorylation of eIF2α29. Similarly, depletion or inhibition of p53 increased phosphorylation of eIF2α (Fig. 6d). siRNA-mediated depletion of IRE-1α from HCT116 (Fig. 6e, f) cells or removal of the ire1α gene from MEFs (Fig. 6f) inhibited autophagy induced by p53 neutralization.
Figure 6.

p53 inhibition induces autophagy by ER stress (a-c) WT HCT116 cells were transiently transfected with GFP-LC3, treated with PFT-α for 2, 4 or 6 h and subjected to immunofluorescence staining to observe the colocalization of GFP-LC3+ puncta with the ER marker ERp57 (a) or a mitochondrial marker (b). Note that cells with GFP-LC3+ puncta often contain scarce amounts of ERp57 (white arrows) after prolonged incubation with PFT-α. The percentage of colocalization (means ± s.d., n = 3 experiments with 50 images per experiment) was determined by confocal microscopy (c). (d) eIF2α phosphorylation in WT or p53-/- HCT116 cells treated with PFT-α (n = 5). (e, f) WT HCT116 cells (e, f) or p53-/- HCT116 cells (f) were transfected with GFP-LC3, plus a control siRNA or the indicated combination of IRE1α- and/or p53-specific siRNA, and treated with PFT-α, thapsigargin, tunicamycin or brefeldin A. (g) GFP-LC3-transfected WT or ire1α-/- MEF were treated with PFT-α. Columns in e-g show means ± s.d. of 3 independent experiments (*P < 0.05).
As well as inducing autophagy, tunicamycin, a specific inhibitor of N-glycosylation, and other autophagy inducers (for example, nutrient depletion, rapamycin and lithium) caused concomitant depletion of p53 (Fig. 7a, b). Both cytoplasmic and nuclear p53 were depleted with similar kinetics (data not shown). Rapamycin treatment or ER stress induced by tunicamycin, thapsigargin or brefeldin A caused HDM2-and proteasome-dependent degradation of p53 (refs 30, 31) as well as autophagy29,32 (Fig. 7a-d). When p53 degradation was prevented by proteasome inhibition with MG132 or siRNA-mediated depletion of HDM2, autophagy was not induced. This effect was observed in HCT116 cells (Fig. 7c, d), as well as in HeLa cells (Supplementary Information, Fig. 6c, d) and non-transformed fibroblasts (data not shown). Two pharmacological inhibitors of HDM2, Nutlin-3 (ref. 33) and RITA34, also stabilized p53 and inhibited autophagy induced by ER stressors, nutrient starvation, lithium and rapamycin (Fig. 7d). In control experiments, HDM2 knockdown, MG132 (Fig. 7e), Nutlin-3 or RITA (Fig. 7f) did not block the constitutively elevated autophagy of p53-/- cells, indicating that these agents do not exert an intrinsic autophagy-inhibitory effect. Overexpression of wild-type p53 and a p53 mutant that is resistant to HDM2-dependent degradation (p53ΔI)35 also inhibited autophagy induced by tunicamycin, thapsigargin and rapamycin in a concentration-dependent manner. In this assay, the p53ΔI mutant was relatively resistant to degradation triggered by autophagy inducers and was more efficient than wild-type p53 in inhibiting GFP- LC3 redistribution (Fig. 7g).
Figure 7.
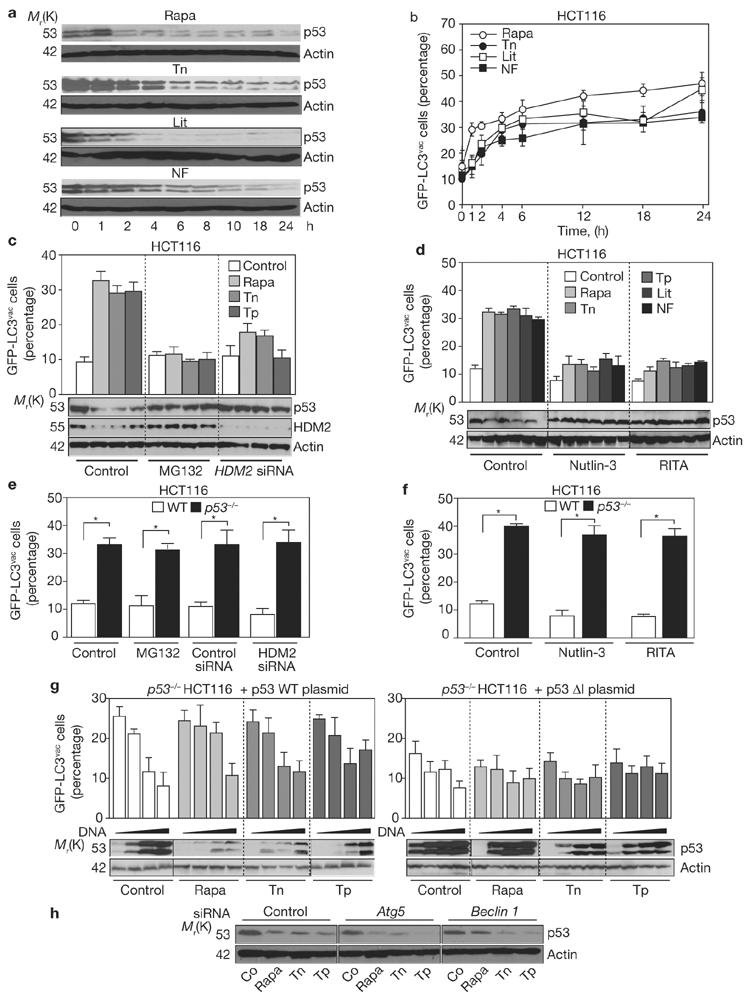
Induction of autophagy requires p53 degradation mediated by HDM2 and the proteasome. Kinetics of p53 degradation (a) and induction of GFP-LC3 puncta (b) in WT HCT116 cells cultured in complete medium in the presence of rapamycin (Rapa), tunicamycin (Tn), lithium (Li) or in nutrient-free medium (NF). (c) WT HCT116 cells transfected with control or HDM2-targeted siRNAs were cultured in the presence or absence of MG132 for 3 h, followed by the treatment with autophagy inducers for 6 h (Tp, thapsigargin; other abbreviations as in b). The abundance of p53 and HDM2 was determined by immunoblotting analysis and the percentage of cells showing accumulation of GFP-LC3 in vacuoles (GFP-LC3vac) was determined. (d) Effects of HDM2 inhibitors on p53 protein levels and autophagy. WT HCT116 were left untreated or treated with Nutlin-3, RITA and/or the indicated autophagy inducers, followed by the p53 immunoblot or immunofluorescence microscopy to visualize GFP-LC3 redistribution. (e) Effect of MG132 and HDM2 siRNA on autophagy of WT and p53-/- HCT116 cells. (f) Failure of HDM2 inhibitors to prevent autophagy induced by the absence of p53. Nutlin-3 and RITA were added to p53-/- HCT116 cells, followed by quantification of GFP-LC3 puncta. (g) Effect of WT and ubiquitination-resistant p53 on autophagy. p53-/- HCT116 cells were transfected with GFP-LC3 alone or in combination with different concentrations of pcDNA3.1, WT p53 or p53 lacking the ubiquitination site at amino acids 13-19 (p53ΔI), then treated with rapamycin, tunicamycin or thapsigargin. Thereafter, p53 expression and GFP-LC3 puncta were quantified. (h) Depletion of Atg5 or Beclin 1 does not prevent p53 degradation triggered by rapamycin or ER stressors. In WT HCT116 cells transfected with siRNA targeting Atg5 or Beclin 1, p53 levels were determined by immunoblot analysis after 6 h of treatment with rapamycin, tunicamycin or thapsigargin. Quantification of GFP- LC3vac cells in b-g are mean ± s.d. of 3 separate experiments, *P < 0.05. The blots in a, c, d, g and h shown are representative of 3-5 different experiments.
These results provide evidence for a link between p53 depletion and induction of autophagy. Notably, under conditions of ER stress, depletion of Atg5 or Beclin 1 did not prevent p53 degradation (Fig. 7h), suggesting that autophagy is not required for depletion of p53. Rather, depletion of p53 seems to be required for induction of autophagy (Fig. 7a-h).
DISCUSSION
Here we show that deletion, depletion or inhibition of p53 induces autophagy in human, mouse and nematode cells. Moreover, various inducers of autophagy cause p53 degradation, whereas inhibition of p53 degradation prevents autophagy, suggesting that p53 inhibition is essential for induction of autophagy. These findings have implications for the spatial regulation of the p53 system, the connections between p53 and autophagy/survival pathways and the role of p53 as a tumour suppressor protein.
At present, the exact mechanisms by which autophagy-inducing stimuli, such as ER stress, cause the cytoplasmic translocation and subsequent degradation of p53 remain unknown. However, this process involves sequential phosphorylation of p53 on Ser 315 and Ser 376, its nuclear export, ubiquitination by HDM2 and proteasome-mediated degradation30,31. As shown here, inhibiting HDM2 or the proteasome prevents degradation of p53 induced by various autophagy triggers, and inhibits autophagy. The present study provides some insight into the mechanisms by which p53 depletion de-represses autophagy. We have shown that p53 inhibits autophagy through a cytoplasmic (non-nuclear) effect, as indicated by the finding that pharmacological inhibition of p53 can trigger autophagy in cytoplasts. Moreover, cytoplasm-targeted, but not nucleus-targeted, variants of p53 suppress autophagy. Importantly, either a point mutation that changes the conformation of p53 (R175H) or the removal of the πPP abolished the autophagy-inhibitory action of p53. Another cytoplasmic function of p53, namely the induction of mitochondrial membrane permeabilization and apoptosis, is abolished by the R175H and ΔPP mutations as well27,36,37. Factors that interact with p53 to induce apoptosis belong to the Bcl-2 family of proteins27,36,38, which also regulate autophagy39. As with pro-apoptotic BH3-only proteins, p53 inhibits the anti-apoptotic effect of Bcl-2 homologues36 and activates Bax38 and Bak40, which are Bcl-2 proteins that promote apoptosis. However, BH3-only proteins are autophagy inducers, not inhibitors41, indicating that the modulatory effect of p53 in autophagy is different from that of BH3-only proteins. Hence, the exact molecular pathway through which cytoplasmic p53 inhibits autophagy remains elusive.
Irrespective of the molecular details, induction of autophagy by p53 has been linked to inhibition of mTOR10,42, whereas autophagy inhibition by p53 correlates with enhanced mTOR activity. Moreover, it appears that induction of autophagy by p53 depends on transactivation of genes such as DRAM14, whereas inhibition is a cytoplasmic effect. Nucleo-cytoplasmic shuttling of p53 is regulated by post-translational modifications of nuclear export signals43 and has a major role in regulation of autophagy.
p53 is activated by various stressors, including agents that affect DNA structure (for example, ultraviolet light, ionizing irradiation and chemotherapeutics) and conditions that induce reactive oxygen species. The activation of p53 by stress involves multiple mechanisms that often lead to stabilization of the p53 protein44. Once activated, p53 mediates several effects ranging from stimulation of DNA repair after transient cell-cycle arrest to irreversible cell-cycle arrest (senescence) or induction of apoptosis. Most of these effects depend on the transactivation of p53 target genes9,45. Determining whether p53 induces adaptive responses (DNA repair) or cellular demise has been linked to distinct transcriptional programmes, correlating with the particular pattern of post-transcriptional modifications of p53 (such as phosphorylation of Ser 46, which is pro-apoptotic). Although DNA-damaging and pro-oxidant conditions activate p53, some cellular stressors inactivate p53, as demonstrated for ER stressors30,31 and starvation (as shown in this study). These findings demonstrate that subtle differences in the nature of the stressors can either induce or inhibit p53.
As mentioned earlier, p53 may potently induce autophagy through transcriptional activation of the autophagy-inducing protein DRAM14, and also through inactivation of the mTOR pathway10. This p53-mediated activation of autophagy has been linked to cell death in a positive fashion (because DRAM is also required for apoptosis induction via p53) or in a negative fashion (because autophagy inhibition can enhance p53-mediated apoptosis). In addition, we show here that physiological levels of p53 repress autophagy. Thus, any perturbation of p53 system — either activation or inhibition — may induce autophagy. It is possible that any kind of cellular stress, caused by either inactivation or overactivation of p53, may induce autophagy, perhaps in the context of genotoxic stress or failure to handle such stress. Autophagy induced by p53 overactivation can either be cytocidal or cytoprotective, as discussed above. In contrast, it seems plausible that autophagy linked to p53 inhibition is cytoprotective, as illustrated by the finding that autophagy is required for the enhanced resistance of p53-/- cells to metabolic stress.
Together, these findings indicate that p53 may be either activated or inhibited by different stressors, that p53 can inhibit or enhance autophagy and that autophagy can increase or reduce cell survival, in a series of binary decisions that are probably determined by cellular events, as well as by the intensity of the signals. For example, reduction of glucose concentration can activate p53 through AMPK-mediated phosphorylation42,46, but the removal of all nutrients from the medium induces p53 degradation, as shown here. Moreover, although p53 can stimulate autophagy by mTOR inhibition10 or DRAM transactivation14 in the context of DNA damage or p53 overexpression, we show here that baseline p53 levels can inhibit autophagy in other cellular contexts.
Undoubtedly, p53 is one of the tumour suppressors most frequently inactivated in cancer. What could be the advantage, in teleological terms, of increasing autophagy in tumour cells? Pharmacological stimulation of autophagy increases the resistance of cells to apoptosis, presumably due to the removal of pro-apoptotic mitochondria47; both resistance to apoptosis and reduction of oxidative phosphorylation are hallmarks of cancer48,49. Autophagy is a process by which cells struggle to survive under conditions of reduced intracellular metabolite concentrations, caused by loss of growth factor signalling that governs the uptake of nutrients24. As shown here, cells lacking p53 are particularly resistant to ATP depletion and cell death induced by metabolic stress caused by hypoxia and nutrient depletion, and this resistance is lost when autophagy is inhibited. Oxygen and glucose supply to cancer cells is often low, especially in non-vascularized areas of tumour nodules. In this tumour microenvironment, an enhanced level of baseline autophagy may improve the fitness of malignant cells and constitute an initial advantage for those cancer cells that lose paracrine or contact-dependent growth signals as they infiltrate normal tissue or metastasize.
Although enhanced autophagy may confer an advantage, at least for stressed cells, it is plausible (yet remains to be proven) that a constant increase in self-cannibalism may reduce cell proliferation rates. In this context, the partial inhibition of autophagy either by loss of one beclin 1 allele (and perhaps other, yet-to-be-discovered genetic or epigenetic modifications) or constitutive activation of the PI-3K/Akt axis may function as a `corrective' measure to dampen autophagy. It will be important to determine in which order p53 and Beclin-1 are inactivated (or the PI-3K/Akt pathway is activated) during the natural history of breast, ovary and prostate cancers and how this correlates with the enhanced or reduced autophagic capacity of premalignant and malignant cells.
METHODS
Cells and in vitro treatments
HCT116 cells were a gift from B. Vogelstein15. MCF7 cells stably transfected with a tetracycline (2 μg ml-1)-repressible Beclin 1 construct were a gift from B. Levine20. Human fetal fibroblasts (HFFF2) and neuroblastoma SH-SY5Y cells were purchased from ATCC. For serum and amino acid starvation, cells were cultured in serum-free Earle's Balanced Salt Solution medium (Sigma), which we refer to as nutrient-free medium7. Metabolic stress was induced by culturing the cells for 48 h in nutrient free medium under hypoxic conditions (0.1% oxygen, 5% CO2).
Cells (3 × 104) were seeded in 24-well plates and grown for 24 h before treatment with rapamycin (1 μM; Tocris Bioscience), PFT-α (30 μM), tunicamycin (2.5 μM), thapsigargin (3 μM; Calbiochem), brefeldin A (20 μM), lithium chloride (10 mM), bafilomycin A1 (1 nM), leupeptin (100 nM), 4-hydroxytamoxifen (4-OHT, 100 nM), methylpyruvate (MetPyr, 10 mM), MG132 (10 μM; Sigma), Nutlin-3 (10 μM) or RITA (10 μM, Alexis Biochemicals) for the indicated period (6 h, unless otherwise stated).
Plasmids, transfection and RNA interference
Cells at 80% confluence were transfected in 6-well plates with Oligofectamine (Invitrogen) and siRNAs (100 nM). siRNA-mediated protein downregulation was controlled by immunoblots or by RT-PCR with specific primers. Transient transfections were performed with Lipofectamine 2000 (Invitrogen), and cells were used 24 h after transfection, unless indicated otherwise. Cells were transfected with an empty vector alone or together with plasmids encoding GFP-LC3 (ref. 50), in the presence or absence of p53 NES-, p53 NLS- (gift from C.G. Maki, Department of Radiation and Cellular Oncology, University of Chicago, IL), p53 ERTam, p53 ERTam ΔPP (gift from J. Chipuk, Department of Immunology, St Jude's Children's Hospital, Memphis TN), p53 ΔI (gift from K. Vousden, The Beatson Institute for Cancer Research, Glasgow, UK), p53WT, p53ER, p53ER R175H or p53 R175H27,36.
Quantification of GFP-LC3 puncta
Autophagy was quantified by counting the percentage of cells showing accumulation of GFP-LC3 in dots or vacuoles (GFP-LC3vac, of a minimum of 100 cells per preparation in three independent experiments). Cells presenting a mostly diffuse distribution of GFP-LC3 in the cytoplasm and nucleus were considered non-autophagic, whereas cells representing several intense punctate GFP-LC3 aggregates with no nuclear GFP-LC3 were classified as autophagic. Each GFP-LC3 staining was read by two independent investigators (E.T. and M.C.M. or E.M.). To study the effect of starvation, 6-week-old mice carrying a GFP-LC3 transgene on a p53+/+ or p53-/- background were deprived of food for 24 h, but had free access to drinking water.
Statistical Analysis
Student's t-distribution probability density function was used for calculation of P values.
Supplementary Material
ACKNOWLEDGEMENTS
We thank N. Mizushima for GFP-LC3-transgenic mice, Levine and B. Vogelstein for cell lines, J. Chipuk, C. G. Maki, M. Oren and K. Vousden for mutant p53 plasmids, E. Zaharioudaki, B. Gardie-Capdeville, C. Ladrou, M.R. Duchen, A. Jalil, F. Fanelli and A. Petrini for expert assistance. The cep-1(gk138) mutant strain (TJ1) was provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). N.T. is funded by EMBO and the EU Sixth Framework Programme. F.C. is funded by Fondazione Telethon, AIRC, Compagnia di San Paolo and the Italian Ministry of Health. E.T. is a recipient of a PhD fellowship from Institut National contre le Cancer (INCa). G.K. is supported by Ligue Nationale contre le Cancer, Agence Nationale pour la Recherche, Cancéropôle Ile-de-France, INCa, Fondation pour la Recherche Médicale, and European Union (Active p53, Apo-Sys, ApopTrain, ChemoRes, TransDeath, RIGHT).
Footnotes
Note: Supplementary Information is available on the Nature Cell Biology website.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J. Clin. Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 5.Matthew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature Rev. Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nature Rev. Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 7.Boya P, et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maiuri C, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature Rev. Mol. Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 9.Vousden KH, Lane DP. p53 in health and disease. Nature Rev. Mol. Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 10.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl Acad. Sci. USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amaravadi RK, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 15.Bunz F, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komarov PG, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 17.Klionsky DJ, al. e. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Criollo A, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–1039. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 20.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007;3 doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 22.Meley D, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 23.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nature Rev. Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 24.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Natue Rev. Mol. Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 25.Talos F, Petrenko O, Mena P, Moll UM. Mitochondrially targeted p53 has tumor suppressor activities in vivo. Cancer Res. 2005;65:9971–9981. doi: 10.1158/0008-5472.CAN-05-1084. [DOI] [PubMed] [Google Scholar]
- 26.Baker SJ, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 27.Tomita Y, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 28.Vater CA, Bartle LM, Dionne CA, Littlewood TD, Goldmacher VS. Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53-/- mouse embryo fibroblasts. Oncogene. 1996;13:739–748. [PubMed] [Google Scholar]
- 29.Ogata M, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu L, et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18:261–277. doi: 10.1101/gad.1165804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pluquet O, Qu LK, Baltzis D, Koromilas AE. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3β. Mol. Cell Biol. 2005;25:9392–9405. doi: 10.1128/MCB.25.21.9392-9405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 34.Issaeva N, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nature Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 35.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 36.Mihara M, et al. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 37.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–381. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 38.Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 39.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nature Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 41.Maiuri MC, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng Z, et al. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 43.Fabbro M, Henderson BR. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp. Cell Res. 2003;282:59–69. doi: 10.1016/s0014-4827(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 44.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nature Rev. Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 45.Lim YP, et al. The p53 knowledgebase: an integrated information resource for p53 research. Oncogene. 2007;26:1517–1521. doi: 10.1038/sj.onc.1209952. [DOI] [PubMed] [Google Scholar]
- 46.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 47.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nature Rev. Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 48.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 49.Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 2007;19:223–229. doi: 10.1016/j.ceb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.