

Iron is required for bacteria survival and is growth limiting at low concentrations present during vertebrate infections.1 In response to a low [Fe3+/2+] microenvironment, bacteria up-regulate the production of enzymes that synthesize and transport iron scavenging small molecules called siderophores.2 These compounds often serve as virulence factors enabling the maintenance of infection for pathogenic bacteria.3 The key functional attribute of siderophores is high affinity for ferric iron (the predominant form in oxidizing environments); Nature utilizes several types of coordinating functionality assembled in small molecule scaffolds to provide multi-dentate Fe3+ ligation.2
A major biosynthetic strategy in pathogenic bacteria is the use of nonribosomal peptide synthetase (NRPS) assembly logic and protein machinery to assemble three types of iron chelating moieties: phenols and catechols, oxazolines and thiazolines, and N-OH amides (hydroxamates). Some siderophores such as enterobactin4 have only catechol functional groups, while desferrioxamine5 displays an arrangement of hydroxamates as ferric iron ligands. The Pseudomonas siderophore pyochelin6 reflects predominantly the dihydrocyclization strategy (thiazoline). Combinatorial biosynthetic evolution has led to siderophores with mixed chelating functional groups, including catechols and oxazolines in vibriobactin7 and phenols and thiazolines in yersiniabactin8 from the bacteria responsible for cholera and plague, respectively. All three functional groups (catechol, oxazoline/thiazoline, and hydroxamate) are present in the reported structure of acinetobactin 1 from the Gram-negative pathogen Acinetobacter baumannii9 and in anguibactin 2 from the fish pathogen Vibrio anguillarum (Figure 1).10 Despite efforts to engineer NRPS pathways, selectivity conferred by adenylation and condensation domains often limits the range of monomers accepted at each stage of assembly.11
Figure 1.
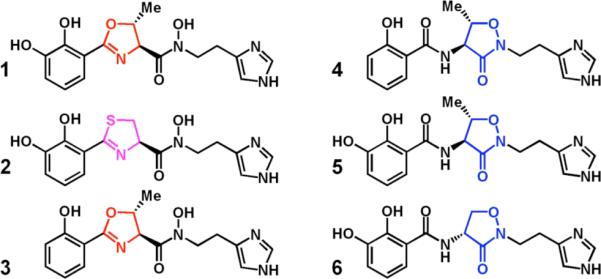
Siderophore natural products contain a variety of iron-chelating functionality.
The biosynthetic gene clusters for acinetobactin and anguibactin have been identified and resemble that of vibriobactin and a structural relative, pseudomonine (4). We recently purified the five-protein pseudomonine biosynthetic assembly line from Pseudomonas entomophila, which uses salicylate, threonine and histamine as building blocks, and we reconstituted in vitro the biosynthesis of isoxazolidinone siderophore 4.12 Notably, histamine is converted to N-OH-histamine by a flavoprotein hydroxylase PmsF, which is then used as the chain-terminating nucleophile to capture a hydroxyphenyl-methyloxazolinyl acyl group tethered covalently on the pantetheinyl arm of a carrier protein domain of the protein PmsG. This yields a close homolog of 1, which we termed pre-pseudomonine (3). Under physiologic buffered conditions, 3 rearranges non-enzymatically to 4, establishing the oxazoline to be a latent electrophile trapped intramolecularly by the hydroxamate oxygen. Detection of the oxazoline as the initial product was facilitated by the use of histamine rather than N-OH-histamine as the chain-termination substrate, yielding a vibriobactin-like oxazolinyl amide 8 (rather than oxazolinyl-N-OH amide), which cannot be processed by intramolecular rearrangement (Figure 2A).
Figure 2.

A) PmsDEG assembly line production of a family of siderophore and “siderophore-like” compounds. B) Time course reconstitution assay with PmsDEG to produce acinetobactin (5) and preacinetobactin (1). C) Reconstitution assay with PmsDEG to produce anguibactin (m/z = 349.20).
Because pre-pseudomonine and structure 1 for acinetobactin are so similar, we investigated whether the purified pseudomonine assembly line proteins (PmsDEG) would utilize 2,3-dihydroxybenzoate (DHB) in place of 2-hydroxybenzoate (salicylate). Purified PmsE generates 2,3-DHB-AMP and then transfers it covalently to the pantetheinyl arm of its thiolation domain. In turn, PmsD transfers the DHB acyl group to threonyl-S-pantetheinyl-PmsG to yield the dihydroxyphenyl-methyloxazolinyl-S-PmsG covalent acyl-enzyme intermediate. The C domain of PmsG can then use synthetic N-OH-histamine as a nucleophilic substrate and releases a siderophore with the correct molecular weight for 1. However, that molecule rapidly rearranges non-enzymatically to the stable compound 5 over a sixty-minute time period (Figure 2B).
This phenomenon is similar to what we have previously observed for conversion of pre-pseudomonine to pseudomonine.12 Indeed, it is likely the same N-OH-oxazoline to isoxazolidinone rearrangement is occurring in the acinetobactin scaffold. This was validated by proton NMR analysis of 5. Comparison of the spectrum of enzymatic 5 with the spectroscopic data reported in the original acinetobactin isolation and structure assignment paper suggests the structure for acinetobactin is isoxazolidinone 5.9 Accordingly, we redefine structure 1 as pre-acinetobactin. To further evaluate the substrate scope of the N-OH oxazoline to isoxazolidinone siderophore scaffold rearrangement under physiologic conditions, we next tested benzoic acid as a surrogate for salicylate and DHB. Surprisingly, the pseudomonine assembly line produced an initial oxazoline that rearranges to the isoxazolidinone (10). Of particular interest was the relative rate of rearrangement, which we found increases in direct proportion to the electron density of the aryl moiety (relative rates of rearrangement: 5 > 4 > 10).
Given the promiscuity of the pseudomonine synthetase assembly line for analogs of 2-hydroxybenzoate and N-OH-histamine, we next evaluated if threonine could be substituted by the amino acids serine and cysteine. Indeed, serine was accepted as a substrate both when salicylate and DHB are acyl donors, and likewise is converted to the stable isoxazolidinone. The serine product, 6, was then targeted for synthesis to positively confirm the structural assignment of the isoxazolidinone scaffold in this desmethyl acinetobactin congener (as cycloserine is commercially available). The synthesis of 6 was completed in four steps and provided material identical to that prepared enzymatically (as indicated by 1H NMR and HPLC analysis, see supporting information).
The use of cysteine as a substrate revealed that the PmsDEG assembly line can make a third known siderophore, anguibactin (2) (Figure 2C). In this case, the initial amide bond between DHB and Cys-S-PmsG is formed, followed by PmsD mediated cyclization, which provides the thiazoline intermediate. Acyl transfer to the nitrogen of N-OH-histamine mediated by the C domain of PmsG then yields anguibactin. Unlike the nascent oxazolinyl N-OH-histamine siderophores that undergo facile intramolecular rearrangement, the thiazolinyl ring of 2 persists in the presence of the hydroxamate group. This may be due to the decreased electrophilicity of Cβ of the thiazoline ring compared to that of the oxazoline ring. Propensity towards rearrangement does not appear to depend on the CH3 substituent at Cβ as both oxazolinyl (from Ser) and methyl-oxazolinyl (from Thr) rearrange to isoxazolidinones 6 and 5, respectively.
Nature commonly generates structural diversity by the differential tailoring of a core scaffold through late stage functional group modification.13 However, a more unusual source of diversification involves the use of different starting monomers to generate new structures. Through in vitro study of this highly dissociated three-component NRPS, we have unmasked a cryptic promiscuity that enables the synthesis of a family of siderophores and siderophore-like products. The products formed include anguibactin and acinetobactin, produced by two pathogens unrelated to Pseudomonas: Vibrio anguillarum and Acinetobacter baumannii, respectively. The inherent capability of these biosynthetic enzymes to produce different molecules depending on substrate availability points to the evolvability of the pseudomonine synthetase.
This work also reassigns the structure of acinetobactin and further underscores the biological relevance of the N-OH-oxazoline to isoxazolidinone internal rearrangement in siderophore diversification. In addition, our study sheds light on functional group compatibility within these densely functionalized natural product small molecule frameworks. The proximal location of hydroxamate and thiazoline for Fe3+ multi-dentate chelation is stable, presumably due to the electronic difference of S vs. O in lowering Cβ electrophilicity in thiazoline vs. oxazoline frameworks.
The rearranged isoxazolidinone scaffold in acinetobactin and pseudomonine is predicted to retain sufficient affinity for ferric iron to provide its producers a selective advantage in iron-limited environments. Hydroxamate and oxazoline functional groups are compatible in siderophore scaffolds if they are distal, and not prone to intramolecular reaction in a five membered transition state, as shown by the structure of the Mycobacterium tuberculosis siderophore mycobactin.14 Because acquisition of iron is such a central survival function, the combinatorial evolution and shuffling of pathogenic bacterial siderophore assembly lines reveals how highly functionalized small molecule chemical manifolds are explored and functionally diversified.
Supplementary Material
Acknowledgement
This work was supported by NIH Grant AI47238 (C.T.W.); Elizabeth Sattely is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1980-08).
Footnotes
Supporting Information Available: Supplemental figures, experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fischbach MA, Lin H, Liu DR, Walsh CT. Nature Chem. Biol. 2006;2:132. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 2.Crosa JH, Walsh CT. Microbiol. Mol. Biol. Rev. 2002;66:223. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haas H, Eisendle M, Turgeon BG. Ann. Rev. Phytopathology. 2008;46:149. doi: 10.1146/annurev.phyto.45.062806.094338. [DOI] [PubMed] [Google Scholar]
- 4.Raymond KN, Dertz EA, Kim SS. Proc. Natl. Acad. Sci. 2003;100:3584. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters G, Keberle H, Schmid K, Brunner H. Biochem. Pharmacol. 1966;15:93. doi: 10.1016/0006-2952(66)90114-6. [DOI] [PubMed] [Google Scholar]
- 6.Cox CD, Rinehart KL, Jr., Moore ML, Cook JC., Jr. Proc. Natl. Acad. Sci. 1981;78:4256. doi: 10.1073/pnas.78.7.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffiths GL, Sigel SP, Payne SM, Neilands JB. J. Biol. Chem. 1984;259:383. [PubMed] [Google Scholar]
- 8.Haag H, Hantke K, Drechsel H, Stojiljkovic I, Jung G, Zahner H. J. Gen. Microbiol. 1993;139:2159. doi: 10.1099/00221287-139-9-2159. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto S, Okujo N, Sakakibara Y. Arch. Microbiol. 1994;162:249. doi: 10.1007/BF00301846. [DOI] [PubMed] [Google Scholar]
- 10.Actis LA, Fish W, Crosa JH, Kellerman K, Ellenberger SR, Hauser FM, Sanders-Loehr J. J. Bacteriology. 1986;167:57. doi: 10.1128/jb.167.1.57-65.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh CT. ChemBioChem. 2003;3:124–134. [Google Scholar]
- 12.Sattely ES, Walsh CT. J. Am. Chem. Soc. 2008;130:12282. doi: 10.1021/ja804499r. [DOI] [PubMed] [Google Scholar]
- 13.Samel SA, Marahiel MA, Essen L-O. Mol. BioSyst. 2008;4:367. doi: 10.1039/b717538h. [DOI] [PubMed] [Google Scholar]
- 14.Barclay R, Ewing DF, Ratledge C. J. Bacteriol. 1985;164:896. doi: 10.1128/jb.164.2.896-903.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.