

Abstract

Ruthenium catalyzed transfer hydrogenation of N-sulfonamido allene 1e in the presence of aromatic aldehydes 2a-2f, α,β-unsaturated aldehydes 2g-2i and aliphatic aldehydes 2j-2l results in reductive coupling to furnish anti-aminoallylation products 3a-3l. Reductive coupling of allenamide 1e to aldehyde 2a conducted using d8-isopropanol as terminal reductant delivers deuterio-3a. The observed pattern of deuterium incorporation suggests reversible allene hydrometallation with incomplete regioselectivity in advance of carbonyl addition. A survey of mono-substituted allenes 1f-1i was conducted. High levels of anti-diastereoselectivity only are observed using tert-butyl allene 1f. This protocol represents an alternative to the use of amino-substituted allylborane reagents in carbonyl amino-allylation and avoids the use of stoichiometric metallic reagents.
Classical protocols for the addition of non-stabilized carbanions to carbonyl compounds and imines rely upon the use of preformed organometallic reagents. Recent studies from our laboratory demonstrate that simple unsaturates (alkenes, alkynes and allenes) serve as non-stabilized carbanion equivalents under the conditions of hydrogenation and transfer hydrogenation.1 This concept has evoked a diverse set of methods for catalytic carbonyl vinylation,2,3 allylation,4,5 propargylation6 and aldol addition.7 Unlike their classical counterparts, such hydrogenative carbonyl additions occur under essentially neutral conditions, avoid generation of stoichiometric metallic byproducts and, in certain cases, may be conducted directly from the alcohol oxidation level.1c,2f,4b-f,5a,b,6
Whereas diastereo- and enantioselective carbonyl allylation and crotylation is achieved under the conditions of iridium catalyzed transfer hydrogenation,4b-f related ruthenium catalyzed allylations lack stereocontrol.1c,5 Here, we report that sulfonamido-allenes engage aldehydes in highly anti-diastereoselective reductive addition to deliver vinyl-substituted 1,2-amino alcohols.8-12 This process represents a new functional group interconversion and an alternative to the use of amino-substituted allylborane reagents in carbonyl amino-allylation.12
Initial studies focused on the reductive coupling of sulfonamido allenes 1a-1e, to p-nitrobenzaldehyde 2a. A range of ruthenium catalysts were assayed: Ru(O2CCF3)2(CO)(PPh3)2, RuHCl(CO)(PPh3)3, RuH2(CO)(PPh3)2, RuCl2(CO)2(PPh3)2 and RuBr(η3-C3H5)(CO)3. In accord with earlier studies on the reductive coupling of 1,1-disubstituted allenes to aldehydes,5c RuBr(η3-C3H5)(CO)3 was unique in its ability to catalyze C-C bond formation. Yet in stark contrast to earlier observations, substantial levels of anti-diastereocontrol were observed (Table 1 entries 1-5). Indeed, using allenamide 1e, which incorporates p-nitrobenzenesulfonyl and 2,4-dimethoxybenzyl protecting groups, aldehyde 2a is converted to the 2-sulfonamido-homoallyl alcohol 3a in 91% isolated yield with complete regio- and anti-diastereoselectivity, as determined by 1H NMR and single crystal X-ray diffraction analysis (Table 1 entry 5).
Table 1.
anti-Diastereoselective aminoallylation of aldehydes via ruthenium catalyzed transfer hydrogenative coupling of N-substituted allenes 1a-1ea
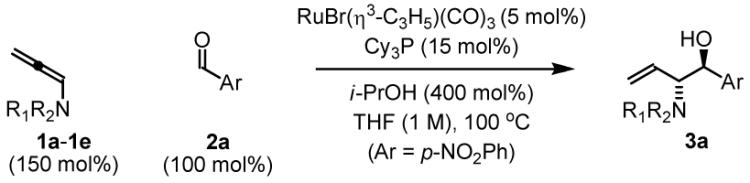 | ||||
|---|---|---|---|---|
| Entry | Allene | R1 | R2 | Yield 3a (dr) |
| 1 | 1a | p-Toluenesulfonyl | Benzyl | 92% (5:1) |
| 2 | 1b | Phthalimido | --- | 37% (3:1) |
| 3 | 1c | Boc | Benzyl | 71% (8:1) |
| 4 | 1d | o-Nitrobenzenesulfonyl | Benzyl | 59% (≥ 20:1) |
| 5 | 1e | p-Nitrobenzenesulfonyl | 2,4-Dimethoxybenzyl | 91% (≥ 20:1) |
In all cases, cited yields are of isolated material. See Supporting Information for detailed experimental procedures.
To explore the scope of this process, allenamide 1e was coupled to structurally diverse aldehydes 2a-2l. Aromatic aldehydes 2a-2f are transformed to adducts 2a-2f as single diastereomers, α,β-unsaturated aldehydes 2g-2i are transformed to adducts 3g-3i as single diastereomers, and aliphatic aldehydes bearing α-heteroatoms 2j-2k are converted to anti-aminoallylation products 3j-3k in good yield and with complete anti-diastereocontrol. Finally, as demonstrated by the conversion of 2l to 3l, simple unactivated aliphatic aldehydes engage in highly anti-diastereoselective reductive coupling (Table 2). In general, it was found that conversion improves upon use of more electrophilic aldehydes. For less activated aldehydes, higher loadings of allene 1e (200 mol%) are required to enforce high conversion. To explore the utility of the aminoallylation products, adduct 3j was converted to the fully protected nonproteinogenic amino acids 4b and 4c. Notably, the p-nitrobenzenesulfonyl and 2,4-dimethoxybenzyl protecting groups are subject to removal under mild conditions (Scheme 1).
Table 2.
Ruthenium catalyzed transfer hydrogenative coupling sulfonamido-allene 1e to aldehydes 2a-2la
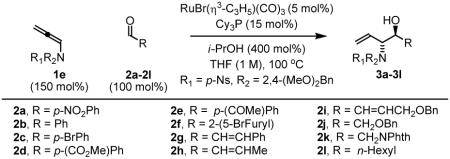 | ||
|---|---|---|
| Coupling to Aryl Aldehydes | ||
 91% Yield, 3a |
 70% Yield, 3bb |
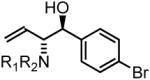 77% yield, 3c |
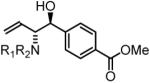 94% yield, 3d |
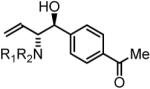 74% Yield, 3e |
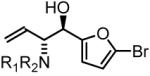 90% Yield, 3f |
| Coupling to Enals | ||
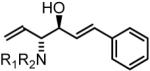 61% Yield, 3gb |
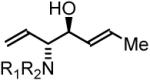 63% Yield, 3hb |
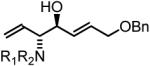 65% Yield, 3ib |
| Coupling to Aliphatic Aldehydes | ||
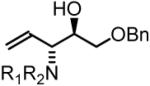 77% Yied, 3j |
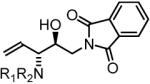 85% Yield, 3k |
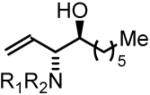 69% Yield, 3l |
In all cases, cited yields are of isolated material and represent the average of two runs. In each case, >20:1 anti-diastereoselectivity is observed, as determined by 1H NMR analysis. See Supporting Information for detailed experimental procedures.
Two equivalents of allene 1e were used.
Scheme 1.
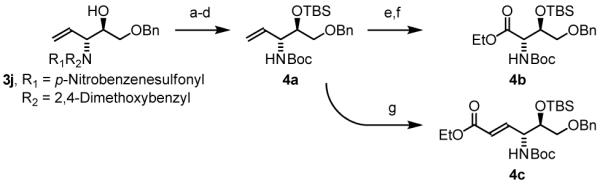
Elaboration of aminoallylation product 3j to nonproteinogenic amino acid esters 4b and 4c.a
Reagents: (a) TFA, PhMe-PhOMe, 25 °C, 89% Yield. (b) TBSOTf, 2,6-Lutidine, DCM, 25 °C, 88% Yield. (c) PhSH, Cs2CO3, MeCN, 50 °C. (d) Boc2O, MeCN, 25 °C, 79% Yield Over 2 Steps. (e) NaIO4, RuCI3(H2O) (5 mol%), MeCN-CCI4-H2O, 25 °C. (f) TMSCHN2, CHCI3-MeOH, 25 °C, 67% Yield Over 2 Steps. (g) MeO2CCH=CH2, Hoveyda-Grubbs-II (5 mol%), DCM, 40 °C, 91% Yield, 20:1 Z:E.
aIn all cases, cited yields are of isolated material. See Supporting Information for detailed experimental procedures.
One possible model to account for the observed branch-regioselectivity and anti-diastereoselectivity involves regio- and stereoselective allene hydrometallation at the π-face distal and opposite to the sulfonamido moiety to provide the primary (Z)-σ-allylruthenium intermediate. Internal chelation to the sulfonamido oxygen13 may stabilize the kinetic (Z)-σ-allyl haptomer, which must then engage the aldehyde through a boat-like transition structure. Alternatively, the kinetic (Z)-σ-allyl haptomer may isomerize to the (E)-σ-allyl haptomer, which must then engage the aldehyde through a chair-like transition structure.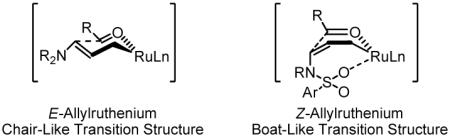
To gain further mechanistic insight, and potentially discriminate between the aforementioned reaction pathways, the coupling of allenamide 1e to aldehyde 2a was conducted using d8-isopropanol as terminal reductant. The product, deuterio-3a, incorporates deuterium at the internal vinylic position (29%) and terminal vinylic positions (9% and 7%). These data suggest reversible allene hydrometallation with incomplete regioselectivity in advance of carbonyl addition.14 Finally, a series of alkyl-substituted allenes 1f-1i were coupled to aldehyde 2a under standard conditions to deliver adducts 3m-3p. Notably, high levels of anti-diastereoselectivity only are observed using tert-butyl allene 1f, as corroborated by single crystal X-ray diffraction analysis of 3m. These data reveal that internal chelation to the sulfonamido oxygen13 is not required for high levels of anti-diastereoselectivity, corroborate intervention of the (E)-σ-allyl haptomer and a chair-like transition structure.
In summary, we report an anti-diastereoselective reductive coupling of sulfonamido-allenes and aldehydes under the conditions of ruthenium catalyzed transfer hydrogenation. This protocol circumvents the use of stoichiometric metallic reagents in carbonyl aminoallylation and represents the first stereocontrolled C-C bond forming hydrogenation based on a ruthenium catalyst. Enantioselective variants of this processes are currently under investigation.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation and the NIH-NIGMS (RO1-GM069445) for partial support of this research. Eduardas Skucas thanks the Livingston Endowment for a graduate fellowship.
References
- (1).For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see:Ngai M-Y, Kong JR, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- (2).For hydrogenative and transfer hydrogenative carbonyl vinylation employing non-conjugated alkynes as vinyl donors, see:Rhee J-U, Krische MJ. J. Am. Chem. Soc. 2006;128:10674. doi: 10.1021/ja0637954.Skucas E, Kong JR, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896.Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432. doi: 10.1021/ja073018j.Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e.Han SB, Kong J-R, Krische MJ. Org. Lett. 2008;10:4133. doi: 10.1021/ol8018874.Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja809456u. On-line at JACS-ASAP.
- (3).For hydrogenative carbonyl vinylation employing 1,3-enynes and 1,3-diynes as vinyl donors, see:Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2004;126:4664. doi: 10.1021/ja0316566.Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269. doi: 10.1021/ja051104i.Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l.Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027.Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745. doi: 10.1021/ol7015548.
- (4).For carbonyl allylation via iridium catalyzed hydrogenative and transfer hydrogenative coupling of dienes, allenes and allyl acetate, see:Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u.Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b.Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han S-B, Krische MJ. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja808857w. On-line at JACS-ASAP.
- (5).For carbonyl allylation via ruthenium catalyzed transfer hydrogenative coupling of dienes and allenes, see:Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x.Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j.Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v.
- (6).For carbonyl propargylation via ruthenium catalyzed transfer hydrogenative coupling of 1,3-enynes, see:Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359.
- (7).For intermolecular aldol and Mannich addition via rhodium catalyzed hydrogenative coupling of enones, see:Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2002;124:15156. doi: 10.1021/ja021163l.Jung C-K, Garner SA, Krische MJ. Org. Lett. 2006;8:519. doi: 10.1021/ol052859x.Jung C-K, Krische MJ. J. Am. Chem. Soc. 2006;128:17051. doi: 10.1021/ja066198q.Han SB, Krische MJ. Org. Lett. 2006;8:5657. doi: 10.1021/ol0624023.Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843. doi: 10.1021/jo070779w.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2747. doi: 10.1021/ja710862u.
- (8).For a review on the use of allenamides in organic synthesis, see:Wei L-L, Xiong H, Hsung RP. Acc. .Chem. Res. 2003;36:773. doi: 10.1021/ar030029i.
- (9).For Ni-catalyzed allene-carbonyl reductive coupling, see:Ng S-S, Jamison TF. J. Am. Chem. Soc. 2005;127:7320. doi: 10.1021/ja0521831.Ng S-S, Jamison TF. Tetrahedron. 2005;61:11405.Ng S-S, Jamison TF. Tetrahedron. 2006;62:11350.(d) Also see reference 11a.
- (10).For intermolecular metal catalyzed alkylative allene-carbonyl 3-component coupling:Anwar U, Grigg R, Rasparini M, Savic V, Sridharan V. Chem. Commun. 2000:645.Takimoto M, Kawamura M, Mori M. Org. Lett. 2003;5:2599. doi: 10.1021/ol034480l.Hopkins CD, Malinakova HC. Org. Lett. 2004;6:2221. doi: 10.1021/ol0492795.Hopkins CD, Guan L, Malinakova HC. J. Org. Chem. 2005;70:6848. doi: 10.1021/jo050886v.Hopkins CD, Malinakova HC. Synthesis. 2007:3558.
- (11).For intramolecular metal catalyzed alkylative allene-carbonyl 3-component coupling:Chevliakov MV, Montgomery J. J. Am. Chem. Soc. 1999;121:11139.Montgomery J, Song M. Org. Lett. 2002;4:4009. doi: 10.1021/ol026670m.Kang S-K, Yoon S-K. Chem. Commun. 2002:2634. doi: 10.1039/b207620a.Ha Y-H, Kang S-K. Org. Lett. 2002;4:1143. doi: 10.1021/ol025557t.Song M, Montgomery J. Tetrahedron. 2005;61:11440.
- (12).For carbonyl amino-allylation employing amino-substituted allylborane reagents, see:Barrett AGM, Seefeld MA. J. Chem. Soc., Chem. Commun. 1993:339.Barrett AGM, Seefeld MA. Tetrahedron. 1993;49:7857.Barrett AGM, Seefeld MA, Williams DJ. J. Chem. Soc., Chem. Commun. 1994:1053.Barrett AGM, Seefeld MA, White AJP, Williams DJ. J. Org. Chem. 1996;61:2677. doi: 10.1021/jo9522062.
- (13).Trost BM, Ferreira EM, Gutierrez AC. J. Am. Chem. Soc. 2008;130:16176. doi: 10.1021/ja8078835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For related observations on ruthenium mediated allene hydrometallation, see:Bai T, Xue P, Sung HH-Y, Williams I, Ma S, Lin Z, Jia G. Organometallics. 2007;26:5581.Bai T, Ma S, Jia G. Coord. Chem. Rev. 2009;253:423.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.