

Abstract
The BCR-ABL oncogene encodes an in-frame fusion protein containing NH2-terminal sequences derived from Bcr and COOH-terminal sequences derived from Abl. Bcr contains a centrally located RhoGEF domain that is retained within p210 Bcr-Abl. Although this domain is subject to autoinhibition in the context of Bcr, here we show that it is constitutively activated in p210 Bcr-Abl. p210 Bcr-Abl can stimulate RhoA activation independently of its tyrosine kinase activity, and mutations within the RhoGEF domain that are predicted to eliminate RhoGEF activity inhibit RhoA activation. The RhoGEF mutant of p210 Bcr-Abl does not affect the tyrosine kinase activity of the molecule, nor the ability of p210 Bcr-Abl to interact with XPB through the RhoGEF domain. Despite retaining normal levels of tyrosine kinase activity, the RhoGEF mutant of p210 Bcr-Abl is impaired in transforming activity as measured by anchorage-independent growth. However, the mutant is still able to confer the phenotype of growth factor independence in myeloid cells, suggesting that some, but not all parameters of p210 Bcr-Abl transformation are dependent upon a catalytically active RhoGEF domain. Collectively, these observations identify a gain-of-function activity attributable to the RhoGEF domain of p210 Bcr-Abl that is required to support the transformed phenotype.
Keywords: Chronic myelogenous leukemia, p210 Bcr-Abl, RhoA, RhoGEF domain, transformation
Introduction
Over 95% of patients with chronic myelogenous leukemia (CML) express an oncogenic fusion protein (p210 Bcr-Abl) that contains NH2-terminal sequences from Bcr and COOH-terminal sequences from Abl (Shtivelman et al., 1985). Although the majority of aberrant cellular activities associated with the expression of p210 Bcr-Abl can be either directly, or indirectly, attributed to a deregulated tyrosine kinase activity that resides within Abl (Lugo et al., 1990), several independent studies have established that sequences that reside within the first exon of Bcr are also required to support transforming activity (Muller et al., 1991; Pendergast et al., 1991; Pendergast et al., 1993). Loss of these sequences is generally associated with diminished tyrosine kinase activity suggesting that one function of the Bcr-encoded sequences is to directly regulate the catalytic activity of Abl.
Centrally located within Bcr (residues 501–870) is a tandem DH/PH domain that is shared by all members of the Rho-specific guanine nucleotide exchange factors (RhoGEF) (Whitehead et al., 1997), and is retained in p210 Bcr-Abl. RhoGEFs activate members of the Rho family of small GTPases by catalyzing the exchange of GDP for GTP (Whitehead et al., 1997). The DH domain represents the catalytic core of RhoGEFs and, consistent with this, a fragment containing the isolated DH domain of Bcr has been shown, in vitro, to catalyze exchange on Rac1, RhoA and Cdc42 (Chuang et al., 1995). We have confirmed this exchange activity for Cdc42 in an in vivo affinity complex assay, but we do not see activation of RhoA and Rac1 (Korus et al., 2002).
Despite the fact that several studies indicate that the isolated RhoGEF domain of Bcr is biologically active, its fate in the context of p210 Bcr-Abl is unclear. In this current study we have introduced point mutations into the RhoGEF domain of Bcr that impair RhoGEF activity. Using these mutations we have identified a RhoGEF activity specific for RhoA in the context of p210 Bcr-Abl and determined that this activity is independent of the Abl-encoded tyrosine kinase activity. Additionally we have been able to demonstrate that the RhoGEF activity is required for some, but not all, parameters of p210 Bcr-Abl transformation.
Results
p210 Bcr-Abl activates Cdc42 in a tyrosine kinase-independent manner
In the context of full-length Bcr the RhoGEF activity is negatively regulated by flanking autoinhibitory sequences (Korus et al., 2002). Since one of these autoinhibitory domains is lost in the chimeric p210 Bcr-Abl molecule, we wondered whether this was sufficient to activate RhoGEF function. For this analysis we constructed mammalian expression constructs that contain full-length, HA-epitope-tagged p210 Bcr-Abl as well as the tyrosine kinase-inactive mutant. Both constructs were expressed in Cos-7 cells, and activated Cdc42 was measured by affinity precipitation assays as we have previously described (Korus et al., 2002). Both p210 Bcr-Abl and p210 Bcr-Abl(KD) were able to activate Cdc42 (Figure 1) to equivalent levels. These results indicate that p210 Bcr-Abl can activate Cdc42 in a tyrosine kinase-independent manner, and suggest that the RhoGEF domain may not be subject to autoinhibition in the context of p210 Bcr-Abl.
Figure 1.

p210 Bcr-Abl activates Cdc42 in a tyrosine kinase-independent manner. Cos-7 cells were transiently transfected with the indicated cDNAs. Lysates were collected at 48 hr and examined by affinity precipitation assays for levels of total (Total-Cdc42), and activated (GTP-Cdc42) Cdc42. Relative levels of activated GTPases were determined by densitometry (lower graphs) and all experiments were performed a minimum of three times.
Identification of residues in the RhoGEF domain of Bcr that are required for Cdc42 activation
A comparison of the DH domain of Bcr with the DH domains of other family members for which structural data were available revealed three residues that are predicted to be solvent exposed, and necessary for substrate interactions (T651, R652 and L655). To examine the contribution of these residues to Bcr RhoGEF activity they were mutated in the context of a catalytically active RhoGEF fragment (Bcr(491–876)) (Korus et al., 2002). In addition to substituting residues for alanines a mutant was constructed in which each residue was substituted for charged lysines. Initially both mutants were expressed in Cos-7 cells to verify stable and equal expression (Figure 2a). Each mutant was then examined for their ability to activate a luciferase-based NF-κB responsive reporter (Figure 2b). Consistent with our previous studies (Korus et al., 2002), the isolated RhoGEF domain of Bcr activated this reporter by 60-fold relative to cognate vector. Whereas the triple alanine mutant still retained 50% of wild-type activity, the triple lysine mutant was completely impaired in its ability to activate the reporter. This latter mutant was designated Bcr(491–876:RD) and selected for further analysis.
Figure 2.

Identification of residues in the RhoGEF domain of Bcr that are required for Cdc42 activation. (A) Equal expression of RhoGEF mutants was demonstrated by Western blot in Cos-7 cells. (B) Activation of an NF-κB responsive promoter element by RhoGEF mutants of Bcr. Cos-7 cells were transiently transfected with the indicated Bcr derivatives along with a luciferase reporter plasmid to determine stimulation of NF-κB-mediated transcriptional activity (NF-κB-luc), and pCMVnlac as an internal control for transfection efficiency. Data shown are representative of at least three independent assays performed on duplicate plates. The error bars indicate standard deviations. (C) The Bcr(491–876:RD) mutant is impaired in Cdc42-specific exchange activity. Cos-7 cells were transiently transfected with the indicated cDNAs. Lysates were collected at 48 hours and examined by affinity precipitation assays for relative levels of activated Cdc42. Assays were performed and data represented as described in the legend of Figure 1.
To further characterize the Bcr(491–876:RD) mutant, we then examined it for its ability to activate Cdc42 in an affinity precipitation assay. Bcr(491–876) and cognate vector were included in the analysis as positive and negative controls respectively. Consistent with the NF-κB data we observed that the Bcr(491–876:RD) mutant is substantially impaired (> 75%) in its ability to activate Cdc42 (Figure 2c).
The Bcr(RD) mutant is structurally intact
Although the residues that were substituted in the RhoGEF domain of Bcr are predicted to be solvent exposed, we were concerned that the substitutions may affect the overall structural integrity of the RhoGEF domain. To address this issue we introduced the mutations into full-length Bcr in the context of a yeast 2-hybrid expression vector (pGBT9-bcr(RD)) and examined binding with known Bcr binding partners. In previous studies it has been shown that yeast 2-hybrid analysis can be used to demonstrate an interaction between full-length Bcr and either XPB or c-Myc (Mahon et al., 2003; Takeda et al., 1999). Additionally, we have observed that ubiquitin binds to the NH2-terminus of Bcr (G.M. unpublished observations). When tested in yeast 2-hybrid analysis full-length Bcr and the Bcr(RD) mutant bind equally well to full-length XPB, c-Myc and ubiquitin (Figure 3a), thus suggesting that the substitutions do not affect the overall fold of the Bcr protein.
Figure 3.
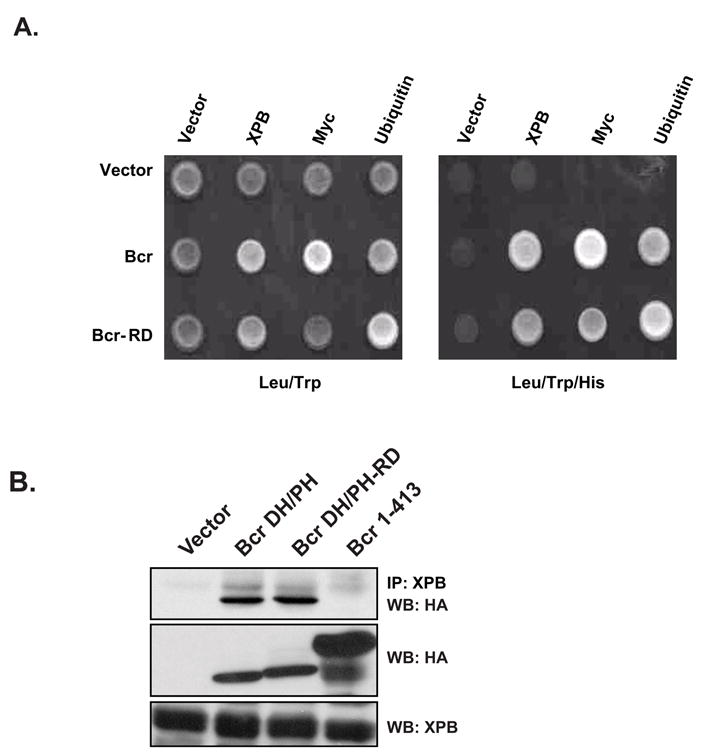
The Bcr(RD) mutant is structurally intact. (A) The Bcr(RD) mutant binds to XPB, ubiquitin and c-Myc in a yeast 2-hybrid analysis. The PJ694A yeast reporter strain was transfected with the indicated combinations of plasmids. The yeast colonies that grew on plates lacking leucine and tryptophan (Leu/Trp) were examined for growth on histidine deficient plates (Leu/Trp/His). Interactions between proteins are demonstrated by the ability to activate the his3 reporter gene. (B) Bcr(RD) interacts with XPB in mammalian cells. IP indicates antibody used in immunoprecipitations and WB indicates antibody used in a western blot to detect an interaction. Cos-7 cells were co-transfected with the indicated combinations of plasmids. Lysates were collected at 48 hr and examined by western blot for expression of XPB (middle panel) or Bcr (lower panel). Immunoprecipitations were then performed with an anti-XPB antibody to detect an interaction (upper panel).
As a second approach we performed co-immunoprecipitations in mammalian cells. For this analysis Cos-7 cells were transiently transfected with mammalian expression vectors that contain HA-epitope-tagged Bcr(491–876), or the Bcr(491–876;RD) mutant. An NH2-terminal fragment of Bcr that lacks the XPB binding site (Bcr(1–413)) was included as a negative control for the immunoprecipitation. Using this approach we were readily able to detect an interaction between XPB and either Bcr(491–876), or Bcr(491–876;RD), but not Bcr(1–413) (Figure 3b). This further confirms the structural integrity of the Bcr(491–876;RD) mutant.
The RhoGEF activity of Bcr is not required to support p210 Bcr-Abl tyrosine kinase activity
In order to examine the activity and fate of the Bcr RhoGEF domain in the context of Bcr-Abl, we introduced the lysine substitutions into full-length HA-epitope-tagged p210 Bcr-Abl (p210 Bcr-Abl(RD)). Initially the p210 Bcr-Abl(RD) cDNA was cloned into the pAX142 mammalian expression vector and then transiently expressed in Cos-7 cells. Cell lysates were then collected and examined by Western blot with an anti-HA antibody. The blots were also examined with an antibody that specifically recognizes an autophosphorylated tyrosine residue within the Abl domain of p210 Bcr-Abl, and an antibody that recognizes phosphorylated CrkL (Tyr207, Cell Signaling), a known substrate for p210 Bcr-Abl transkinase activity (Figure 4a) (ten Hoeve et al., 1994). Both p210 Bcr-Abl and p210 Bcr-Abl(RD) showed equivalent levels of expression, autokinase activity, and transkinase activity suggesting that the RhoGEF activity of Bcr is not required to support the tyrosine kinase activity of p210 Bcr-Abl. To confirm that the mutant is still able to interact with XPB, we also performed a co-immunoprecipitation in Cos-7 cells (Figure 4b). Using this approach we were readily able to detect an interaction between endogenous XPB and either p210 Bcr-Abl of p210 Bcr-Abl(RD).
Figure 4.
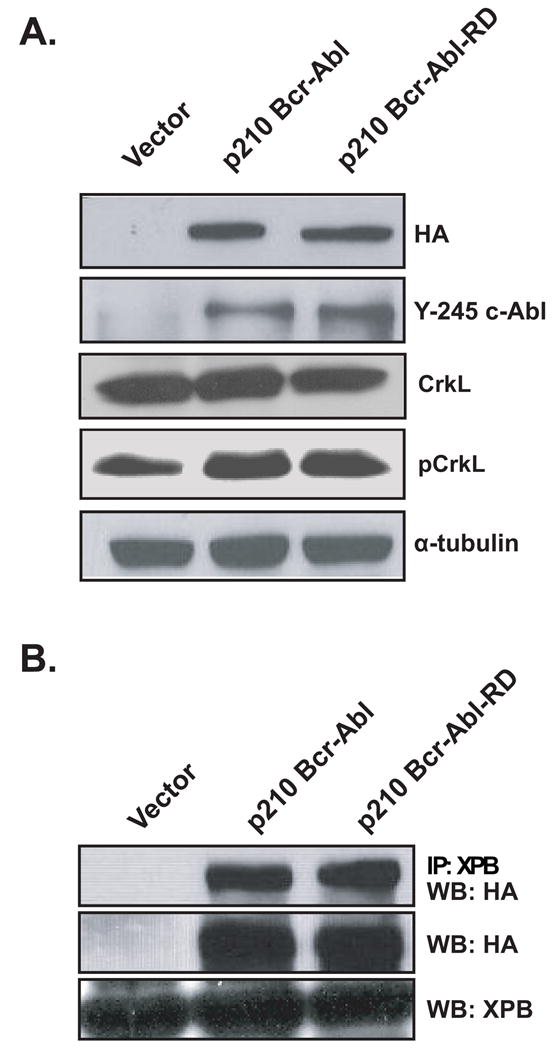
The RhoGEF activity of Bcr is not required to support p210 Bcr-Abl tyrosine kinase or XPB-binding activity. Cos-7 cells were transiently transfected with the indicated combinations of constructs. (A) At 48 hours post-transfection lysates were collected and examined by western blot for expression (HA), autophosphorylation (Y-245), and transphosphorylation (p-CrkL) of the p210 Bcr-Abl derivatives. Data shown are representative of three independent experiments. (B) Lysates were collected at 48 hr and examined by western blot for expression of XPB (middle panel) or Bcr (lower panel). Immunoprecipitations were then performed with an anti-XPB antibody to detect an interaction (upper panel).
The p210 Bcr-Abl(RD) mutant is impaired in RhoA activation
Next we determined whether the ability of p210 Bcr-Abl to activate RhoGTPases is dependant on the catalytic activity of the RhoGEF domain. Thus, we compared the ability of p210 Bcr-Abl and p210 Bcr-Abl(RD) to activate Cdc42 using in vivo affinity precipitation assays (Figure 5). Since it has been previously reported that p210 Bcr-Abl can activate RhoA and Rac1, these GTPases were also included in our analysis (Harnois et al., 2003; Skorski et al., 1998). Surprisingly, the p210 Bcr-Abl(RD) mutant was only impaired in RhoA activation. We conclude that the RhoGEF domain of p210 Bcr-Abl is constitutively active in Cos-7 cells, and exhibits exchange activity that is specific for RhoA.
Figure 5.

The p210 Bcr-Abl(RD) mutant is impaired in RhoA activation. Cos-7 cells were transiently transfected with the indicated cDNAs. Lysates were collected at 48 hr and examined by affinity precipitation assays for levels of total and activated (A) RhoA, (B) Cdc42, and (C) Rac1. Assays were performed and data represented as described in the legend of Figure 1.
The RhoGEF activity of p210 Bcr-Abl is required for transformation of Rat1 cells
The phenotype of p210 Bcr-Abl expressing cells includes anchorage- and growth factor-independent growth (Daley & Baltimore, 1988; Lugo & Witte, 1989). Since such properties have also been attributed to deregulated Rho expression, we wondered whether the RhoGEF activity of p210 Bcr-Abl is required for the transformed phenotype. For this analysis cDNAs encoding p210 Bcr-Abl and the RD mutations were introduced into the MSCV-GFP retroviral vector. High titer retroviral stocks were then generated using Phoenix ecotropic packaging cells, and then used to infect Rat1a(mycER) cells. GFP-expressing cells were then selected by FACS and examined for growth in soft agar. When compared to non-infected cells, vector infected cells showed no increase in colony formation while p210 Bcr-Abl cells showed an increase greater than 90% (Figure 6a). In contrast, cells that were infected with the p210 Bcr-Abl(RD) mutant showed only a 20% increase in colony formation relative to vector or non-infected cells suggesting that the mutant is substantially impaired in its ability to induce anchorage-independent growth. Both p210 Bcr-Abl and p210 Bcr-Abl(RD) showed equivalent levels of expression and autokinase activity in the Rat1a(mycER) cells suggesting that the reduced transforming activity was attributable to the RhoGEF mutation (Figure 6b). To determine whether reduced transformation was attributable to increased cell death quantitative assessment of apoptosis was performed by flow cytometry. No differences in apoptosis were observed between cells that express vector, p210 Bcr-Abl or the mutant (supplemental Figure s1).
Figure 6.

The RhoGEF activity of p210 Bcr-Abl is required for anchorage independent but not IL-3 independent growth. Rat1a(mycER) (A and B) or 32D (C and D) cells were infected with retroviral particles that contain MSCV-bcr-abl/p210-IRES-gfp, MSCV-bcr-abl/p210(RD)-IRES-gfp, or cognate vector. (A) At 48 hr post-infection Rat1a(mycER) cells that express GFP were sorted by FACS and examined for growth in soft agar as described in methods. Colonies were counted on day 14 and data are represented as total number of colonies per dish. (B) Lysates were collected and examined by western blot for expression and autokinase activity. (C) At 48 hr post-infection 32D cells that express GFP were sorted by FACS and then seeded (1 × 106 cells per 3.5 cm dish) in media with and without 10% IL-3. Cells were counted on day 3 using a Vi-cell analyzer (Beckman Coulter). (D) Lysates were collected and examined by western blot for expression and autokinase activity. All data shown are the average of three independent experiments.
The RhoGEF activity of p210 Bcr-Abl is not required for growth factor independence, in myeloid cells
Since 32D cells that express p210 Bcr-Abl exhibit IL-3 independent growth (Daley & Baltimore, 1988), we determined whether the RhoGEF domain also contributes to this phenotype. Thus, our high titer retroviral stocks were also used to infect 32D cells. GFP-expressing cells were selected by FACS and examined for growth in the presence or absence of IL-3 (Figure 6c), as well as expression and autokinase activity (Figure 6d). As expected cells infected with cognate vector were unable to proliferate in the absence of IL-3. In contrast cells that express p210 Bcr-Abl or the p210 Bcr-Abl(RD) mutant both grow in the absence of IL-3 and achieve cell densities of approximately 50% of cells cultured with Il-3 alone. Thus, whereas the RhoGEF activity is required for anchorage-independent growth, it is not required for growth factor independence.
Discussion
Several independent studies have established that kinase inactive mutants of p210 Bcr-Abl are able to induce abnormalities in hematopoietic cells. Thus, both p210 Bcr-Abl and a kinase-inactive mutant are able to increase adhesion between 32D cells and fibronectin (Wertheim et al., 2002), and defects in migration and adhesion are improved, but not reversed in CD34+ primary cells that express the kinase-inactive p210 Bcr-Abl (Ramaraj et al., 2004). These observations may be important for the pathogenesis of CML since altered motility and adhesion to extracellular matrix proteins are likely to contribute to the aberrant release of CML cells from the bone marrow. These observations also suggest that p210 Bcr-Abl may encode kinase-independent activities that are required for transformation, but are not sufficient to initiate CML. Since over 70% of p210 Bcr-Abl is in complex with actin (McWhirter & Wang, 1991), and mutations that impair this interaction are able to completely repair the defects in adhesion and motility associated with p210 Bcr-Abl expression (Renshaw et al., 1995), it is likely that these additional activities are related to the function of p210 Bcr-Abl in this cellular compartment. In the current study we have determined that the RhoGEF domain of Bcr is constitutively activated in the context of p210 Bcr-Abl, and that the RhoGEF and tyrosine kinase activities of p210 Bcr-Abl are mutually independent of each other. In addition, we have identified RhoA as at least one target for the RhoGEF activity. Since members of the Rho family, including RhoA, have been best described for their role in modifying the actin cytoskeleton (Ridley, 1995), our observations may account for some of the defects in cytoskeletal function that are associated with p210 Bcr-Abl expression (Salgia et al., 1997).
Although previous in vitro studies using the isolated RhoGEF fragment of Bcr revealed that it could utilize Cdc42, RhoA, and Rac1 as substrates (Chuang et al., 1995), substrate utilization is restricted to Cdc42 when the fragment is expressed in Cos-7 cells (Korus et al., 2002). Although we and others have shown that p210 Bcr-Abl can also activate Cdc42 in cell-based assays (Harnois et al., 2003), this activity cannot be attributed to the RhoGEF domain. Others have shown that p190 Bcr-Abl, which lacks the RhoGEF domain, can activate Cdc42 to the same extent as p210 Bcr-Abl (Harnois et al., 2003), and in the current study we show that the p210 Bcr-Abl(RD) mutant is not impaired in Cdc42 activation. Thus, the isolated RhoGEF domain of Bcr appears to be activating Cdc42 through a different mechanism than p210 Bcr-Abl. One possibility is that p210 Bcr-Abl is activating Cdc42 indirectly through its association with Vav. Others have shown that p210 Bcr-Abl can activate Rac1 (Skorski et al., 1998, 2002; Harnois et al., 2003), and it is thought that this occurs through its ability to interact with, and activate, Vav (Bassermann et al., 2002; Harnois et al., 2003). Vav is a RhoGEF family member that can activate both Rac1 and Cdc42 if phosphorylated by Src family tyrosine kinases (Han et al., 1997). Vav can bind directly to Abl sequences and is phosphorylated on tyrosine by p210 Bcr-Abl (Bassermann et al., 2002).
Although p210 Bcr-Abl can activate Rac1, Cdc42 and RhoA (Harnois et al., 2003; Skorski et al., 1998), the only substrate that we have been able to identify for the RhoGEF domain is RhoA. This is consistent with a previous study that showed that p190 Bcr-Abl, which does not contain the RhoGEF domain, is able to activate Rac1 and Cdc42, but not RhoA (Harnois et al., 2003). Since the isolated RhoGEF domain of Bcr does not activate RhoA in Cos-7 cells (Korus et al., 2002), while p210 Bcr-Abl does (current study), substrate utilization by p210 Bcr-Abl appears to be determined by sequences that lie outside this domain. One possibility is that Bcr and p210 Bcr-Abl localize to discrete cellular compartments, and thus, are sequestered with discrete rosters of Rho substrates. We have recently determined that Bcr is a component of the endosomal sorting machinery and colocalizes with endomembranes (Olabisi et al., 2006). In contrast, p210 Bcr-Abl contains a COOH-terminal F-actin binding domain which determines its cellular distribution (McWhirter & Wang, 1993). Since one of the known cellular functions of RhoA is to stimulate actin polymerization, as well as the assembly of actin microfilaments, it is likely that RhoA and p210 Bcr-Abl would have overlapping cellular distributions, and that RhoA may mediate changes in cytoskeletal function that are associated with p210 Bcr-Abl expression (Salgia et al., 1997).
Materials and methods
Molecular constructs
The pAX142 mammalian expression vector along with the pAX142-bcr(1–1271) and pAX142-bcr(491–876) constructs have been previously described (Korus et al., 2002). pAX142-bcr-abl and pAX142-bcr-abl(KD) contain HA-epitope tagged full-length p210 Bcr-Abl, and a tyrosine kinase dead mutant of p210 Bcr-Abl respectively. pAX-bcr(491–876;RD), and pAX142-bcr-abl(RD) encode the isolated RhoGEF domain of Bcr, and full-length p210 Bcr-Abl respectively, with lysine substitutions at residues 651, 652 and 655. The full-length yeast 2-hybrid construct, pGBT9-bcr(1–1271), has been previously described (Olabisi et al., 2006). pGBT9-bcr(1–1271;RD) contains lysine substitutions at residues 651, 652 and 655. The yeast 2-hybrid construct for full-length myc (pGAD-myc), has been previously described (Olabisi et al., 2006). pGAD-xpb and pGAD-ubq contain full-length cDNAs for XPB and ubiquitin respectively. The MSCV-IRES-gfp retroviral vector is as described by the manufacturer (Addgene). MSCV-bcr-abl/p210-IRES-gfp and MSCV-bcr-abl/p210(RD)-IRES-gfp contain full-length p210 Bcr-Abl and the p210 Bcr-Abl RhoGEF mutant respectively. pAX-HA-cdc42 has been previously described (Korus et al., 2002).
Cell culture and reporter assays
Cos-7 cells were maintained in Dulbecco’s modified Eagle medium (DMEM; high glucose) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MI). 32D cells were cultured in RPMI supplemented with 10% fetal bovine serum (Sigma, St. Louis, MI) and 10% interleukin (IL)-3. Rat1a(mycER) cells were cultured in phenol-red free media supplemented with 10% fetal bovine serum (Sigma, St. Louis, MI). The luciferase-coupled transcriptional assays were performed in Cos-7 cells exactly as we have previously described (Korus et al., 2002)
Yeast 2-hybrid analysis
Yeast 2-hybrid analysis was performed using the PJ694A reporter strain as previously described (Olabisi et al., 2006).
Co-immunoprecipitations and GTPase activation assay
Co-immunoprecipitations were performed in Cos-7 cells exactly as we have previously described (Olabisi et al., 2006). Affinity purification assays to measure the levels of endogenous GTP-bound RhoA, Rac1 and Cdc42 were performed using the Rho-binding domain of Rhotekin (GST-C21), and the Cdc42- and Rac-binding domain of Pak (GST-PAK) as previously described (Korus et al., 2002)
Anchorage-independent growth
Rat1a(mycER) cells were infected by retroviral particles that contain MSCV-bcr-abl/p210-IRES-gfp, MSCV-bcr-abl/p210(RD)-IRES-gfp, or cognate vector. At 48 hr post-infection cells that express GFP were sorted by FACS and seeded (2 × 104 cells per 6 cm dish) in 0.3% agar over a base layer of 0.6%. Cells were fed on day 7 with media supplemented with 2 μM β-estradiol to induce c-Myc expression. Colonies were scored visually on day 14.
IL-3 independent growth
32D cells were infected by retroviral particles that encode MSCV-bcr-abl/p210-IRES-gfp, MSCV-bcr-abl/p210(RD)-IRES-gfp, or cognate vector. At 48 hr post-infection cells that express GFP were sorted by FACS and then seeded (1 × 106 cells per 3.5 cm dish) in media with and without 10% IL-3. Cells were counted on day 3 using a Vi-cell analyzer (Beckman Coulter).
Apoptosis assay
Apoptosis in Rat1a(mycER) cells stably expressing MSCV-bcr-abl/p210-IRES-gfp, MSCV-bcr-abl/p210(RD)-IRES-gfp or the cognate vector was analysed using the Annexin V-Biotin Apoptosis Detection Kit (Calbiochem). Quantitative assessment of apoptosis was performed by flow cytometry.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service grant CA097066 (IPW) from the National Cancer Institute and AG04821 (HLO) from the National Institute of Aging. PL Rodriguez and NL Pannucci are recipients of fellowships from the New Jersey Commission for Cancer research
References
- Bassermann F, Jahn T, Miething C, Seipel P, Bai RY, Coutinho S, et al. Association of Bcr-Abl with the proto-oncogene Vav is implicated in activation of the Rac-1 pathway. J Biol Chem. 2002;277:12437–12445. doi: 10.1074/jbc.M112397200. [DOI] [PubMed] [Google Scholar]
- Chuang TH, Xu X, Kaartinen V, Heisterkamp N, Groffen J, Bokoch GM. Abr and Bcr are multifunctional regulators of the Rho GTP-binding protein family. Proc Natl Acad Sci U S A. 1995;92:10282–10286. doi: 10.1073/pnas.92.22.10282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell-line by the Chronic Myelogenous Leukemia-specific p210 Bcr-Abl protein. Proc Natl Acad Sci U S A. 1998;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Das B, Wei W, Van Aelst L, Mosteller RD, Khosravi-Far R, et al. Lck regulates Vav activation of members of the Rho family of GTPases. Mol Cell Biol. 1997;17:1346–1353. doi: 10.1128/mcb.17.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnois T, Constantin B, Rioux A, Grenioux E, Kitzis A, Bourmeyster N. Differential interaction and activation of Rho family GTPases by p210bcr-abl and p190bcr-abl. Oncogene. 2003;22:6445–6454. doi: 10.1038/sj.onc.1206626. [DOI] [PubMed] [Google Scholar]
- Korus M, Mahon GM, Cheng L, Whitehead IP. p38 MAPK-mediated activation of NF-B by the RhoGEF domain of Bcr. Oncogene. 2002;21:4601–4612. doi: 10.1038/sj.onc.1205678. [DOI] [PubMed] [Google Scholar]
- Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- Lugo TG, Witte ON. The Bcr-Abl oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol. 1989;9:1263–1270. doi: 10.1128/mcb.9.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon GM, Wang Y, Korus M, Kostenko E, Cheng L, Sun T, et al. The c-Myc oncoprotein interacts with Bcr. Curr Biol. 2003;13:437–441. doi: 10.1016/s0960-9822(03)00090-3. [DOI] [PubMed] [Google Scholar]
- McWhirter JR, Wang JY. Activation of tyrosine kinase and microfilament-binding functions of c-abl by bcr sequences in bcr/abl fusion proteins. Mol Cell Biol. 1991;11:1553–1565. doi: 10.1128/mcb.11.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter JR, Wang JY. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. Embo J. 1993;12:1533–1546. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Young JC, Pendergast AM, Pondel M, Landau NR, Littman DR, et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabisi OO, Mahon GM, Kostenko EV, Liu Z, Ozer HL, Whitehead IP. Bcr interacts with components of the endosomal sorting complex required for transport-I and is required for epidermal growth factor receptor turnover. Cancer Res. 2006;66:6250–6257. doi: 10.1158/0008-5472.CAN-06-0536. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON. BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell. 1991;66:161–171. doi: 10.1016/0092-8674(91)90148-r. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–85. [PubMed] [Google Scholar]
- Ramaraj P, Singh H, Niu N, Chu S, Holtz M, Yee JK, et al. Effect of mutational inactivation of tyrosine kinase activity on BCR/ABL-induced abnormalities in cell growth and adhesion in human hematopoietic progenitors. Cancer Res. 2004;64:5322–5331. doi: 10.1158/0008-5472.CAN-03-3656. [DOI] [PubMed] [Google Scholar]
- Renshaw MW, McWhirter JR, Wang JY. The human leukemia oncogene bcr-abl abrogates the anchorage requirement but not the growth factor requirement for proliferation. Mol Cell Biol. 1995;15:1286–1293. doi: 10.1128/mcb.15.3.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho-related proteins: actin cytoskeleton and cell cycle. Curr Opin Genet Dev. 1995;5:24–30. doi: 10.1016/s0959-437x(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Salgia R, Li JL, Ewaniuk DS, Pear W, Pisick E, Burky SA, et al. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100:46–57. doi: 10.1172/JCI119520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic melogenous leukaemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- Skorski T, Wlodarski P, Daheron L, Salomoni P, Nieborowska-Skorska M, Majewski M, et al. BCR/ABL-mediated leukemogenesis requires the activity of the small GTP-binding protein Rac. Proc Natl Acad Sci U S A. 1998;95:11858–11862. doi: 10.1073/pnas.95.20.11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Shibuya M, Maru Y. The BCR-ABL oncoprotein potentially interacts with the xeroderma pigmentosum group B protein. Proc Natl Acad Sci U S A. 1999;96:203–207. doi: 10.1073/pnas.96.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hoeve J, Kaartinen V, Fioretos T, Haataja L, Voncken JW, Heisterkamp N, et al. Cellular interactions of CRKL, an SH2–SH3 adaptor protein. Cancer Res. 1994;54:2563–2567. [PubMed] [Google Scholar]
- Wertheim JA, Forsythe K, Druker BJ, Hammer D, Boettiger D, Pear WS. BCR-ABL-induced adhesion defects are tyrosine kinase-independent. Blood. 2002;99:4122–4130. doi: 10.1182/blood.v99.11.4122. [DOI] [PubMed] [Google Scholar]
- Whitehead IP, Campbell S, Rossman KL, Der CJ. Dbl family proteins. Biochim Biophys Acta. 1997;1332:F1–23. doi: 10.1016/s0304-419x(96)00040-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.