

Abstract
The chemokine receptor CXCR4 regulates neuronal survival and differentiation and is involved in a number of pathologies, including cancer and human immunodeficiency virus (HIV). Recent data suggest that chemokines act in concert with neurotransmitters and neuropeptides, such as opioids. This study aimed to determine whether μ-opioid agonists alter the effect of CXCL12 (the specific CXCR4 ligand) on central neurons. Neuronal expression of CXCR4 and μ-opioid receptors (MORs) was analyzed by Western blot, immunostaining, and flow cytometry. Single-cell studies showed that all CXCR4-positive neurons coexpress MORs. Treatment of neuronal cultures with the selective MOR agonist DAMGO or the endogenous peptide endomorphin-1 inhibited intracellular signaling pathways (ERK1/2 and Akt) activated by CXCL12. Furthermore, DAMGO abolished the neuroprotective effect of CXCL12 in N-methyl-d-aspartate (NMDA) neurotoxicity studies. The effects of DAMGO and endomorphin-1 were inhibited by a general or a μ-specific opioid receptor antagonist, and not caused by changes in neuronal CXCR4 levels. DAMGO did not affect CXCL12-induced internalization of CXCR4. The authors propose that interactions between MOR and CXCR4 signaling can modulate the action of CXCL12 on neuronal survival—which may have important implications to neuroAIDS as well as other neuroinflammatory disorders.
Keywords: Akt, chemokine, CXCL12, HIV, neuronal survival, signaling
Introduction
Opioid and chemokine receptors are G-protein–coupled receptors playing important roles in the immune and nervous system. Both groups of receptors and their ligands are widely expressed in the brain throughout life and implicated in neuronal development and in neuroimmune responses (Law et al, 2000; Tran and Miller, 2003; Klein and Rubin, 2004; Cartier et al, 2005). Furthermore, chemokine receptors, namely CCR5 and CXCR4, mediate infection of target cells by human immunodeficiency virus (HIV)-1 and are directly involved in the neuronal injury/death caused by the HIV envelope protein gp120 (Miller and Meucci, 1999; Kaul et al, 2005). The natural CXCR4 ligand is SDF-1 (recently renamed CXCL12), one of the best-characterized chemokines in the central nervous system (CNS) that regulates neural migration, differentiation, and survival (Lazarini et al, 2003). In vitro, CXCL12 promotes survival of different types of neurons (Meucci et al, 1998; Chalasani et al, 2003; Khan et al, 2004), but can also induce neurotoxicity (Kaul and Lipton, 1999)—likely depending on cleavage by proteases and/or other unknown factors (Zhang et al, 2003). The crucial role of this chemokine/receptor pair in the central nervous system (CNS) in vivo was demonstrated by studies on animals lacking CXCL12 or CXCR4, showing serious deficits in hippocampus/cerebellum development (Zou et al, 1998; Lu et al, 2002). Interestingly, morphine and other μ-opioid receptor (MOR) agonists inhibit neurogenesis in these brain areas (Eisch et al, 2000; Hauser et al, 2000).
Crosstalk between opiate and chemokine receptors in the immune and peripheral nervous systems has been implicated in the immunosuppressive effects of opiates and regulates pain perception during inflammation. Indeed, a bidirectional heterologous desensitization of opioid and chemokine receptors in immune cells has been proposed (Rogers et al, 2000; Szabo et al, 2002). However, little is known about the interactions of opioids and chemokines in the CNS. Thus, it is not known whether opioids alter chemokine receptor function in central neurons—a hypothesis initially raised by the observations that opiates abuse accelerates HIV neuropathology and that opioid agonists enhance HIV-1 expression in human brain cultures (Bell et al, 1998; Peterson et al, 1999, 2004; Bell et al, 2002; Nath et al, 2002; Steele et al, 2003; Chuang et al, 2005).
This study examines the interaction between MOR and CXCR4 in primary neuronal cultures. The results show that pretreatment with μ-opioid agonists inhibits CXCL12 signaling and prosurvival effects without affecting CXCR4 protein levels. These findings suggest that long-term cellular adaptations induced by opioids may regulate the effect of CXCL12 in the brain and provide evidence for a novel mechanism of CXCR4 regulation in neurons.
Results
Cortical neurons coexpress MOR and CXCR4
We first determined whether MOR and CXCR4 are coexpressed by individual neurons in our culture model. Pure cultures of cortical neurons were grown as described in Methods in the presence of a separate glial feeder layer for 7 to 9 days in vitro. These neurons are all positive for the neuronal markers β-tubulin III (Figure 1A) and MAP-2 (not shown). Expression of MOR and CXCR4 in the neuronal cultures was initially detected by Western blot (Figure 1B). Differentiated and undifferentiated SH-SY5Y neuroblastoma cells were used as positive control, as these cells express both receptors (Geminder et al, 2001; Iglesias et al, 2003; Kivell et al, 2004). A detailed analysis of the immunostaining in cortical neurons shows that MOR and CXCR4 receptors coexist in individual neurons (Figure 1C–E). Specifically, about 80% of the neuronal population was positive to MOR and over 50% of these neurons were also positive to CXCR4 (MOR+ = 611; CXCR4+ = 377; MOR+/CXCR4+ = 355; total = 786). These receptors are functionally coupled to neuronal signal transduction mechanisms (Meucci et al, 1998; Iglesias et al, 2003). In line with these studies, acute stimulation of neurons (in the absence of glia) with DAMGO, a selective μ-opioid peptide, or with CXCL12, the natural CXCR4 ligand, induced phosphorylation of both ERK1/2 and Akt (Figures 1F and 2). As previously reported, time-course and dose-response experiments indicated that peak responses occurred around 5 min after treatment with DAMGO (1 to 10 μM; Figure 2) and 15 min with CXCL12 (20 nM; Khan et al, 2004, Meucci et al, 1998).
Figure 1.
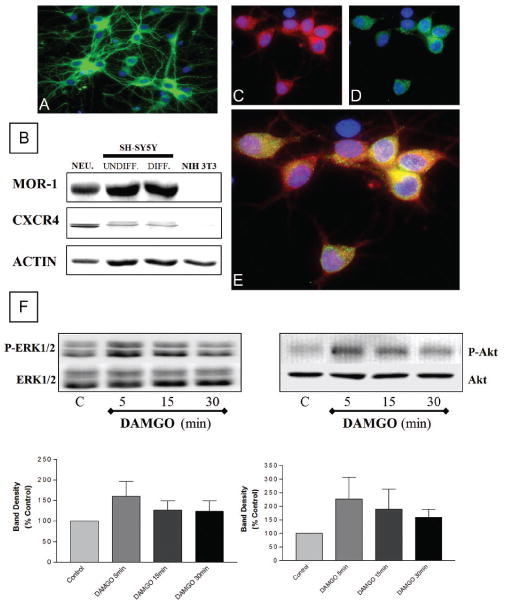
Coexpression of MOR and CXCR4 in cortical neurons. Expression of β-tubulin III (A, green), MOR (C, red), and CXCR4 (D, green) in cortical neurons was detected by immunocytochemistry and fluor escence microscopy. Nuclei are stained with Hoechst 33342 (A-E, blue). Double staining (E) shows coexpression of MOR and CXCR4 on individual neurons. Immunoblots (B) confirm presence of both receptors in cortical neurons. SH-SY5Y and NIH-3T3 cells were used as positive and negative control, respectively; actin levels were determined to verify protein loading. DAMGO (10 μM, 5–30 min) increased phosphorylation of ERK1/2 and Akt in neuronal extracts (F). Graph shows average data from four experiments (top: phosphorylated ERK1/2 [P-ERK] or Akt [P-Akt]; bottom: total ERK1/2 or Akt). However, the kinetics of response for both ERK and Akt is not identical among different experiments (which is expected in primary cultures) and this reflected by the data in the graph showing mean ± SEM for ERK (n = 3) and Akt (n = 4), respectively. Thus, for statistical analysis purposes, peak responses from these experiments (5′ and 15′) were combined and compared to controls (P < .05).
Figure 2.
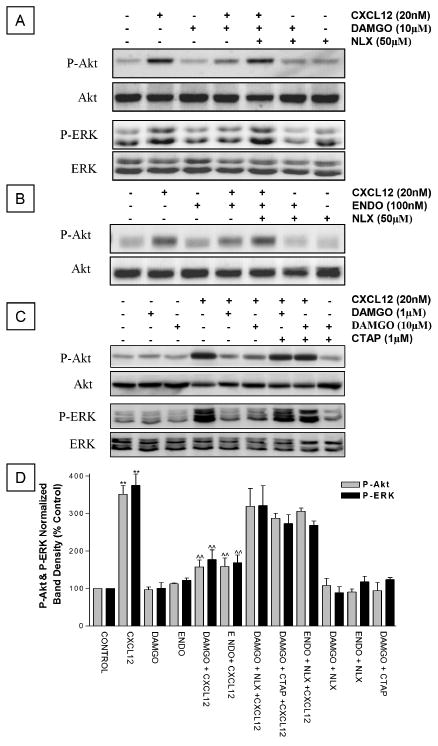
MOR agonists inhibit CXCL12-mediated Akt and ERK phosphorylation in cortical neurons. The effect of DAMGO (1 or 10 μM) or endomorphin-1 (100 nM) on Akt and ERK1/2 phosphorylation induced by CXCL12 (20 nM, 15 min) was studied by Western blot. A general opioid receptors antagonist, naloxone (NALX, 50 μM) (A, B), or a MOR-selective antagonist, CTAP (1 μM, D) was used to determine receptors involved. After blotting with phosphospecific antibodies, membranes were stripped and reblotted using antibodies against total Akt or total ERK1/2. Graphs report average ± SEM of P-Akt or P-ERK band density from three independent experiments. ** P < .001 versus control; ˆˆP < .001 versus CXCL12 alone.
Pretreatment with MOR agonists down-regulates CXCL12-induced phosphorylation of neuronal Akt and ERK
Next, we asked whether long-term treatment of cortical cultures with μ-opioid peptides alters CXCL12 action on neurons. We tested the effect of a 24-h pre-exposure to DAMGO on the acute stimulation of neuronal survival pathways (i.e., PI3K/Akt and ERK1/2) by CXCL12. These kinases are normally activated by chemokines in different cell types and play crucial roles in the regulation of cell survival and proliferation. Indeed, we have previously shown that Akt activation is instrumental for the prosurvival action of chemokines on neurons (Meucci et al, 1998, 2000). In order to study neuronal responses to CXCL12 independently of the glia, which also express CXCR4, neurons were separated from the glia before stimulation with CXCL12. We found that DAMGO pretreatment (Figure 3A, C, 1 and 10 μM) significantly reduced activation of neuronal Akt and ERK1/2 by CXCL12, as determined by immunoblotting with phosphospecific antibodies. Similar results were observed with an endogenous MOR agonist, endomorphin-1 (Figure 3B)—suggesting that this effect may also occur naturally. Furthermore, activation of MOR is necessary to inhibit CXCL12 signaling in neurons (Figure 2). Indeed, the inhibitory action of DAMGO on CXCL12 signaling was blocked by the general opioid antagonist naloxone (50 μM)—which inhibits μ, δ, and κ opioid receptors—as well as by the highly selective MOR antagonist, CTAP (1 μM). CTAP was chosen for its action as a neutral antagonist (i.e., it does not affect basal receptor activity) and its long half-life (>8 h), which makes it suitable for long-term experiments (Wang et al, 2001). Under these conditions, no significant changes in Akt or ERK1/2 phosphorylation were observed when cultures were treated with either agonist alone, or with DAMGO + naloxone, and DAMGO + CTAP (Figure 3). Similarly, levels of ERK or Akt were not significantly affected by either antagonist alone (not shown).
Figure 3.
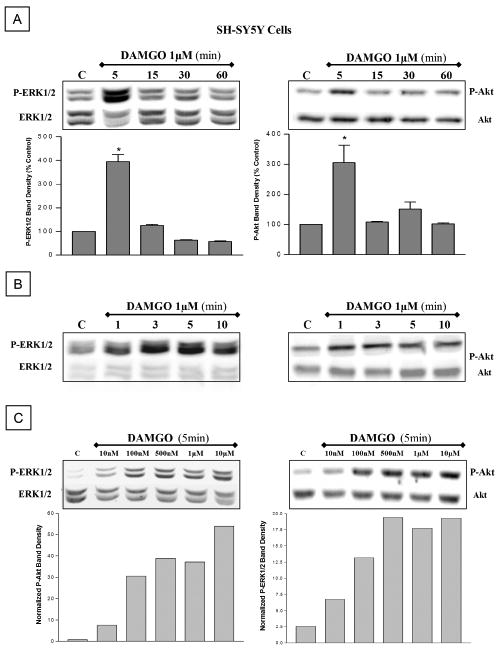
DAMGO transiently phosphorylates Akt and ERK in SH-SY5Y neuroblastoma cells. Acute DAMGO treatment phosphorylates Akt and ERK in SH-SY5Y cells, in a time- (A, 5–60 min; B, 1–10 min) and dose (C, 10 nM to 10 μM) dependent manner. Peak activation occurs at 3 to 5 min. After blotting wit phosphospecific antibodies, membranes were stripped and reblotted using antibodies against total Akt or total ERK1/2.
DAMGO does not change surface or total expression of CXCR4 in neurons
We next determined whether changes in CXCL12 responses induced by MOR activation were due to inhibition of CXCR4 expression. First, we measured CXCR4 levels in rat cortical neurons. As for the previous experiments, neurons were treated with DAMGO (or vehicle) in the presence of glia. After treatment, the glial feeder layer was removed; neurons were lysed and processed for Western blot utilizing a polyclonal anti-CXCR4 antibody. The levels of CXCR4 in neurons treated with DAMGO were comparable to control neurons (Figure 4, 1 to 48 h), suggesting that the peptide does not down-regulate CXCR4 protein expression. To determine whether an increased internalization of CXCR4 receptors is responsible for the inhibition of CXCL12-induced responses by DAMGO, we studied the expression of CXCR4 on the surface of cortical neurons by confocal microscopy, and found no differences between control and DAMGO-treated cultures (Figure 4). We also used the SH-SY5Y cells to address this issue in other cells by flow cytometry (Figure 5). As mentioned above, these cells have been previously used for studies on both MOR and CXCR4. Indeed, we found that stimulation of SH-SY5Y by DAMGO or CXCL12 induces responses resembling those observed in cortical neurons SH-SY5Y cells treated with CXCL12 were used as a positive control of receptor internalization (Figure 5A, B). These experiments showed no effect of DAMGO (10 μM, 24 h) on CXCR4 expression in SH-SY5Y cells as found in rat neurons. Similar results were observed after differentiation of SH-SY5Y cells with retinoic acid, which halts proliferation and induces a more mature neuronal phenotype (not shown). Shorter treatments (1 h) or different concentrations of DAMGO (1 nM to 10 μM) also did not affect surface levels of CXCR4 in SH-SY5Y cells (not shown). In addition, cotreatment of DAMGO and CXCL12 for 1 h did not alter the internalization of CXCR4 caused by CXCL12 alone (Figure 5B). Overall, these findings show that, under these conditions, μ-opioid agonists do not influence total or membrane expression of neuronal CXCR4.
Figure 4.
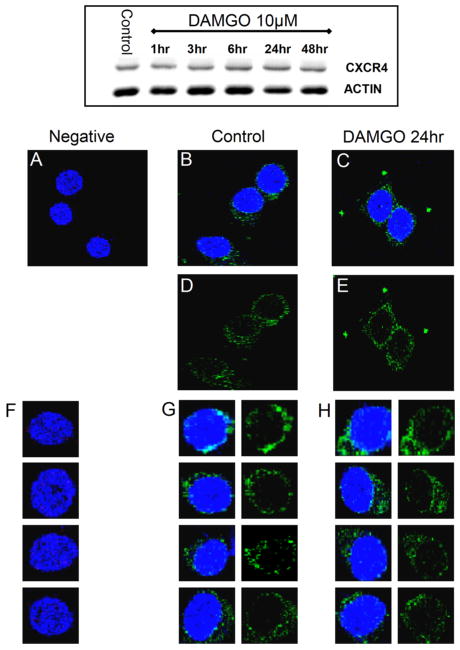
Effect of DAMGO on total and plasma membrane levels of CXCR4 in cortical neurons. CXCR4 protein levels in cortical neurons were studied by Western blot (top panel) and confocal microscopy (A–H) as described in Methods. As shown by the representative gel in the top panel (n = 3), cortical neurons were treated with DAMGO up to 48 h. The confocal microscopy studies were preformed on cultures treated with DAMGO (1 μM) for 24 h. In both sets of experiments, control and DAMGO-treated neurons appeared to have similar levels of CXCR4. For the confocal studies, neurons were stained with an antibody against CXCR4 (green) and the nuclear dye Hoechst 33342 (blue) as detailed in methods section, and visualized using a 63X 1.4 oil immersion objective mounted on a Leica confocal microscope. Images were collected by using a step size of 0.3 μm along the z axis (512 × 512; 8 bits/pixel). A and F: Negative control (i.e., without primary antibody). B, D, and G: Control. C, E, and H: DAMGO-treated. A total of 100 cells per treatment were studied from two independent experiments (three coverslips/group).
Figure 5.
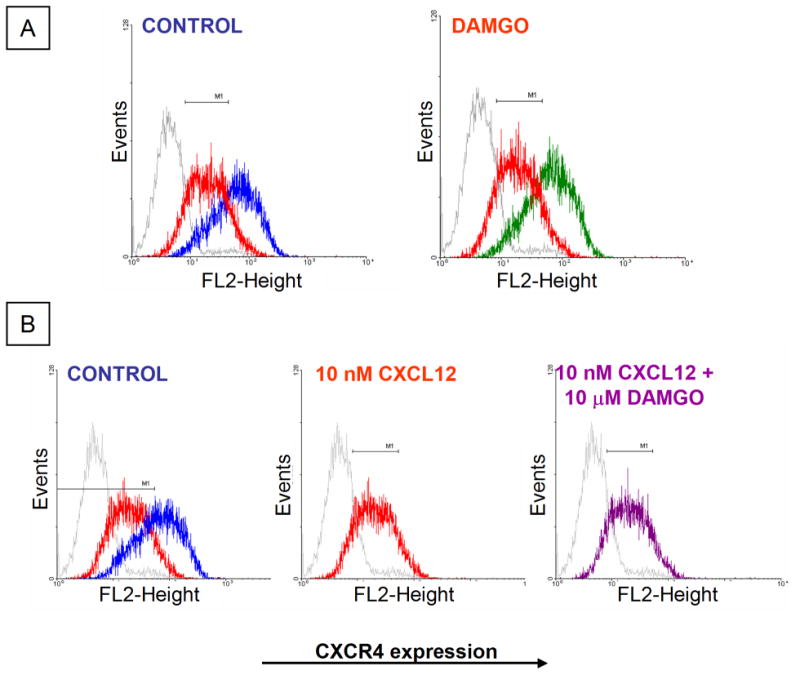
Effect of DAMGO on surface expression of CXCR4 in SH-SY5Y cells. Surface expression of CXCR4 in SH-SY5Y cells was assessed by flow cytometry (all experiments repeated twice). (A) Untreated = blue; DAMGO 10 μM = green; CXCL12 (1 h) = red; IgG2B-PE isotype control = grey. (B) Cells cotreated with DAMGO+CXCL12 are shown in purple.
DAMGO inhibits neuronal survival promoted by CXCL12
Regulation of neuronal response to CXCL12 is essential for shaping of developing and mature CNS (Tran and Miller, 2003). To establish whether MOR agonists alter the effect of CXCL12 on neuronal survival, the ability of CXCL12 to protect neurons from N-methyl-d-aspartate (NMDA)-induced death was examined in the presence and absence of DAMGO. Cortical cultures were treated with DAMGO (1 and 10 μM) as indicated above. Neurons were subsequently exposed to NMDA (20 min) and/or CXCL12 (added 10 min before NMDA) in the absence of glia and then returned to the original culture dishes with the glial feeder layer. Neuronal death was evaluated after 24 h. As expected, CXCL12 prevented NMDA neurotoxicity in control neurons (Figure 6A), but not in neurons treated with DAMGO (Figure 6A). Addition of 1 μM CTAP to the culture medium, before and during DAMGO treatment (both 1 and 10 μM), prevented DAMGO inhibitory effect (Figure 6B). Notably, DAMGO did not affect the prosurvival action of another trophic factor, i.e., Brain Derived Neurotrophic Factor (BDNF)—suggesting that this effect is specific for CXCL12.
Figure 6.
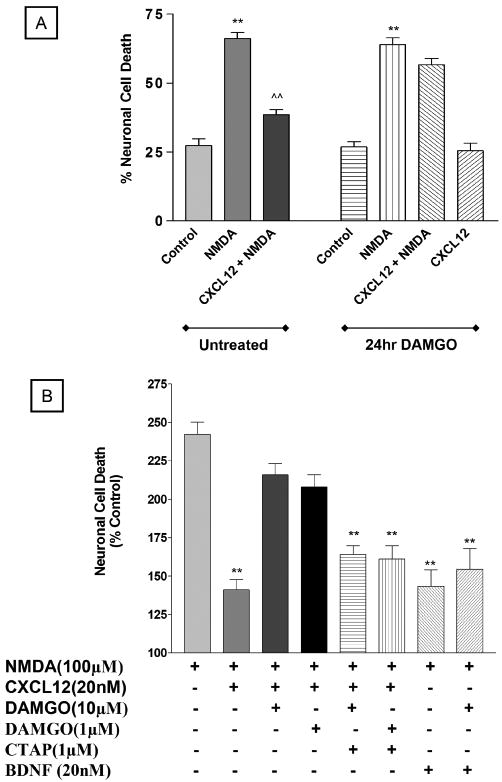
DAMGO prevents CXCL12-induced neuroprotection. Cortical neurons were treated with DAMGO (24 h) and subsequently exposed to NMDA (100 μM) and/or CXCL12 (20 nM) as detailed in the text. Results from three independent experiments with 10μMDAMGO are reported in A (** P < .001versus respective control; ˆˆP < .001 versus NMDA alone). Data shown in B include two additional experiments where neurons were exposed to DAMGO (both 1 and 10 μM) in the presence of the MOR-specific antagonist CTAP—added to the cultures before addition of DAMGO and maintained during DAMGO treatment—and BDNF, which was used as a control (** P < .001 versus NMDA alone). Each experiment was run in triplicate (i.e., three coverslips/treatment).
Discussion
Recently, novel and important effects of chemokines in the CNS have been reported. Although their role in the CNS is not completely understood, these proteins regulate fundamental neuronal and glial functions in normal and disease states, affecting cell survival and differentiation (Tran and Miller, 2003; Woerner et al, 2005). In order to explain their pleiotropic—and sometimes, contradictory (i.e., neurotrophic versus neurotoxic)—nature, the final effect of chemokines in the CNS must be modulated by other neuropeptides and/or neurotransmitters. Opioids are likely candidates for this regulation as they control a number of neuroimmune functions and were reported to interact with chemokines in vivo (Salzet et al, 2000; Szabo et al, 2002). This study demonstrates that opioids modulate the action of the chemokine CXCL12 on central neurons. Specifically, MOR agonists inhibited the prosurvival effects of CXCL12 on cortical cultures. Considering the role of MOR and CXCR4 in neuronal survival and differentiation, as well as the reported effect of morphine on HIV progression, it is possible that interactions between these neuropeptides also occur in the brain. Such interactions could be normally involved in the regulation of chemokine effects on neurons and/or explain the faster progression to neuroAIDS (acquired immunodeficiency syndrome) (or other neurological deficits) in drug abusers (Nath et al, 2002).
Our data exclude a direct effect of μ-opioids on CXCR4 expression, but rather indicate that MOR agonists induce complex neurochemical changes in neuronal signaling, which result in the inhibition of CXCL12 neurotrophic effects. One such effect is the activation of the antiapoptotic kinase Akt that is required for the neuroprotective action of chemokines (Meucci et al, 2000; Khan et al, 2004). The ability of μ-opioid peptides to compromise stimulation of mitogen-activated protein (MAP) kinases by CXCL12, though, suggests that proteins regulating crucial steps of CXCR4 signaling, such as those involved in receptor desensitization and recycling, might be affected by opioids. However, the confocal and flow cytometry studies show that DAMGO does not alter levels of CXCR4 on the neuronal surface, which rules out the hypothesis that increased receptor internalization is responsible for the inhibitory action of the peptide. Our results also exclude that a reduced synthesis/increased degradation of CXCR4 is implicated in the down-regulation of CXCL12 activity, because total receptor levels are unchanged in neurons treated with DAMGO. Based on these findings, one could speculate that activation of MOR may result in uncoupling of CXCR4 from G-proteins signaling—also supported by previous studies showing heterologous desensitization between chemokine (CCR5) and opioid (μ) receptors in immune cells, in the absence of receptor internalization (Rogers et al, 2000; Szabo et al, 2003). This phenomenon could be caused by rearrangement of the receptors and signaling molecules within the lipid drafts. These plasma membrane microdomains, enriched in cholesterol and sphingolipids, concentrate signaling molecules by related receptors (Pike, 2003; Chini and Parenti, 2004). Their composition strongly regulates G-Protein Coupled Receptor (GPCR) function, including that of opioid and chemokine receptors, independently of changes in total protein receptor levels (Popik et al, 2002; Schabath et al, 2006; Xu et al, 2006; Zhao et al, 2006). Importantly, chronic exposure to opioid agonists has been proposed to alter the localization of these signaling molecules (i.e., caveolins, β-arrestins, G proteins and their effectors) within the lipid rafts/caveolae microdomains (Zhao et al, 2006). Disruption of the lipid rafts may significantly affect the behavior of opioid receptors and other GPCRs in different manner, depending on the receptor and the cell type (Pike, 2003). As opioid and chemokine receptors activate similar intracellular pathways, stimulation of neurons with MOR agonists could affect CXCL12 function indirectly, by recruiting/rearranging proteins involved in MOR signaling. Additionally, a recent study in pre-B lymphocytes and breast carcinoma cells (Schabath et al, 2006) demonstrates that exclusion of CXCR4 from membrane lipid rafts, due to reduction in cholesterol levels, impairs the ability of CXCL12 to trigger cell motility via ERK phosphorylation and attenuates tumor growth. An intriguing alternative (that would also depend on colocalization of the two receptors in lipid rafts) is that stimulation with DAMGO or endomorphin-1 induces heterodimerization of MOR with CXCR4, leading to a reduced CXCL12 function as suggested by Fluorescence Resonance Energy Transfer (FRET) studies in cell lines (i.e., HEK293 cells) (Toth et al, 2004). Although CXCR4 receptors seem to exist primarily as homodimer, this study showed a specific association of CXCR4 with other GPCRs (namely μ-opioid, κ-opioid, and m3-muscarininc receptors) that could impair CXCR4 function. Similarly, dimerization of MOR with CCR5 has been reported in Chinese hamster ovarian (CHO) cells (Chen et al, 2004). Further investigation is needed to test these and additional hypotheses in primary neurons, and determine whether a physical interaction between the two receptors naturally occurs. Finally, other investigators reported no changes in CXCR4 function in monocytes exposed to DAMGO (Szabo et al, 2003). Although DAMGO treatments were relatively short in this study as compared to ours (1 versus 24 h), this would indicate that inhibition of CXCR4 responses by MOR agonists might be a neuron-specific mechanism. This is a very intriguing scenario in light of the emerging role of CXCR4 in neuronal development. To this end, it first needs to be established whether glia are involved in the modulatory action of opioids on CXCL12.
Independently of the mechanisms involved, the inhibitory effect of opioids on CXCL12 activity has very important implications for the effects of chemokines in the CNS. For instance, it may represent a natural way to adjust chemokine signaling to the physiological demands. Thus, endogenous levels of endomorphin (or other MOR agonists) may contribute to the fine-tuning of CXCL12 activity in the brain and control CXCL12-mediated responses during inflammatory states. Indeed, other examples of CXCR4 controlling mechanisms have been reported in brain cells, such as the ability of tumor necrosis factor (TNF)-α to down-regulate CXCR4 mRNA in astroglia (Han et al, 2001). These are critical issues because CXCL12 regulates neuronal function in both developing and mature neurons and affects multiple behaviors of neuronal and non-neuronal cells (Lazarini et al, 2003). Of note, CXCL12 plays a crucial role in the proliferation and migration of neural progenitor cells (Klein et al, 2001; Stumm et al, 2003; Belmadani et al, 2005), whereas opiates—including morphine—are known to inhibit neurogenesis in vivo and in vitro (Eisch et al, 2000; Hauser et al, 2000). Furthermore, the activation of MOR by drugs of abuse may exacerbate neurotoxicity of viral proteins, such as the HIV-1 gp120 that acts as a partial CXCR4 agonist (Khan et al, 2004). Under the experimental conditions reported here, DAMGO increases gp120 neurotoxicity in vitro (Patel et al, unpublished data); morphine exerts similar effects in human fetal brain cultures (Hu et al, 2005). Opiates in general are potential cofactors in AIDS progression (Bell et al, 1998; Donahoe and Vlahov, 1998; Bell et al, 2002; Nath et al, 2002).
In conclusion, this study reports the first example of a functional crosstalk between CXCL12 and μ-opioids in cultures of central neurons. Though these findings will have to be validated in vivo, they suggest a novel mechanism of CXCR4 regulation in the CNS and indicate that levels of endogenous or exogenous neuropeptides in the brain can affect the actions of CXCL12 on neurons. Such regulation could take place in healthy as well as pathological conditions, including neuroAIDS.
Methods
Cultures
Rat cortical neurons were prepared and cultured as previously described (Meucci et al, 2000; Khan et al, 2005) using a bilaminar cell culture system, i.e., a feeder layer of glia supporting the pure neuronal layer. Neurons were separated from the glia before treatment with CXCL12 and maintained in the original culture medium or in saline solution.
SH-SY5Y human neuroblastoma cells were obtained from ATCC and cultured in minimal essential medium (MEM)/Hams F-12 (1:1) containing 10% fetal calf serum and 50 μg/ml gentamycin. When indicated, cells were differentiated by treatment with 10 μM all-trans-retinoic acid as reported in (Encinas et al, 1999).
Western blots
Cells were washed with saline solution and scraped in lysis buffer (Meucci et al, 1998, 2000). Protein concentration was determined by bicinchoninic acid protein assay (Pierce). The following antibodies were used: anti-CXCR4 (H-118 and G-19, raised against amino acids 176 to 293 and N-terminus, respectively) (1:500); anti-MOR (H-80) (1:500) from Santa Cruz Biotech; anti-Akt, anti-phosphoAkt (Ser473), anti-ERK, and anti-phosphoERK from Cell Signaling (1:2,000); anti-β-actin from Sigma-Aldrich (1:5000). An image acquisition/analysis system (ChemiDoc System; BioRad) and the UN-SCAN IT software (Silk Scientific) were used for detection of chemiluminescent bands and densitometric analysis. Data are reported as mean ± SEM. Intensity values from actin bands or total Akt and ERK bands were used to normalize phosphoprotein signals and compensate for possible variations in protein loading. Data are expressed as percentage of control after normalization. Statistical analysis of band densities was performed by one-way analysis of variance (ANOVA) followed by Newman-Keuls test.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde and incubated with anti-CXCR4 (G-19, raised against the N-terminus) and anti-MOR antibodies (H-80, raised against amino acids 1 to 80). Both antibodies were from Santa Cruz Biotech and used at a dilution of 1:50. Secondary antibodies conjugated to Cy3 or Cy2 (Jackson ImmunoResearch Labs; 1:333) were used for detection. Neurons were stained with anti-CXCR4 or anti-MOR prior to permeabilization with 0.1% Triton X-100 for staining of the neuronal marker β tubulin III (1:500; Covance). This sequence of staining allowed the detection of only surface CXCR4 and MOR. Hoechst 33342 (3 μg/ml) was used for nuclear counterstaining. Negative controls for the studies included no primary and fibroblasts; furthermore, specificity of the antibody is also supported by the presence of neurons that did not show staining for CXCR4 and/or MOR (in the same microscopic field of stained neurons). However, to validate results obtained with the H-80 anti-MOR, antibodies directed against an internal region of the receptor (Chemicon AB1580; 1:1000) were also used. For these experiments cells were permeabilized before incubation with the primary antibody (the percentage of MOR-positive neurons was 85.2 ± 4.6, mean ± SEM, N = 315). Coverslips were mounted on an inverted microscope (Olympus IX70) connected to a CCD camera (Micromax YS1300; Princeton Instruments) and a computer, or a confocal microscope (Leica TCS SP2). Images were acquired/analyzed using the software Metamorph (Universal Imaging) and the Leica confocal software.
Cell survival and apoptosis
Neurons (9 days in vitro) were treated with DAMGO for 24 h in their original culture dish, subsequently transferred to a dish containing Mg2+-free saline with glycine (15 μM), and exposed to NMDA (100 μM; Tocris, UK) and/or CXCL12 (20 nM; R&D System, Minneapolis, MN) in absence of glia. After treatments, neurons were moved back into the original culture dishes containing glia. Neuronal death was evaluated after 24 h. Hoechst 33342 (3 μg/ml) combined with cleaved caspase-3 (1:100; Cell Signaling) staining was used to identify normal and apoptotic cells (Meucci et al, 1998). Five microscopic fields/coverslip were counted, and 3 coverslips/treatment were used for each experiment.
Flow cytometry
SH-SY5Y cells were washed and re-suspended in cold Ca2+-free phosphate-buffered saline (PBS), and then incubated in PBS supplemented with 10% horse serum (30 min on ice). Cells were then resuspended in fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin [BSA]/PBS) and incubated with 12.5 μg/ml phycoerytrin-conjugated antibodies (FAB173P from R&D System). A mouse monoclonal anti-human CXCR4-PE and a mouse IgG2B-PE (isotype control) were used. After incubation cells were washed and fixed with 1% paraformaldehyde in FACS buffer. Samples acquisition was performed with a FACS Calibur (BD).
Acknowledgments
The authors thank Alessandro Fatatis and Robert Nichols for discussion and critical reading of the manuscript, Randi Hippen-steel for technical assistance with the cultures, and NIH-NIDA for support (grants DA-15014 and DA-19808 to O.M.; NRSA fellowship DA-020234 to JP).
Footnotes
Publisher's Disclaimer: Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article maybe used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
References
- Bell JE, Arango JC, Robertson R, Brettle RP, Leen C, Simmonds P. HIV and drug misuse in the Edinburgh cohort. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S35–S42. doi: 10.1097/00126334-200210012-00003. [DOI] [PubMed] [Google Scholar]
- Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain. 1998;121(Pt 11):2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Tran PB, Ren D, Assimacopoulos S, Grove EA, Miller RJ. The chemokine stromal cell-derived factor-1 regulates the migration of sensory neuron progenitors. J Neurosci. 2005;25:3995–4003. doi: 10.1523/JNEUROSCI.4631-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Chalasani SH, Baribaud F, Coughlan CM, Sunshine MJ, Lee VM, Doms RW, Littman DR, Raper JA. The chemokine stromal cell-derived factor-1 promotes the survival of embryonic retinal ganglion cells. J Neurosci. 2003;23:4601–4612. doi: 10.1523/JNEUROSCI.23-11-04601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY. Heterodimerization and cross-desensitization between the mu-opioid receptor and the chemokine CCR5 receptor. Eur J Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol. 2004;32:325–338. doi: 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- Chuang RY, Suzuki S, Chuang TK, Miyagi T, Chuang LF, Doi RH. Opioids and the progression of simian AIDS. Front Biosci. 2005;10:1666–1677. doi: 10.2741/1651. [DOI] [PubMed] [Google Scholar]
- Donahoe RM, Vlahov D. Opiates as potential cofactors in progression of HIV-1 infections to AIDS. J Neuroimmunol. 1998;83:77–87. doi: 10.1016/s0165-5728(97)00224-5. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci U S A. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, Ben-Baruch A. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol. 2001;167:4747–4757. doi: 10.4049/jimmunol.167.8.4747. [DOI] [PubMed] [Google Scholar]
- Han Y, Wang J, He T, Ransohoff RM. TNF-alpha down-regulates CXCR4 expression in primary murine astrocytes. Brain Res. 2001;888:1–10. doi: 10.1016/s0006-8993(00)02924-3. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Houdi AA, Turbek CS, Elde RP, Maxson W., 3rd Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: differential impact of mu and delta receptor activation on proliferation and neurite elongation. Eur J Neurosci. 2000;12:1281–1293. doi: 10.1046/j.1460-9568.2000.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Lokensgard JR, Peterson PK. Morphine potentiates HIV-1 gp120-induced neuronal apoptosis. J Infect Dis. 2005;191:886–889. doi: 10.1086/427830. [DOI] [PubMed] [Google Scholar]
- Iglesias M, Segura MF, Comella JX, Olmos G. Muopioid receptor activation prevents apoptosis following serum withdrawal in differentiated SH-SY5Y cells and cortical neurons via phosphatidylinositol 3-kinase. Neuropharmacology. 2003;44:482–492. doi: 10.1016/s0028-3908(03)00024-8. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Patel JP, Huynh N, Wang J, Huang Z, Fatatis A, Meucci O. Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Res Hum Retroviruses. 2004;20:1063–1071. doi: 10.1089/aid.2004.20.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Shimizu S, Patel JP, Nelson A, Le MT, Mullen-Przeworski A, Brandimarti R, Fatatis A, Meucci O. Regulation of neuronal P53 activity by CXCR 4. Mol Cell Neurosci. 2005;30:58–66. doi: 10.1016/j.mcn.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell BM, Day DJ, McDonald FJ, Miller JH. Mu and delta opioid receptor immunoreactivity and mu receptor regulation in brainstem cells cultured from late fetal and early postnatal rats. Brain Res Dev Brain Res. 2004;149:9–19. doi: 10.1016/j.devbrainres.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25:306–314. doi: 10.1016/j.it.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, Luster AD. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–1981. doi: 10.1242/dev.128.11.1971. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–148. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci U S A. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RJ, Meucci O. AIDS and the brain: is there a chemokine connection? Trends Neurosci. 1999;22:471–479. doi: 10.1016/s0166-2236(99)01408-3. [DOI] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Cabral G, Lokensgard JR. Cannabinoids and morphine differentially affect HIV-1 expression in CD4(+) lymphocyte and microglial cell cultures. J Neuroimmunol. 2004;147:123–126. doi: 10.1016/j.jneuroim.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Lokensgard J, Portoghese PS, Chao CC. Endomorphin-1 potentiates HIV-1 expression in human brain cell cultures: implication of an atypical mu-opioid receptor. Neuropharmacology. 1999;38:273–278. doi: 10.1016/s0028-3908(98)00167-1. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol. 2002;76:4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ, Steele AD, Howard OM, Oppenheim JJ. Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann N Y Acad Sci. 2000;917:19–28. doi: 10.1111/j.1749-6632.2000.tb05369.x. [DOI] [PubMed] [Google Scholar]
- Salzet M, Vieau D, Day R. Crosstalk between nervous and immune systems through the animal kingdom: focus on opioids. Trends Neurosci. 2000;23:550–555. doi: 10.1016/s0166-2236(00)01642-8. [DOI] [PubMed] [Google Scholar]
- Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J Cell Sci. 2006;119:314–325. doi: 10.1242/jcs.02741. [DOI] [PubMed] [Google Scholar]
- Steele AD, Henderson EE, Rogers TJ. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. 2003;309:99–107. doi: 10.1016/s0042-6822(03)00015-1. [DOI] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A. 2002;99:10276–10281. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–1082. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1alpha and the HIV-1 coat protein gp120: a fluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Ther. 2004;310:8–17. doi: 10.1124/jpet.103.064956. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003;4:444–455. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- Wang D, Raehal KM, Bilsky EJ, Sadee W. Inverse agonists and neutral antagonists at mu opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77:1590–1600. doi: 10.1046/j.1471-4159.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- Woerner BM, Warrington NM, Kung AL, Perry A, Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 2005;65:11392–11399. doi: 10.1158/0008-5472.CAN-05-0847. [DOI] [PubMed] [Google Scholar]
- Xu W, Yoon SI, Huang P, Wang Y, Chen C, Chong PL, Liu-Chen LY. Localization of the kappa opioid receptor in lipid rafts. J Pharmacol Exp Ther. 2006;317:1295–1306. doi: 10.1124/jpet.105.099507. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM, Power C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6:1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- Zhao H, Loh HH, Law PY. Adenylyl cyclase super-activation induced by long-term treatment with opioid agonist is dependent on receptor localized within lipid rafts and is independent of receptor internalization. Mol Pharmacol. 2006;69:1421–1432. doi: 10.1124/mol.105.020024. [DOI] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]