

Abstract
Objective
To determine whether APOE ε4 predicts rate of cognitive change in incident and prevalent AD.
Methods
Individuals were recruited from two longitudinal cohort studies - the Washington Heights and Inwood Columbia Aging Project (WHICAP; population-based) and the Predictors Study (clinic-based), and were followed for an average of four years. Three samples of participants diagnosed with Alzheimer’s disease, with diverse demographic characteristics and baseline cognitive functioning were studied: 1) 199 (48%) of the incident WHICAP cases; 2) 215 (54%) of the prevalent WHICAP cases; and 3)156 (71%) of the individuals diagnosed with AD in the Predictors Study. Generalized estimating equations (GEE) were used to test whether rate of cognitive change, measured using a composite cognitive score in WHICAP and the Mini-Mental Status Exam in Predictors, varied as a function of ε4 status in each sample.
Results
The presence of at least one ε4 allele was associated with faster cognitive decline in the incident population-based AD group (p = .01). Parallel results were produced for the two prevalent dementia samples only when adjusting for disease severity or excluding the most impaired participants from the analysis.
Conclusion
APOE ε4 may influence rate of cognitive decline most significantly in the earliest stages of AD.
INTRODUCTION
The rate of cognitive deterioration during the course of Alzheimer’s disease (AD) varies markedly across individuals (1, 2) and is likely driven by a combination of genetic and environmental factors. Given its role in disease onset (3), the apolipoprotein E (APOE) ε4 allele may contribute to differential rates of cognitive decline in AD; however, this hypothesis has been explored for over ten years with no resolution. ε4 carriers have been found to demonstrate both slower (4–6) and faster rates of decline (7–13), potentially reflecting methodological differences in recruitment techniques, the measurement of cognition, follow-up time, and participants’ stage of dementia. A subset of studies has also demonstrated non-differential rates of decline in relation to ε4 status (14–24); however, most of these studies had relatively small sample sizes which may have limited their ability to detect differential change over time.
The current study was undertaken to comprehensively evaluate the effect of ε4 on rate of cognitive decline in three separate samples of participants with AD: incident cases from a population-based study, prevalent cases from a population-based study, and prevalent cases recruited from the clinic. Few if any prior studies have examined incident cases in which rates of cognitive decline can be observed from the earliest stages of dementia. More typically, analyses have included cases with prevalent disease frequently drawn from clinic samples, a recruitment approach that may lead to biased sampling. The simultaneous study of these three samples, which represent both incident and prevalent disease, as well as diverse demographic characteristics, recruitment techniques, and assessment procedures, allowed for one of the most comprehensive studies to date of the role of ε4 in cognitive decline following diagnosis of AD. Although analyses of the population based incident sample likely have the best internal and external validity, in order to understand the discrepancies in prior studies, it is valuable to repeat the analyses across an array of samples reflecting methodological differences in prior work.
MATERIALS AND METHODS
Participants
Three samples of participants with AD (population-based incident, population-based prevalent, and clinic-based) were drawn from two different research studies as follows:
1. Population-Based Study (WHICAP)
The first two samples of AD participants were incident and prevalent samples drawn from the Washington Heights and Inwood Columbia Aging Project (WHICAP), a prospective, population-based study of aging and dementia in 4,309 Medicare recipients, 65 years and older, residing in northern Manhattan (Washington Heights, Hamilton Heights, Inwood) that has been described in detail in earlier work (25, 26). Briefly, a stratified random sample of 50% of all persons older than 65 years was obtained from the Health Care Finance Administration (HCFA). All persons were sent a letter from HCFA explaining that they had been selected to participate in a study of aging by investigators at Columbia University. Each participant underwent an in-person interview of general health and functional ability at the time of study entry followed by a standardized assessment, including medical history, physical and neurological examination and a neuropsychological battery. Ethnic group was classified by participant’s self-report using the format of the 1990 US Census. Participants were asked if they considered themselves white, black or other, and then asked if they were Hispanic. Participants were recruited at two time points (1992–1994 and 1999–2002). They have been followed at approximately 18-month intervals with similar assessments at each interval. Evaluations were conducted in either English or Spanish, based on the primary language or preference of the participant. Recruitment, informed consent and study procedures were approved by the Institutional Review Boards of Columbia Presbyterian Medical Center and Columbia University Health Sciences and the New York State Psychiatric Institute.
Consensus diagnoses of “dementia” or “no dementia” were based on physician-administered physical and neurological examinations in conjunction with a standardized neuropsychological battery (27) according to criteria outlined in the Diagnostic and Statistical Manual of Mental Disorders, Revised Third Edition (DSM-III-R) and determined at a consensus conference attended by neurologists and neuropsychologists. Evidence of social or occupational function deficits was required and results from the neuropsychological battery were considered. All available ancillary information, including medical charts and imaging studies, was considered in the evaluations. Medical diagnoses were assigned when applicable. The type of dementia was subsequently determined. Diagnosis of probable or possible AD was made based on criteria of the National Institute of Neurological and Communicative Disorders and Stroke – Alzheimer’s Disease and Related Disorders Association (28). The study was reviewed and approved by the Columbia University institutional review board, and written informed consent was obtained from all subjects.
The first sample was comprised of 199 (48%) of the 411 incident AD cases in WHICAP. All cases were mild at diagnosis defined by a score of 1 on the Clinical Dementia Rating (CDR) scale (29). 56 (14%) cases were excluded due to unavailable APOE genotype. 156 (38%) additional cases were ineligible due to lack of follow-up cognitive data; many of these were diagnosed relatively recently and had not yet been re-evaluated despite ongoing participation in the study. The 212 incident cases from WHICAP excluded from the current study did not differ significantly from the final incident sample in terms of sex (p = .08) or cognitive score at diagnosis (p = .09). However, excluded participants were approximately three years older at diagnosis (M = 84.60; SD = 6.42) than the final sample (M = 81.97; SD = 6.50), F (1, 409) = 16.84, p < .01, and slightly more educated (M = 8.02; SD = 4.79) than the final sample (M = 6.30; SD = 4.50), F (1, 407) = 13.56, p < .01. The final sample of 199 individuals was followed for an average of 2.9 (1.3) visits (beginning with the incident visit). Approximately 20% of participants who did not die were lost to follow up after each of the first five visits. The proportion of individuals lost to follow up increased once individuals participated at least five times.
The second sample was comprised of 215 (54%) of the 397 prevalent AD cases WHICAP who met criteria for mild AD (CDR of 1) at baseline. 120 (30%) participants were excluded due to unavailable APOE genotyping, and an additional 55 (14%) were excluded due to lack of follow-up data. The 182 prevalent cases of mild AD from WHICAP excluded from the current study did not differ significantly from the final prevalent sample in terms of sex (p = .09) or baseline cognitive score (p = .47). However, excluded participants were approximately four years older at baseline (M = 84.13; SD = 7.82) than the final sample, (M = 80.70; SD = 7.10), F (1, 395) = 21.43, p < .01, and slightly more educated (M = 7.14; SD = 4.30) than the final sample (M = 5.90; SD = 4.20), F (1, 395) = 8.26, p < .01. The final sample of 215 individuals was followed for an average of 3.6 (1.6) visits. Approximately 12% of participants who did not die were lost to follow up after each of the first six visits. The proportion of individuals lost to follow up increased once individuals participated at least six times.
2. Clinic-Based Study (Predictors II)
The final sample included individuals with AD drawn from the second cohort of the Predictors Study. Recruitment for this cohort began in 1997 using methods established for the first Predictors cohort (described in detail in earlier work (30)). Briefly, participants in the Predictors study were recruited and studied at 3 US sites: Columbia University, New York, Johns Hopkins University, Baltimore, and Massachusetts General Hospital/Harvard Medical School, Boston. All participants met DSM-III-R criteria for primary degenerative dementia of the Alzheimer type and National Institute of Neurological Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association criteria for probable AD at enrollment. Enrollment required a modified Mini-Mental State Examination score of ≥ 30 (maximum = 57) which is equivalent to a score of approximately 16 or more on the Folstein Mini-Mental State Exam (MMSE) (31, 32). Exclusion criteria were diagnosis of Parkinson’s disease or parkinsonism at any time prior to the onset of intellectual decline, clinical or historical evidence of stroke, history of alcohol abuse or dependence, any electroconvulsive treatment (ECT) within 2 years of recruitment or 10 or more ECT sessions at any time, and history or current clinical evidence of schizophreniaor schizoaffective disorder that started before the onset of intellectual decline.
Each consecutive patient who met the criteria of the study was included, except for those who did not consent to participate or who lived too far away and were unable to return to the study site for regular follow-up. Neurologic and mental status examinations were conducted at study entry and at 6-month intervals thereafter. The cognitive function measure used for the analysis was the MMSE. Onset of illness was estimated by the treating neurologist based on discussion with the family. The study was reviewed and approved by the participating institutions’ review boards, and written informed consent was obtained from all subjects.
The current study included 156 (71%) of the 221 individuals diagnosed with AD in the second cohort of the Predictors Study. 62 (28%) participants from the original sample were excluded due to unavailable genotyping and 3 (1%) subjects were excluded due to lack of follow-up cognitive data. The 65 cases excluded from the current study did not differ significantly from the final sample in terms of years of education (p = .09), sex (p = .11), or baseline cognition (p = .39). However, excluded participants were approximately three years older at baseline (M = 78.57; SD = 8.33) than the final sample (M = 75.30; SD = 7.70), F (1, 219) = 7.72, p < .01. The final sample of 156 individuals was followed for an average of 3.6 (1.6) visits. Approximately 3% of participants who did not die were lost to follow up after each of the first seven visits. The proportion of individuals lost to follow up increased once individuals participated at least seven times.
Predictors
Presence of at least one ε4 allele was the primary predictor tested. The pattern of each subject’s APOE isoforms was determined using the method of Hixson and Vernier (33). Standard demographic variables (age at diagnosis in the incident sample and age at first visit for the other cohorts, sex, ethnicity, and years of education) were included as covariates in statistical models. We chose to use age at the first visit in prevalent cases because these cases were selected early in the disease course (CDR = 1) and age of first visit was more clearly defined than an estimate of time of disease onset.
Outcome Measures
The primary outcome in this analysis was rate of cognitive decline from baseline. Baseline is defined as the diagnostic visit for the incident cases, and the point of study entry for the other two samples. For the clinic-based sample, cognition was measured with the MMSE. For the two population-based samples, a composite cognitive z-score was developed to summarize performance on a variety of tests assessing five cognitive domains (27). These included the following: 1) Memory: total and delayed recall of the Selective Reminding Test, and the recognition component of the Benton Visual Retention Test; 2) Abstract Reasoning: WAIS-R Similarities subtest, and the Identities and Oddities subtest of the Dementia Rating Scale; 3) Visual-Spatial: five selected items from the Rosen drawing test and the matching component of the Benton Visual Retention Test; 4) Language: 15-item Boston Naming Test, the eight high probability items from the Repetition subtest of the Boston Diagnostic Aphasia Examination (BDAE), and the first six items of the BDAE Comprehension subtest; and 5) Executive-Speed: average scores for phonemic fluency assessed by the Controlled Oral Word Association Test, and category fluency (Animals, Food, Clothing).
The composite cognitive measure was derived as follows: each of the above 12 raw scores were transformed into z-scores using means and standard deviations of scores from 272 non-demented controls in WHICAP with a similar distribution of age, education, and ethnicity to the AD patients. Z-scores for individual tests were then averaged to create a z-score for each cognitive domain. If more than half of the tests in a given domain were missing, the domain score was considered missing and was excluded from the analysis. The composite score was derived by averaging the five domain scores, with missing data treated in the manner described above.
Statistical analyses
One-way analyses of variance (ANOVA) and chi square tests were used to examine between-group differences across the population-based samples, as well as within-group differences across APOE genotype in each sample. Generalized estimating equations (GEE) (34) were used to test whether APOE ε4 was associated with a differential rate of cognitive change. GEE takes into account the multiple visits per subject and the fact that the characteristics of the same individual over time are likely to be correlated. The repeated measures for each subject are treated as a cluster. GEE models in each sample examined the extent to which time (years from diagnosis in the incident sample and years from baseline in the prevalent cases), ε4 status (present or absent), and the time × ε4 interaction predicted global cognition. A significant time effect would indicate that global cognition changed significantly over time in non-carriers (the reference group). A significant ε4 effect would indicate that cognitive performance at baseline (diagnosis for the incident sample and study entry for there remaining two samples) varied as a function of ε4 status. Finally, a significant interaction term would indicate that rate of cognitive decline varied as a function of ε4 status. To evaluate the contribution of other variables potentially related to cognitive change, GEE models also included demographic variables such as age at diagnostic visit, sex, years of education, and ethnicity as covariates. Supplementary analyses were conducted to examine the effect of baseline disease severity on ε4-related decline, and to investigate potential differences in ε4-related decline across demographically stratified groups.
RESULTS
Descriptive Statistics
Table 1 outlines follow-up, demographic, and clinical data in each of the three samples.
Table 1.
Demographic and clinical data by ε4 status in population and clinic AD samples
| WHICAP: Population Incident | WHICAP: Population Prevalent | Predictors II: Clinic Based | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ε4 | −ε 4 | All | ε4 | −ε 4 | All | ε4 | −ε 4 | All | |
| Years Followed | 3.9 (2.8) | 3.9 (2.4) | 3.9 (2.5)* | 4.4 (2.9) | 5.2 (3.3) | 5.0 (3.1)* | 3.2 (1.7) | 3.1 (1.5) | 3.2 (1.6) |
| Visits | 2.9 (1.3) | 2.9 (1.2) | 2.9 (1.3)* | 3.4 (1.6) | 3.8 (1.7) | 3.6 (1.6)* | 7.2 (2.8 ) | 6.8 (2.5) | 7.1 (2.6) |
| Baseline Cognition | −1.1 (.5) | −1.1 (.5) | −1.0 (.5)* | −1.3 (.5) | −1.3 (.6) | −1.3 (.6)* | 21.9 (3.6) | 22.7 (3.5) | 22.2 (3.6) |
| % original sample with gentoyping | NA | NA | 86* | NA | NA | 70* | NA | NA | 71 |
| % ε4 | NA | NA | 33 | NA | NA | 36 | NA | NA | 56 |
| % ε4 homozygous | 4 | NA | 3 | 5 | NA | 5 | 11 | NA | 11 |
| Baseline Age | 81.6 (6.2) | 82.1 (6.7) | 82.0 (6.5) | 81.3 (7.1) | 80.3 (7.1) | 80.7 (7.1) | 73.7 (7.5)^ | 77.4 (7.6)^ | 75.3 (7.7) |
| Education (Years) | 6.4 (4.9) | 6.3 (4.3) | 6.3 (4.5) | 5.9 (4.2) | 5.9 (4.3) | 5.9 (4.2) | 15.0 (3.0) | 14.3 (3.5) | 14.7 (3.3) |
| % Women | 79 | 71 | 73 | 79 | 74 | 76 | 55 | 61 | 58 |
| % Caucasian | 5 | 9 | 8 | 7 | 12 | 10 | 91 | 97 | 94 |
| % Hispanic | 59 | 62 | 61 | 61 | 62 | 62 | 0 | 0 | 0 |
| % AA | 36 | 28 | 31 | 33 | 26 | 28 | 8 | 1 | 5 |
| % HTN | 62 | 66 | 65 | 73 | 63 | 67 | 32 | 37 | 34 |
| % Diabetes | 27 | 23 | 24 | 18 | 24 | 21 | 8 | 8 | 8 |
| % Stroke | 23 | 13 | 17 | 18 | 19 | 19 | NA | NA | NA |
Note. Mean (SD) are reported for continuous data. Years followed and number of visits were calculated from the first study visit in prevalent cases and the diagnostic visit in incident cases. Baseline = First visit for prevalent cases and diagnostic visit for incident cases. Baseline cognition is Mini Mental State Examination in the clinic sample and composite cognitive score in WHICAP. AA = African American; HTN = Hypertension. History of vascular risk factors was assessed at baseline. NA = Not applicable.
Significant across Prevalent and Incident Cases (p < .05).
Significant within sample differences across ε4 group.
GEE: Main Effects
Table 2 details the predictive value of time, ε4 status, and the time × ε4 interaction in each of the three samples using several models. As expected, there was a significant time effect in all models, indicating that cognitive scores declined over time in the non-carriers. There was no main effect of the ε4 allele on baseline performance in any model.
Table 2.
GEE Models examining the predictive value of ε4 for rate of global cognitive decline in three AD samples
| WHICAP: Population Incident | WHICAP: Population Prevalent | Predictors II: Clinic Based | ||||
|---|---|---|---|---|---|---|
| Demographic Adjusted | Beta | p | Beta | p | Beta | p |
| Time | −.05 | <.01 | −.04 | <.01 | −1.4 | <.01 |
| ε 4 | .11 | .16 | <.01 | .96 | −.61 | .38 |
| ε 4 × Time | −.06 | .01 | −.02 | .39 | −.58 | .12 |
| High Baseline Cognition | Beta | p | Beta | p | Beta | p |
| Time | −.08 | .00 | −.05 | .00 | −1.1 | .00 |
| ε 4 | .07 | .42 | .00 | .98 | −.05 | .94 |
| ε 4× Time | −.07 | .02 | −.06 | .04 | −1.0 | .03 |
| Low Baseline Cognition | Beta | p | Beta | p | Beta | p |
| Time | −.02 | .33 | −.03 | .04 | −2.2 | .00 |
| ε4 | .12 | .16 | −.02 | .85 | .28 | .71 |
| ε 4 × Time | −.06 | .04 | .00 | .95 | .38 | .47 |
Note. All adjusted model includes age at diagnosis (baseline age for prevalent cases), sex, and education. Models in the population samples are also adjusted for African American and Hispanic ethnicities. Beta values are not directly comparable across the clinic and population studies as the dependent variable differs across studies. Baseline = First visit for prevalent cases and diagnostic visit for incident cases High Baseline Cognitive Score = Analyses restricted to individuals performing in the top 50th percentile for composite cognitive score at baseline. Low Baseline Cognitive Score = Analyses restricted to individuals performing in the bottom 50th percentile for composite cognitive score at baseline. NA = Analysis not applicable.
Unavailable = Data not collected.
GEE: Rate of Change by APOE genotype
Incident Sample
Demographically adjusted models reveal that cognitive scores in ε4 carriers declined more quickly than non-carriers by an additional 6% of a z-score per year (p = .01). See Figure 1.
Figure 1. Predicted cognitive decline by ε4 status in Incident AD.
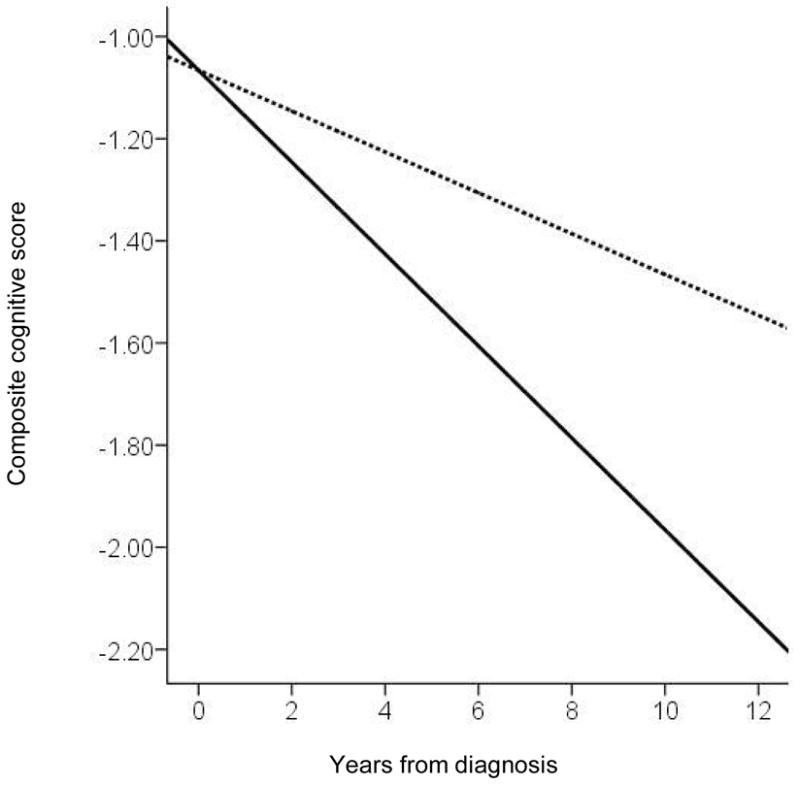
Dotted line = non-carriers
Solid line = ε4 carriers
Prevalent Sample
Rate of cognitive change did not differ significantly as a function of ε4 status in this sample. We thus hypothesized that the relationship between ε4 and rate of cognitive decline may attenuate after greater disease duration or severity of illness. When we restricted analyses to subjects with cognitive scores above the 50th percentile at baseline, there was a significant interaction between time and ε4 comparable to that seen in the incident sample. In contrast, when analyses were restricted to subjects with cognitive scores below the 50th percentile at baseline, ε4 was unrelated to rate of decline in the prevalent samples. (See Table 2).
Clinic Sample
Results for all GEE models were comparable to those reported for the prevalent population-based sample such that rate of cognitive change did not differ significantly as a function of ε4 status until analyses were restricted to subjects with cognitive scores above the 50th percentile at baseline. However, in this sample, we were also able to adjust the primary GEE model for duration of illness (ε4 carriers: M (SD) = 4.25 (2.21) years; non-carriers: M (SD) = 3.84 (2.12) years, p = .25) which revealed that ε4 carriers declined an additional 1.4 points on the MMSE than non-carriers per year (p <.01). This statistical correction was possible only in the clinic-based sample in which the treating neurologist estimated the onset of illness based on discussion with the family.
Stratified Models
Demographically stratified analyses were conducted in the incident sample to further explore the effect of ε4 on cognitive decline (Table 3). Analyses were restricted to the incident sample in which there was a clear effect of ε4 on rate of decline in primary analyses. The ε4 allele was associated with faster rates of cognitive decline in women, Hispanics, Caucasians, and more highly educated participants. However, follow-up GEE analyses examining the interaction between these demographic variables, ε4 status and time, provided strong support only for faster decline in Caucasian ε4 carriers (β = −.46, p < .01) and marginal support for faster decline in more highly educated ε4 carriers (β = −.12, p = .05).
Table 3.
Stratified GEE Models examining the influence of ε4 for rate of decline in the Incident sample
| Stratification Sample | n | Beta | p |
|---|---|---|---|
| Young (< 82) | 101 | −.05 | .08 |
| Old (>= 82) | 98 | −.07 | .05 |
| Women | 146 | −.07 | <.01 |
| Men | 53 | −.01 | .87 |
| Hispanic | 121 | −.07 | .01 |
| African American | 61 | −.02 | .68 |
| Caucasian | 15 | −.52 | <.01 |
| Low Education (<= 6) | 105 | −.04 | .14 |
| High Education (> 6) | 93 | −.11 | <.01 |
Note. Beta values represent the rate of change over time as a function of ε4 status. Age and education groups were based on a median split. Models within a demographic category included the remaining demographic variables as covariates.
DISCUSSION
The factors which determine rate of progression in Alzheimer’s disease remain incompletely understood. It has been proposed that carriers of the ε4 allele may demonstrate accelerated cognitive decline (3) due to the allele’s cumulative impact on beta amyloid and neurofibrillary tangle biochemical pathways (20, 35, 36). However, studies directly examining decline as a function of APOE genotype have produced markedly inconsistent findings (4–6, 8–10, 12, 14, 17–21). In the current study, we had a unique opportunity to examine the influence of APOE- ε4 on cognitive decline in both population- and clinic-based samples of participants with AD.
The population-based samples were primarily Hispanic and African American, and were on average older and less educated than the predominantly Caucasian participants in the clinic-based sample. There was also a lower proportion of ε4 carriers in the population-based samples than in the clinic sample, consistent with the older age of the population samples (37), and the fact that participants in the population samples were primarily Hispanic and African American, ethnic groups in which there may be a weaker association between ε4 and AD onset leading to an under-representation of the ε4 allele in demented participants (38). Overall, however, rates of ε4 in all samples were comparable to other studies of AD and higher than rates in the general population (38).
The influence of ε4 on rate of cognitive decline varied across samples. In the incident sample, we were able to examine rate of change from the earliest stages of clinically observable AD. In this sample, the presence of the ε4 allele was associated with more rapid cognitive decline, even after adjustment for potential demographic contributors to disease course, suggesting a robust relationship between the ε4 allele and faster cognitive decline in incident AD. In contrast, ε4 was not associated with rate of change in either of the prevalent AD samples (population or clinic-based). As a possible explanation for this inconsistency, we hypothesized that more advanced illness among prevalent cases may have limited our ability to detect ε4-associated decline. In fact, when we accounted for disease severity by adjusting for disease duration or by restricting analyses to those with better cognitive function at baseline we were able to demonstrate faster decline in ε4 carriers in both prevalent samples.
Our interpretation of the overall pattern of results is that ε4 influences cognitive decline most clearly in the earliest stages of disease and less so, or not at all, in the moderate to severe stages. There may be several explanations for this. First, to the extent that our cognitive batteries are less sensitive to change as the disease progresses, we may be unable to detect a differential role for ε4 on rate of change in the later stages. It is possible that a different cognitive assessment tool or a functional measure would better capture differential rates of decline further into the disease. A second possibility is that the influence of the ε4 allele may be time limited, until some neurobiological threshold is crossed and after which medical, social, or disease related variables other than ε4 become more prominent in determining rate of decline.
Investigations documenting faster rates of decline in ε4 carriers (8–10, 12, 13) included two of the largest studies to date. Convincingly, a recent study detected faster rates of decline using both a linear and non-linear mixed-effect statistical model (8). Faster rates of cognitive decline in ε4 carriers is consistent with the well established role of APOE ε4 as a risk factor for developing AD (35, 39), and parallels findings from imaging studies suggesting accelerated rates of neurodegenerative change in ε4 carriers (40, 41).
Three studies, including an early study from our group, found slower decline among ε4 carriers than non-carriers. However, the earliest study retrospectively estimated baseline MMSE score based on age and education predicted values (4), and is thus less compelling than the remaining studies which used growth curve analysis (GCA) to determine rate of progression (5, 6). The GCA study from our group likely suffered from selective attrition as APOE genotyping was done 6 years after the study began. It is thus possible that ε4 carriers progressed more quickly in the early years of the study, and were more likely to have been lost to follow up by the time ε4 data was collected. Additionally, as genotyping was completed on 42% of participants in the first cohort compared with 71% in the second (current) cohort, the current study better represents the population of ε4 carriers. In fact, re-examination of the Predictors I data with additional follow-up data using the GEE model described in the current paper found no relationship between ε4 status and rate of decline in that cohort (data not shown).
In the current study, stratified analyses revealed that ε4 influenced rate of change differentially across a number of demographic subgroups. Specifically, ε4 appears to influence rate of decline primarily in Caucasians rather than Hispanics and African Americans. This pattern of results is largely consistent with the literature examining the risk for developing AD (38) although there is some evidence for ε4 as a factor in Hispanic (42, 43) and African American populations as well (44).
APOE status may also interact with educational level; results offered marginal support for the ε4 allele predicting faster decline in those with higher education (greater than six years) only. The relevance of this finding is unclear, and further work is needed to more thoroughly investigate this potential interaction. One possibility is that ε4 exacerbates the increased rate of decline previously observed in individuals with higher education (25).
Although initial stratified analyses revealed a differential effect of ε4 by sex, follow-up GEE analyses did not support this finding. A sex-specific role of ε4 has been noted in previous studies reporting increased 4-related entorhinal atrophy in early AD most prominently in women (45), and increased risk for age-related cognitive decline in women carrying the ε4 allele (46). Studies examining the development of late-onset familial AD have also revealed a sex-associated role of APOE genotype with the ε4 allele conferring greater risk in women (47). In contrast, a recent study linked ε4 to mortality rate in men only (48); however, the processes contributing to disease development and rate of decline early in the disease course may differ from those contributing to mortality at a later stage. Future work is needed to directly examine this potential discrepancy.
The current study was limited by the fact that ε4 genotyping was not available for 100% of individuals, introducing the possibility that participants do not accurately represent the distribution of ε4 in the population. Individuals excluded from the study were largely similar to the analysis sample with regard to basic demographic and clinical variables, except that excluded individuals were generally older and more highly educated than participants in each of the final samples. A third limitation to the current study may be the use of a linear rather than non-linear approach to model cognitive decline, a technique that may have restricted our ability to observe differential rates of decline in the more impaired participants.
Acknowledgments
Dr. Cosentino was responsible for the preparation of this manuscript. Drs. Cosentino, Scarmeas, Helzner, Glymour, and Stern were involved in the data analysis, interpretation, and critical review of this manuscript. Drs. Stern, Brandt, Albert, and Blacker were involved in the critical review of this manuscript as well as the conceptualization of this research question and study design. This research was supported by federal grants AG0732, AG00261, RR00645, and R01AG007370. A portion of this work was presented at the 10th International Conference on Alzheimer’s Disease and Related Disorders in Madrid, Spain (July 2006). The authors wish to thank Dr. Nicole Schupf for her thorough review of this manuscript and her insightful contributions.
Footnotes
Dr. Cosentino conducted the statistical analyses at Columbia University Medical Center.
Drs. Cosentino, Scarmeas, Helzner, Glymour, Brandt, Albert, and Blacker have no relevant disclosures. Dr. Stern provides consultation to pharmaceutical companies unrelated to this study including Elan, Esai, and Wyeth.
References
- 1.Hui JS, Wilson RS, Bennett DA, Bienias JL, Gilley DW, Evans DA. Rate of cognitive decline and mortality in Alzheimer’s disease. Neurology. 2003;61(10):1356–1361. doi: 10.1212/01.wnl.0000094327.68399.59. [DOI] [PubMed] [Google Scholar]
- 2.Storandt M, Grant EA, Miller JP, Morris JC. Rates of progression in mild cognitive impairment and early Alzheimer’s disease. Neurology. 2002;59(7):1034–1041. doi: 10.1212/wnl.59.7.1034. [DOI] [PubMed] [Google Scholar]
- 3.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 4.Frisoni GB, Govoni S, Geroldi C, Bianchetti A, Calabresi L, Franceschini G, et al. Gene dose of the epsilon 4 allele of apolipoprotein E and disease progression in sporadic late-onset Alzheimer’s disease. Ann Neurol. 1995;37(5):596–604. doi: 10.1002/ana.410370509. [DOI] [PubMed] [Google Scholar]
- 5.Stern Y, Brandt J, Albert M, Jacobs DM, Liu X, Bell K, et al. The absence of an apolipoprotein epsilon4 allele is associated with a more aggressive form of Alzheimer’s disease. Ann Neurol. 1997;41(5):615–620. doi: 10.1002/ana.410410510. [DOI] [PubMed] [Google Scholar]
- 6.Hoyt BD, Massman PJ, Schatschneider C, Cooke N, Doody RS. Individual growth curve analysis of APOE epsilon 4-associated cognitive decline in Alzheimer disease. Arch Neurol. 2005;62(3):454–459. doi: 10.1001/archneur.62.3.454. [DOI] [PubMed] [Google Scholar]
- 7.Marra C, Bizzarro A, Daniele A, De Luca L, Ferraccioli M, Valenza A, et al. Apolipoprotein E epsilon4 allele differently affects the patterns of neuropsychological presentation in early- and late-onset Alzheimer’s disease patients. Dement Geriatr Cogn Disord. 2004;18(2):125–131. doi: 10.1159/000079191. [DOI] [PubMed] [Google Scholar]
- 8.Martins CA, Oulhaj A, de Jager CA, Williams JH. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology. 2005;65(12):1888–1893. doi: 10.1212/01.wnl.0000188871.74093.12. [DOI] [PubMed] [Google Scholar]
- 9.Dal Forno G, Rasmusson DX, Brandt J, Carson KA, Brookmeyer R, Troncoso J, et al. Apolipoprotein E genotype and rate of decline in probable Alzheimer’s disease. Arch Neurol. 1996;53(4):345–350. doi: 10.1001/archneur.1996.00550040085017. [DOI] [PubMed] [Google Scholar]
- 10.Craft S, Teri L, Edland SD, Kukull WA, Schellenberg G, McCormick WC, et al. Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer’ disease. Neurology. 1998;51(1):149–153. doi: 10.1212/wnl.51.1.149. [DOI] [PubMed] [Google Scholar]
- 11.Wilson RS, Bennett DA, Bienias JL, Aggarwal NT, Mendes De Leon CF, Morris MC, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910–1914. doi: 10.1212/01.wnl.0000036905.59156.a1. [DOI] [PubMed] [Google Scholar]
- 12.Hirono N, Hashimoto M, Yasuda M, Kazui H, Mori E. Accelerated memory decline in Alzheimer’s disease with apolipoprotein epsilon4 allele. J Neuropsychiatry Clin Neurosci. 2003;15(3):354–358. doi: 10.1176/jnp.15.3.354. [DOI] [PubMed] [Google Scholar]
- 13.Kanai M, Shizuka M, Urakami K, Matsubara E, Harigaya Y, Okamoto K, et al. Apolipoprotein E4 accelerates dementia and increases cerebrospinal fluid tau levels in Alzheimer’s disease. Neurosci Lett. 1999;267(1):65–68. doi: 10.1016/s0304-3940(99)00323-7. [DOI] [PubMed] [Google Scholar]
- 14.Jonker C, Schmand B, Lindeboom J, Havekes LM, Launer LJ. Association between apolipoprotein E epsilon4 and the rate of cognitive decline in community-dwelling elderly individuals with and without dementia. Arch Neurol. 1998;55(8):1065–1069. doi: 10.1001/archneur.55.8.1065. [DOI] [PubMed] [Google Scholar]
- 15.Murphy GM, Jr, Taylor J, Kraemer HC, Yesavage J, Tinklenberg JR. No association between apolipoprotein E epsilon 4 allele and rate of decline in Alzheimer’s disease. Am J Psychiatry. 1997;154(5):603–608. doi: 10.1176/ajp.154.5.603. [DOI] [PubMed] [Google Scholar]
- 16.Holmes C, Levy R, McLoughlin DM, Powell JF, Lovestone S. Apolipoprotein E: non-cognitive symptoms and cognitive decline in late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1996;61(6):580–583. doi: 10.1136/jnnp.61.6.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurz A, Egensperger R, Haupt M, Lautenschlager N, Romero B, Graeber MB, et al. Apolipoprotein E epsilon 4 allele, cognitive decline, and deterioration of everyday performance in Alzheimer’s disease. Neurology. 1996;47(2):440–443. doi: 10.1212/wnl.47.2.440. [DOI] [PubMed] [Google Scholar]
- 18.Basun H, Grut M, Winblad B, Lannfelt L. Apolipoprotein epsilon 4 allele and disease progression in patients with late-onset Alzheimer’s disease. Neurosci Lett. 1995;183(1–2):32–34. doi: 10.1016/0304-3940(94)11107-t. [DOI] [PubMed] [Google Scholar]
- 19.Growdon JH, Locascio JJ, Corkin S, Gomez-Isla T, Hyman BT. Apolipoprotein E genotype does not influence rates of cognitive decline in Alzheimer’s disease. Neurology. 1996;47(2):444–448. doi: 10.1212/wnl.47.2.444. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol. 1996;39(1):62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 21.Kleiman T, Zdanys K, Black B, Rightmer T, Grey M, Garman K, et al. Apolipoprotein E epsilon4 allele is unrelated to cognitive or functional decline in Alzheimer’s disease: retrospective and prospective analysis. Dement Geriatr Cogn Disord. 2006;22(1):73–82. doi: 10.1159/000093316. [DOI] [PubMed] [Google Scholar]
- 22.Asada T, Kariya T, Yamagata Z, Kinoshita T, Asaka A. ApoE epsilon 4 allele and cognitive decline in patients with Alzheimer’s disease. Neurology. 1996;47(2):603. doi: 10.1212/wnl.47.2.603. [DOI] [PubMed] [Google Scholar]
- 23.Farlow MR, Cyrus PA, Nadel A, Lahiri DK, Brashear A, Gulanski B. Metrifonate treatment of AD: influence of APOE genotype. Neurology. 1999;53(9):2010–2016. doi: 10.1212/wnl.53.9.2010. [DOI] [PubMed] [Google Scholar]
- 24.Slooter AJ, Houwing-Duistermaat JJ, van Harskamp F, Cruts M, Van Broeckhoven C, Breteler MM, et al. Apolipoprotein E genotype and progression of Alzheimer’s disease: the Rotterdam Study. J Neurol. 1999;246(4):304–308. doi: 10.1007/s004150050351. [DOI] [PubMed] [Google Scholar]
- 25.Scarmeas N, Albert SM, Manly JJ, Stern Y. Education and rates of cognitive decline in incident Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2006;77(3):308–316. doi: 10.1136/jnnp.2005.072306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R. Influence of education and occupation on the incidence of Alzheimer’s disease. Jama. 1994;271(13):1004–1010. [PubMed] [Google Scholar]
- 27.Stern Y, Andrews H, Pittman J, Sano M, Tatemichi T, Lantigua R, et al. Diagnosis of dementia in a heterogeneous population. Development of a neuropsychological paradigm-based diagnosis of dementia and quantified correction for the effects of education. Arch Neurol. 1992;49(5):453–460. doi: 10.1001/archneur.1992.00530290035009. [DOI] [PubMed] [Google Scholar]
- 28.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 29.Morris JC, Edland S, Clark C, Galasko D, Koss E, Mohs R, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part IV Rates of cognitive change in the longitudinal assessment of probable Alzheimer’s disease. Neurology. 1993;43(12):2457–2465. doi: 10.1212/wnl.43.12.2457. [DOI] [PubMed] [Google Scholar]
- 30.Stern Y, Folstein M, Albert M, Richards M, Miller L, Bylsma F, et al. Multicenter study of predictors of disease course in Alzheimer disease (the “predictors study”). I Study design, cohort description, and intersite comparisons. Alzheimer Dis Assoc Disord. 1993;7(1):3–21. doi: 10.1097/00002093-199307010-00002. [DOI] [PubMed] [Google Scholar]
- 31.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 32.Stern Y, Sano M, Paulson J, Mayeux R. Modified mini-mental state examination: validity and reliability. Neurology. 1987;37(suppl 1):179. [Google Scholar]
- 33.Hixson JE. Apolipoprotein E polymorphisms affect atherosclerosis in young males. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb. 1991;11(5):1237–1244. doi: 10.1161/01.atv.11.5.1237. [DOI] [PubMed] [Google Scholar]
- 34.Liang KY, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrica. 1986;73:13–22. [Google Scholar]
- 35.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohm TG, Kirca M, Bohl J, Scharnagl H, Gross W, Marz W. Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer-type neurofibrillary tangle formation. Neuroscience. 1995;66(3):583–587. doi: 10.1016/0306-4522(94)00596-w. [DOI] [PubMed] [Google Scholar]
- 37.Meyer MR, Tschanz JT, Norton MC, Welsh-Bohmer KA, Steffens DC, Wyse BW, et al. APOE genotype predicts when--not whether--one is predisposed to develop Alzheimer disease. Nat Genet. 1998;19(4):321–322. doi: 10.1038/1206. [DOI] [PubMed] [Google Scholar]
- 38.Mayeux R. Apolipoprotein E, Alzheimer disease, and African Americans. Arch Neurol. 2003;60(2):161–163. doi: 10.1001/archneur.60.2.161. [DOI] [PubMed] [Google Scholar]
- 39.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 40.Mori E, Lee K, Yasuda M, Hashimoto M, Kazui H, Hirono N, et al. Accelerated hippocampal atrophy in Alzheimer’s disease with apolipoprotein E epsilon4 allele. Ann Neurol. 2002;51(2):209–214. doi: 10.1002/ana.10093. [DOI] [PubMed] [Google Scholar]
- 41.Lehtovirta M, Kuikka J, Helisalmi S, Hartikainen P, Mannermaa A, Ryynanen M, et al. Longitudinal SPECT study in Alzheimer’s disease: relation to apolipoprotein E polymorphism. J Neurol Neurosurg Psychiatry. 1998;64(6):742–746. doi: 10.1136/jnnp.64.6.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olarte L, Schupf N, Lee JH, Tang MX, Santana V, Williamson J, et al. Apolipoprotein E epsilon4 and age at onset of sporadic and familial Alzheimer disease in Caribbean Hispanics. Arch Neurol. 2006;63(11):1586–1590. doi: 10.1001/archneur.63.11.1586. [DOI] [PubMed] [Google Scholar]
- 43.Rippon GA, Tang MX, Lee JH, Lantigua R, Medrano M, Mayeux R. Familial Alzheimer disease in Latinos: interaction between APOE, stroke, and estrogen replacement. Neurology. 2006;66(1):35–40. doi: 10.1212/01.wnl.0000191300.38571.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murrell JR, Price B, Lane KA, Baiyewu O, Gureje O, Ogunniyi A, et al. Association of apolipoprotein E genotype and Alzheimer disease in African Americans. Arch Neurol. 2006;63(3):431–434. doi: 10.1001/archneur.63.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juottonen K, Lehtovirta M, Helisalmi S, Riekkinen P, Soininen H. Major decrease in the volume of the entorhinal cortex in patients with Alzheimer’s disease carrying apolipoprotein E e4 allele. Journal of Neurology, Neurosurgery, and Psychiatry. 1998;65:322–327. doi: 10.1136/jnnp.65.3.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mortensen EL, Hogh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001;57(1):89–95. doi: 10.1212/wnl.57.1.89. [DOI] [PubMed] [Google Scholar]
- 47.Martinez M, Campion D, Brice A, Hannequin D, Dubois B, Didierjean O, et al. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol. 1998;55(6):810–816. doi: 10.1001/archneur.55.6.810. [DOI] [PubMed] [Google Scholar]
- 48.Dal Forno G, Carson KA, Brookmeyer R, Troncoso J, Kawas CH, Brandt J. APOE genotype and survival in men and women with Alzheimer’s disease. Neurology. 2002;58(7):1045–1050. doi: 10.1212/wnl.58.7.1045. [DOI] [PubMed] [Google Scholar]