

Abstract
The asialoglycoprotein (ASGP) receptor is an abundant hepatocyte-specific receptor involved in receptor-mediated endocytosis. This receptor's abundance and function is decreased by chronic ethanol administration prior to the appearance of pathology such as necrosis or inflammation. Hence, this study aimed to determine if ASGP receptor function is required to protect against liver injury by utilizing a knockout mouse model lacking functional ASGP receptor in the setting of carbon tetrachloride (CCl4) hepatotoxicity. Briefly, ASGP receptor-deficient (RD) mice and wild-type (WT) mice were injected with 1 ml/ kg body weight of CCl4. In the subsequent week, mice were monitored for liver damage and pathology (aspartate transaminase (AST), alanine transaminase (ALT) and light microscopy). The consequences of CCl4 injection were examined by measuring α-smooth muscle actin (α-SMA) deposition, contents of malondialdehyde and the percentage of apoptotic hepatocytes. After CCl4 injection, RD mice showed increased liver pathology together with significantly increased activities of AST and ALT compared to that in WT mice. There were also significantly more apoptotic bodies, lipid peroxidation and deposition of α-SMA in RD mice versus WT mice following CCl4 injection. Since these two mouse strains only differ in whether or not they have the ASGP receptor, it can be concluded that proper ASGP receptor function exerted a protective effect against CCl4 toxicity. Thus, receptor-mediated endocytosis by the ASGP receptor could represent a novel molecular mechanism that is responsible for subsequent liver health or injury.
Keywords: Carbon Tetrachloride, Asialoglycoprotein Receptor, Asialoglycoprotein Receptor Knockout, Liver injury, Receptor-mediated Endocytosis
1. Introduction
Chronic ethanol consumption causes major health problems by leading to liver injuries such as fatty liver, alcoholic hepatitis and cirrhosis [1,2]. In recent years, many pathways have been studied in search of potential mechanisms involved in the promotion of pathological features associated with alcoholic liver disease. Work from our laboratory has studied how ethanol administration affects key hepatocellular functions involved in protein trafficking, many of which utilize the abundant asialoglycoprotein (ASGP) hepatocyte receptor.
The ASGP receptor is an abundantly expressed hepatocyte-specific receptor [3]. This is a well characterized receptor [4] that has served as an excellent model to study receptor trafficking events such as receptor-mediated endocytosis. During the receptor-mediated endocytosis process, the ASGP receptor binds to its ligand and the receptor-ligand complex is then internalized by a clathrin-coated pathway. Intracellularly, these vesicles lose their clathrin coats and the resulting endosomes are acidified leading to uncoupling of the receptor-ligand complexes. The separated receptors and ligands can then be recycled back to the cell surface or trafficked to the lysosomes for degradation [5]. Ligands recognized by the ASGP receptor include glycoproteins bearing terminal galactose or N-galactosamine residues [3]. Furthermore, it has been determined that the process of apoptosis or programmed cell death, results in the formation of apoptotic bodies, which have an increased amount of desialylated glycoconjugates [6,7,8,9]. Importantly, it has been shown that the ASGP receptor recognizes and facilitates the timely removal of apoptotic bodies [6,10,11].
Previous work in our laboratory has shown that many facets of ASGP receptor-mediated endocytosis were impaired in hepatocytes isolated from animals after ethanol administration. These impairments range from decreased binding, internalization, degradation and receptor cycling [12,13,14,15,16,17]. In addition, chronic ethanol administration also decreases ASGP receptor-specific mRNA and hence the receptor content of the ASGP receptor [18]. Given that the ASGP receptor is required to remove potentially harmful desialylated glycoconjugates, the decreased receptor content caused by ethanol administration could result in increased liver damage. Thus, to determine whether ASGP receptor function is protective against liver injury, a knockout mouse model lacking the ASGP receptor (RD) was employed.
The RD mouse was generated by disrupting the MHL-2 gene that encodes the protein for the minor subunit of the ASGP receptor [19]. In the murine system, the ASGP receptor is comprised of 2 subunits that are required for a functional receptor [4]. These knockout mice show a complete lack of MHL-2 protein (minor subunit) and a substantial reduction in the expression of the MHL-1 gene (major subunit). In spite of this deletion, RD mice remain viable and fertile, have a normal lifespan and do not display any obvious phenotypic abnormalities [19,20].
In order to determine whether or not ASGP receptor provides protection from liver injury, the animal model was challenged by an acute injection of CCl4 (1 mg/ kg body weight). Carbon tetrachloride is used as a model to study hepatotoxic effects and causes liver damage through a number of mechanisms. Briefly, CCl4 can induce liver damage through the formation of reactive free radicals that can bind covalently to cellular macromolecules forming nucleic acid, protein and lipid adducts; through the induction of hypomethylated ribosomal RNA, resulting in inhibition of protein synthesis; and finally, CCl4 can affect hepatocellular calcium homeostasis [21,22]. Overall, CCl4 treatment can result in centrilobular steatosis, inflammation, apoptosis and necrosis [21,22,23]. If the damage exceeds the repair capacity of the liver, the liver will progress to fibrosis and cirrhosis [21,22].
In the present study, we administered an acute dose of CCl4 to WT and RD mice. The importance of ASGP receptor function was then ascertained by comparing the hepatic responses of these two mouse strains. Using this model, we conclude that the ASGP receptor exerts a protective effect against toxic liver injury.
2. Materials and methods
2.1. Animals
Female WT (C57BL/6 – 129SV) and RD (B6, 129SV-Asgpr2) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Animals were housed in the Animal Research Facility at the Omaha Veterans Affairs Medical Center, which has been approved by the American Association for the Accreditation of Laboratory Animal Care. Mice (20 – 25 g) were acclimated for 1 week prior to use in experiments and were allowed free access to water and fed a Purina chow diet through the experiment.
2.2. Treatment conditions
Experimental animals were given an intraperitoneal injection of CCl4 (1.0 ml/ kg body weight in olive oil; ∼200 μl/ mouse; CCl4 from Sigma-aldrich, St Louis, MO). Animals were then sacrificed at 12 h, 24 h, 48 h, 72 h, 96 h or 7-day after injection. Control animals were injected with an equivalent amount of olive oil intraperitoneally. Since the injection of olive oil did not affect the results of any assays carried out in this study, the values for the various time points were averaged and the control was presented as a single value in the subsequent results.
2.3. Collection of Tissues
Mice were sacrificed by exsanguination after being anaesthetized with sodium pentobarbital (50 ng/ g body weight; Ovation Pharmaceuticals Inc, Deerfield, IL). Serum was collected and stored at -70°C until analysis. Following removal, a portion of the liver was saved for histologic analysis and the remaining portion was freeze clamped and stored at -70°C until analysis.
2.4. Histopathological Analysis
Haematoxylin-eosin staining, followed by light microscopy analysis, was performed on the liver samples that were formalin fixed at the time of animal death. At least three different sections were examined per liver sample, with the pathologist blinded to the identity of treatment group when assessing the histology.
2.5. Measurement of Activities of AST and ALT
Activities of AST and ALT in the serum were assessed spectrophotometrically using a commercially available kit (Sigma-aldrich, St Louis, MO).
2.6. Measurement of Malonaldehyde
The content of malondialdehyde in the livers was determined by the method of Uchiyama and Mihara [24]. The standard curve (0 – 12.5 nmol) was produced using malondialdehyde that was a gift from Dr. Geoffrey Thiele (VA Hospital, Omaha, NE). Protein contents were determined using a BCA Protein Reagent Assay Kit (Pierce, Rockford, IL).
2.7. Western Blot Analysis of α-Smooth Muscle Actin
Liver homogenates (20% w/ v in 0.1 M Tris/ 0.25 M Sucrose, pH 7.5) were adjusted to 2 – 5 mg ml-1 in Laemmli denaturing sample buffer [25]. Aliquots of the suspension were resolved on a 10% SDS-PAGE gel and proteins were transferred to a nitrocellulose membrane. After electrotransfer, the blots were blocked for one hour at room temperature in blocking buffer containing 0.01 M Tris, 0.1 M NaCl, 0.1 % Tween 20 and 5% milk (pH 7.5). The blots were then incubated with a 1:5000 dilution of monoclonal anti-α smooth muscle actin antibody (clone 1A4, Sigma-aldrich, St Louis, MO) in blocking buffer (overnight at 4°C). Following several washes in buffer containing 0.01 M Tris, 0.1 M NaCl and 0.1% Tween 20 (pH 7.5), the blots were incubated in a 1:2500 dilution of alkaline phosphatase-conjugated mouse IgG secondary antibody (Sigma-aldrich, St Louis, MO) diluted in blocking buffer (1 h at room temperature). Following several washes in buffer, the immunoreactive proteins were visualized colorometrically and quantified using a Bio-Rad FluorS-MultiImager and Quantity One software (Bio-Rad, Hercules, CA). All other reagents were reagent grade and obtained from commercially available sources.
2.8. Immunohistochemical Analysis of Apoptotic Hepatocytes
The presence of apoptotic hepatocytes was determined qualitatively using the Deadend™ colorimetric TUNEL system (Promega, Madison, WI) according to the manufacturer's instructions. An average of 3000 total cells per animal was counted in order to determine the percent of apoptotic hepatocytes.
2.9. Statistical Analysis
Results are presented as means ± standard error of the mean (S.E.M.). Comparisons between the control and various time points were carried out using one-way analysis of variance followed by Bonferroni's multiple range test to evaluate differences between means. Comparisons between WT and RD mice were carried out using independent t-tests. Arcsine transformation was applied to all percentage data before statistical analysis. Differences with P < 0.05 were regarded as statistically significant.
3. Results
3.1. Injection of CCl4 increased activities of AST and ALT and increased liver pathology to a greater extent in RD versus WT mice
Activities of AST and ALT showed a similar trend after CCl4 injection (Fig. 1). AST (Fig. 1A) and ALT (Fig. 1B) activities were significantly increased 48 h after CCl4 injection for both WT and RD. At this time point, AST activities for WT and RD mice were 63- and 79-fold increased over the control value respectively. For ALT, activities for WT and RD mice were 311- and 668-fold increased over the control value respectively. In addition, for activities of AST and ALT 48 h after CCl4 injection, RD values were significantly greater than WT values by 1.3- and 1.8-fold respectively.
Fig. 1.

Activities (U l-1) of (A) aspartate transaminase (AST) and (B) alanine transaminase (ALT), in the serum of wild-type (WT) and ASGP receptor-deficient (RD) mice after injection with CCl4. WT values are represented in open bars and RD values in filled bars. Controls were injected with an equivalent amount of olive oil. Values represent means ± S.E.M. with N = 3 – 7. *Significantly different from the corresponding WT condition, P < 0.05. #Significantly different from the corresponding control condition, P < 0.05.
Liver damage was also assessed by histology. Control livers for WT (Fig. 2A) and RD (Fig. 2D) mice were similar in appearance and showed no pathology. However, by 48 h after CCl4 injection, centrilobular liver damage was apparent in both WT (Fig. 2B) and RD (Fig. 2E) mice. The damage was more severe in RD mice, which showed a greater number of neutrophilic inflammatory infiltrates. Although AST and ALT activities returned to normal by 72 h, damage could still be seen histologically up to 96 h. At this point, RD mice still showed more damage than WT mice.
Fig. 2.
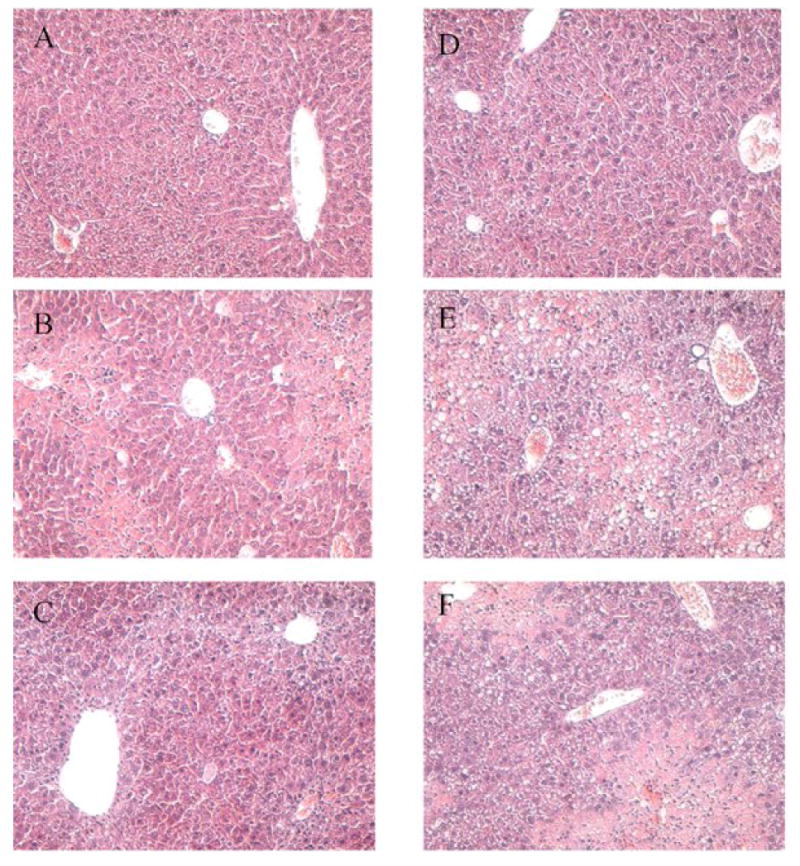
Histology of livers from wild-type (WT) and ASGP receptor-deficient (RD) mice after injection with CCl4. Paraffin embedded sections were prepared and stained with H & E. Controls were injected with an equivalent amount of olive oil. Photomicrographs show representative liver sections at 100 times magnification. (A) WT, control, (B) WT, 48 h after CCl4 injection, (C) WT, 96 h after CCl4 injection, (D) RD, control, (E) RD, 48 h after CCl4 injection and (F) RD, 96 h after CCl4 injection. Note, the more severe necrosis in the RD (E) versus WT (B) mice at 48 h and even at 96 h after CCl4 injection (F versus C).
3.2. Lipid peroxidation was increased in RD but not WT livers following injection of CCl4
The amount of malondialdehyde in liver tissue serves as an indicator of lipid peroxidation, which is a noted occurrence in liver injury due to the generation of reactive oxygen species. The levels of malondialdehyde in WT mice were not significantly different through the course of the experiment (Fig. 3). However, malondialdehyde in RD mice increased significantly over control values at 24 h and 48 h after CCl4 injection by 2.5- and 2.6-fold respectively. These values were also significantly different from that of the WT mice. Subsequently, malondialdehyde decreased by 72 h and continued to decrease till 7 days following CCl4 injection to reach levels reminiscent of the control condition (Fig. 3).
Fig. 3.

Content (nmol mg protein-1) of malondialdehyde (MDA) in the liver of wild-type (WT) and ASGP receptor-deficient (RD) mice after injection with CCl4. WT values are represented in open bars and RD values in filled bars. Controls were injected with an equivalent amount of olive oil. Values represent means ± S.E.M. with N = 3 – 7. *Significantly different from the corresponding WT condition, P < 0.05. #Significantly different from the corresponding control condition, P < 0.05.
3.3. α-SMA levels were increased to a greater extent in RD versus WT mice after CCl4 injection
α- SMA levels were increased significantly in WT and RD mice compared to control (Fig. 4). WT mice had increased levels 72 h after CCl4 injection (5.1-fold), whilst RD mice had a more prolonged increase at 48 and 72 h after injection (3.6- and 3.2-fold respectively). In addition, α-SMA levels in RD mice were increased significantly over WT mice at 48 h, 96 h and 7 day after injection by 1.9-, 2.1- and 1.7-fold respectively.
Fig. 4.

Content (O.D. μg DNA-1) of α-smooth muscle actin (α-SMA) in the liver of wild-type (WT) and ASGP receptor-deficient (RD) mice after injection with CCl4. WT values are represented in open bars and RD values in filled bars. Controls were injected with an equivalent amount of olive oil. Values represent means ± S.E.M. with N = 3 – 9. *Significantly different from the corresponding WT condition, P < 0.05. #Significantly different from the corresponding control condition, P < 0.05.
3.4. Injection of CCl4 increased hepatocyte apoptosis to a greater extent in RD versus WT mice
The presence of apoptotic hepatocytes as detected by TUNEL, was found to be significantly increased in both WT and RD mice at 48 h after CCl4 injection (Fig. 5). However, mice lacking a functional ASGP receptor displayed 2.4-fold more apoptotic hepatocytes than WT mice.
Fig. 5.

Apoptotic hepatocytes (%) in the liver of wild-type (WT) and ASGP receptor-deficient (RD) mice after injection with CCl4. WT values are represented in open bars and RD values in filled bars. Controls were injected with an equivalent amount of olive oil. Values represent means ± S.E.M. with N = 3 – 4. *Significantly different from the corresponding WT condition, P < 0.05. #Significantly different from the corresponding control condition, P < 0.05.
4. Discussion
It is well documented that alcohol perturbs many aspects of liver homeostasis. Clinically, fatty liver is observed in the early stage followed by apoptosis, necrosis, alcoholic hepatitis, fibrosis and cirrhosis in the later stages. Our laboratory has been interested for several years in receptor-mediated endocytosis (particularly that mediated by the asialoglycoprotein (ASGP) receptor) and its role in furthering hepatocellular damage in alcoholic liver disease.
We have found that chronic ethanol administration markedly decreased ASGP receptor mRNA expression and receptor content prior to the appearance of pathology such as necrosis or inflammation [18,26]. This suggests that impairments in ASGP receptor function may initiate a cascade of events leading to subsequent liver injury. In order to examine the ASGP receptor's role in the progression of pathological disease, knockout mice that lack the ASGP receptor (RD) were used in the setting of CCl4-induced hepatotoxicity. Differences in responses between the mouse strains can be attributed to the loss of ASGP receptor function.
The results of this study demonstrated that RD mice had significantly higher activities of AST and ALT. These results indicate that RD mice sustained greater liver injury than wild-type mice, since AST and ALT are sensitive indicators of liver cell injury [27]. This was further supported by histological evidence showing that RD mice had larger necrotic foci and more severe neutrophilic inflammatory infiltrates. Since these mice differed only in the presence or absence of the ASGP receptor, it is proposed that proper receptor function may exert a protective effect during toxicant-induced liver damage.
Another aspect that the WT and RD mice differed on was the levels of malondialdehyde. Malondialdehyde is a reactive aldehyde that is cytotoxic [21]. Since it is formed as a by-product of lipid peroxidation, it can also be used as an indicator of the amount of lipid peroxidation. Lipid peroxidation occurs when free radicals produced by the metabolism of CCl4 attack polyunsaturated fatty acids in the cellular membrane, thereby initiating the subsequent lipid peroxidation and resulting ultimately in a loss in membrane integrity [21,22]. In this study, the earliest significant change observed in RD mice as compared to WT mice was the enhanced level of malondialdehyde content measured at 24 h. Thus, this observation suggests that lipid peroxidation is an early event in liver injury and that the free radicals generated may play a role in subsequent damage, leading to necrosis, apoptosis and extracellular matrix deposition. Presently, evidence is accumulating to suggest that free radicals, (1) can cause oxidative mitochondrial damage (that directly causes hepatocyte death or favors alcohol-induced sensitization to the pro-apoptotic action of TNF-α); (2) can regulate gene expression contributing to the development of fibrosis; (3) can perpetuate chronic inflammatory processes [28].
Although the reason(s) behind how the ASGP receptor exerts its protection is unclear, it is recognized that rat hepatocytes undergo apoptosis after injection of CCl4 [23]. Our results also showed that apoptotic hepatocytes are significantly increased at 48 h and this increase is significantly greater in RD mice compared to WT mice. Increased accumulation of apoptotic cells has also been shown during alcohol-induced injury in a variety of species including humans and is thought to play an important role in the progression of liver injury [29,30,31,32,33,34,35]. Since the ASGP receptor has been shown to play a key role in the clearance of apoptotic bodies [11], we speculate that the ASGP receptor exerts a protection through timely removal of apoptotic bodies, preventing secondary necrosis and subsequent leakage of potentially harmful materials. Interestingly, despite the absence of functional ASGP receptor, apoptotic cell numbers in RD mice decrease back to baseline by 72 h as in WT mice. The removal of apoptotic bodies in this case is probably carried out by redundant pathways that recognize exposed terminal sialic acids, externalized phosphatidylserine or modifications that have occurred to membrane lipid asymmetry [36]. Depending on the cell type, the receptors responsible could include several members, such as the scavenger receptor family, the αvβ3 vitronectin receptor or CD14 [36,37]. However, it is important to note that these cannot totally compensate for the ASGP receptor's role since there is a significant increase in apoptotic cell number at 48 h in RD compared to WT mice. A further consequence of this impaired uptake in RD mice could be the enhanced phagocytosis of apoptotic bodies by non parenchymal cells such as kupffer cells or hepatic stellate cells. Uptake of apoptotic bodies by kupffer cells stimulates the generation of death ligands, including Fas ligand and tumor necrosis factor α, leading to increased hepatocyte apoptosis, increased neutrophil infiltration, and markers of stellate cell activation [38]. Uptake of apoptotic bodies by stellate cells induces transforming growth factor β expression and NADPH oxidase activation (which results in the upregulation of procollagen α1) [39]. Thus, uptake of apoptotic cells by non parenchymal cells such as kupffer cells and stellate cells, could contribute to the progression of liver disease.
Another possible way by which the ASGP receptor may be protective is through its role in regulating the turnover of extracellular matrix. We showed here that RD mice displayed increased α-SMA deposition 24 h earlier than WT and that α-SMA deposition remained increased up to 7 days. This was in contrast to the WT mice, which only showed a spike at 72 h. Thus, RD mice were compromised in their ability to adjust their extracellular matrix turnover, putting them at an increased risk of developing fibrosis. Similarly, it is attractive to speculate that the deposition of other matrix proteins, such as the glycoprotein fibronectin, may also be enhanced as a consequence of altered ASGP receptor function. Particularly, the insoluble form of fibronectin, cellular fibronectin, is deposited as filaments in the extracellular matrix and is one of the first extracellular matrix proteins to accumulate during fibrosis [40,41,42]. Cellular fibronectin displays a high density of terminal galactose residues, which make it a potential ligand for the ASGP receptor [42,43]. Indeed, studies have shown that the pattern of uptake and degradation of 125I-cellular fibronectin from preloaded liver slices is very similar to that of 125I-asialofetuin (a selective ligand of the ASGP receptor) [42]. Furthermore, the prior infusion of excess asialofetuin or the addition of a terminal sialic acid to the cellular fibronectin inhibits the removal of cellular fibronectin [42,43]. Thus, this suggests that the ASGP receptor is involved in the uptake of cellular fibronectin and as such decreased ASGP receptor function could lead to increased extracellular matrix deposition and hence lead to fibrosis and cirrhosis.
In summary, this study demonstrates that the absence of ASGP receptor led to increased liver pathology and indices of liver damage. Additional consequences of the missing receptor were deposition of α-SMA, increased apoptosis and increased lipid peroxidation. Thus, we conclude that proper ASGP receptor function is essential for protection against liver injury. At present, the exact mechanism by which damage occurs in the absence of ASGP receptor is not known. However, utilizing the knockout mouse model employed in this study may enhance the delineation of potential mechanisms. Overall, understanding how impaired receptor-mediated endocytosis leads to liver injury could represent a novel molecular mechanism through which the ASGP receptor protects against liver injury. This new line of inquiry may provide therapeutic leads for protection and possibly treatment of liver disease.
Acknowledgments
This work was supported by grants from the National Institute on Alcohol Abuse and Alcoholism (AA07846 and KAA015577) and the Department of Veterans Affairs.
Abbreviations
- ASGP
asialoglycoprotein
- CCl4
carbon tetrachloride
- RD
asialoglycoprotein receptor-deficient
- WT
wild-type
- AST
aspartate transaminase
- ALT
alanine transaminase
- α-SMA
α-smooth muscle actin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dufour MC, Stinson FS, Caces MF. Trends in cirrhosis morbidity and mortality: United States, 1979-1988. Semin Liver Dis. 1993;13:109–25. doi: 10.1055/s-2007-1007343. [DOI] [PubMed] [Google Scholar]
- 2.French SW. Ethanol and hepatocellular injury. Clin Lab Med. 1996;16:289–306. [PubMed] [Google Scholar]
- 3.Ashwell G, Harford J. Carbohydrate-specific receptors of the liver. Annu Rev Biochem. 1982;51:531–54. doi: 10.1146/annurev.bi.51.070182.002531. [DOI] [PubMed] [Google Scholar]
- 4.Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS, Anderson RGW, Russell DW, Schneider WJ. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annu Rev Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 6.Dini L, Autuori F, Lentini A, Oliverio S, Piacentini M. The clearance of apoptotic cells in the liver is mediated by the asialoglycoprotein receptor. FEBS Lett. 1992;296:174–8. doi: 10.1016/0014-5793(92)80373-o. [DOI] [PubMed] [Google Scholar]
- 7.Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest. 1989;84:1518–27. doi: 10.1172/JCI114328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in a neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–75. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savill JS, Dransfield I, Hogg N, Haslett C. Vitronectin receptor-mediated endocytosis of cells undergoing apoptosis. Nature. 1990;343:170–3. doi: 10.1038/343170a0. [DOI] [PubMed] [Google Scholar]
- 10.Dini L, Lentini A, Diez GD, Rocha M, Falasca L, Serafino L, et al. Phagocytosis of apoptotic bodies by liver endothelial cells. J Cell Sci. 1995;108:967–73. doi: 10.1242/jcs.108.3.967. [DOI] [PubMed] [Google Scholar]
- 11.McVicker BL, Tuma DJ, Kubik JA, Hindemith AM, Baldwin CR, Casey CA. The effect of ethanol on asialoglycoprotein receptor-mediated phagocytosis of apoptotic cells by rat hepatocytes. Hepatology. 2002;36:1478–87. doi: 10.1053/jhep.2002.37137. [DOI] [PubMed] [Google Scholar]
- 12.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Chronic ethanol administration impairs the binding and endocytosis of asialo-orosomucoid in isolated hepatocytes. J Biol Chem. 1987;262:2704–9. [PubMed] [Google Scholar]
- 13.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Ethanol-induced impairments in receptor-mediated endocytosis of asialoorosomucoid in isolated rat hepatocytes: time course of impairments and recovery after ethanol withdrawal. Alcohol Clin Exp Res. 1989;13:258–63. doi: 10.1111/j.1530-0277.1989.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 14.Casey CA, Volentine GD, Jankovich CJ, Kragskow SL, Tuma DJ. Effect of chronic ethanol administration on the uptake and degradation of asialoglycoproteins by the perfused rat liver. Biochem Pharmacol. 1990;40:1117–23. doi: 10.1016/0006-2952(90)90501-b. [DOI] [PubMed] [Google Scholar]
- 15.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Zonal differences in ethanol-induced impairments in receptor-mediated endocytosis of asialoglycoproteins in isolated rat hepatocytes. Hepatology. 1991;13:260–6. [PubMed] [Google Scholar]
- 16.Casey CA, Tuma DJ. Receptors and endocytosis. In: LeBouton AV, editor. Molecular and Cell Biology of the Liver. Boca Raton: CRC; 1993. pp. 117–41. [Google Scholar]
- 17.Casey CA, Wiegert RL, Tuma DJ. Chronic ethanol administration impairs ATP-dependent acidification of endosomes in the rat liver. Biochem Biophys Res Commun. 1993;195:1127–33. doi: 10.1006/bbrc.1993.2161. [DOI] [PubMed] [Google Scholar]
- 18.Tworek BL, Tuma DJ, Casey CA. Decreased binding of asialoglycoproteins to hepatocytes from ethanol-fed rats; consequence of both impaired synthesis and inactivation of the asialoglycoprotein receptor. J Biol Chem. 1996;271:2531–8. doi: 10.1074/jbc.271.5.2531. [DOI] [PubMed] [Google Scholar]
- 19.Ishibashi S, Hammer RE, Herz J. Asialoglycoprotein receptor deficiency in mice lacking the minor receptor subunit. J Biol Chem. 1994;269:27803–6. [PubMed] [Google Scholar]
- 20.Braun JR, Willnow TE, Ishibashi S, Ashwell G, Herz J. The major subunit of the asialoglycoprotein receptor is expressed on the hepatocellular surface in mice lacking the minor subunit. J Biol Chem. 1996;271:21160–6. doi: 10.1074/jbc.271.35.21160. [DOI] [PubMed] [Google Scholar]
- 21.Manibusan MK, Odin M, Eastmond DA. Postulated carbon tetrachloride mode of action: a review. J Environ Sci Health C. 2007;25:185–209. doi: 10.1080/10590500701569398. [DOI] [PubMed] [Google Scholar]
- 22.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–36. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 23.Shi J, Aisaki K, Ikawa Y, Wake K. Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am J Pathol. 1998;153:515–25. doi: 10.1016/S0002-9440(10)65594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchiyama M, Mihara M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–8. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Casey CA, McVicker BL, Donohue TM, McFarland MA, Wiegert RL, Nanji AA. Liver asialoglycoprotein receptor levels correlate with severity of alcoholic liver damage in rats. J Appl Physiol. 2004;96:76–80. doi: 10.1152/japplphysiol.00375.2003. [DOI] [PubMed] [Google Scholar]
- 27.Pratt DS, Kaplan MM. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N Engl J Med. 2000;342:1266–71. doi: 10.1056/NEJM200004273421707. [DOI] [PubMed] [Google Scholar]
- 28.Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 29.Benedetti A, Brunelli E, Risicato R, Cilluffo T, Jezequel AM, Orlandi F. Subcellular changes in apoptosis induced by ethanol in rat liver. J Hepatol. 1988;6:137–43. doi: 10.1016/s0168-8278(88)80024-2. [DOI] [PubMed] [Google Scholar]
- 30.Deaciuc I, Fortunato F, D'Souza N, Hill D, Schmidt J, Lee E, McClain C. Modulation of caspase-3 activity and Fas ligand mRNA expression in rat liver cells in vivo by alcohol and lipopolysaccharide. Alcohol Clin Exp Res. 1999;23:349–56. [PubMed] [Google Scholar]
- 31.Goldin RD, Hunt NC, Clark J, Wickramasinghe SN. Apoptotic bodies in a murine model of alcoholic liver disease: reversibility of ethanol-induced changes. J Pathol. 1993;171:73–6. doi: 10.1002/path.1711710115. [DOI] [PubMed] [Google Scholar]
- 32.Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34:248–53. doi: 10.1016/s0168-8278(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 33.Yacoub LK, Fogt F, Griniuviene B, Nanji AA. Apoptosis and bcl-2 protein expression in experimental alcoholic liver disease in the rat. Alcohol Clin Exp Res. 1995;19:854–9. doi: 10.1111/j.1530-0277.1995.tb00958.x. [DOI] [PubMed] [Google Scholar]
- 34.Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, et al. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol. 2001;34:254–60. doi: 10.1016/s0168-8278(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 35.Zhao M, Laissue JA, Zimmermann A. TUNEL-positive hepatocytes in alcoholic liver disease. A retrospective biopsy study using DNA nick end-labelling. Virchows Arch. 1997;431:337–44. doi: 10.1007/s004280050108. [DOI] [PubMed] [Google Scholar]
- 36.Dini L, Pagliara P, Carla EC. Phagocytosis of apoptotic cells by liver: a morphological study. Microsc Res Tech. 2002;57:530–540. doi: 10.1002/jemt.10107. [DOI] [PubMed] [Google Scholar]
- 37.Platt N, da Silva RP, Gordon S. Recognizing death: the phagocytosis of apoptotic cells. Trends Cell Biol. 1998;8:365–372. doi: 10.1016/s0962-8924(98)01329-4. [DOI] [PubMed] [Google Scholar]
- 38.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 39.Zhan S, Xiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 40.Owens MR, Cimino CD. Synthesis of fibronectin by the isolated perfused rat liver. Blood. 1982;59:1305–9. [PubMed] [Google Scholar]
- 41.Ruoslahti E, Engvall E, Hayman EG. Fibronectin: current concepts of its structure and functions. Coll Relat Res. 1981;1:95–128. doi: 10.1016/s0174-173x(80)80011-2. [DOI] [PubMed] [Google Scholar]
- 42.Rotundo RF, Rebres RA, McKeown-Longo PJ, Blumenstock FA, Saba TM. Circulating cellular fibronectin may be a natural ligand for the hepatic asialoglycoprotein receptor: possible pathway for fibronectin deposition and turnover in the rat liver. Hepatology. 1998;28:475–85. doi: 10.1002/hep.510280227. [DOI] [PubMed] [Google Scholar]
- 43.Rotundo RF, Vincent PA, McKeown-Longo PJ, Blumenstock FA, Saba TM. Hepatic fibronectin matrix turnover in rats: involvement of the asialoglycoprotein receptor. Am J Physiol. 1999;40:1189–1199. doi: 10.1152/ajpgi.1999.277.6.G1189. [DOI] [PubMed] [Google Scholar]