

Abstract
Human life expectancy and welfare has decreased because of the increase in environmental stressors in the air. An environmental stressor is a natural or human-made component present in our environment that upon reaching an organic system produces a coordinated response. This response usually involves a modification of the metabolism and physiology of the system. Inhaled environmental stressors damage the airways and lung parenchyma, producing irritation, recruitment of inflammatory cells, and oxidative modification of biomolecules. Oxidatively modified biomolecules, their degradation products, and adducts with other biomolecules can reach the systemic circulation, and when found in higher concentrations than normal they are considered to be biomarkers of systemic oxidative stress and inflammation. We classify them as metabolic stressors because they are not inert compounds; indeed, they amplify the inflammatory response by inducing inflammation in the lung and other organs. Thus the lung is not only the target for environmental stressors, but it is also the source of a number of metabolic stressors that can induce and worsen pre-existing chronic inflammation. Metabolic stressors produced in the lung have a number of effects in tissues other than the lung, such as the brain, and they can also abrogate the mechanisms of immunotolerance. In this review, we discuss recent published evidence that suggests that inflammation in the lung is an important connection between air pollution and chronic inflammatory diseases such as autoimmunity and neurodegeneration, and we highlight the critical role of metabolic stressors produced in the lung. The understanding of this relationship between inhaled environmental pollutants and systemic inflammation will help us to: 1) understand the molecular mechanism of environment-associated diseases, and 2) find new biomarkers that will help us prevent the exposure of susceptible individuals and/or design novel therapies.
Keywords: environmental stressor, lung, oxidative stress, inflammation, metabolic stressor, chronic inflammatory disease
Introduction
Biological systems are continuously exposed to oxidants, which may be generated either endogenously (e.g., from mitochondrial electron transport during respiration or during activation of phagocytes) or exogenously (e.g., pollutants, nanoparticles, dust microorganisms, ozone or cigarette smoke) [1,2]. When these oxidants are inhaled, their main target is the lung, where they may cause chronic inflammation. However, the link between our environment, the lung, and chronic inflammation has not received the attention it deserves. During the past few years, it has become clear that inhaled pollutants also cause adverse effects outside the respiratory tract, and these effects may in some cases become more important than the respiratory effects [3].
One of the most important topics of environmental research in recent years is the genetic-environment interplay as a determinant of disease susceptibility, progression, and outcome [4-6]. Although the social and economic impact of chronic inflammatory diseases in our societies is great, the role of the lung in worsening or inducing disease is poorly appreciated, particularly in connection with environmental-genetic interactions. However, there is a clear and parallel increase in chronic inflammatory disease (for example, cardiovascular diseases, autoimmunity, neurodegeneration, and cancer) associated with an increase in air pollution [1,6-8]. Air pollution and the concomitant inhalation of environmental stressors have now been associated with the worsening of pre-existing chronic inflammatory diseases such as type II diabetes [8], rheumatic autoimmune diseases [6] and neurodegenerative disorders [9,10]. In this regard, the indirect effect of inhaled stressors on cell metabolism and genetic and epigenetic factors has gained importance, and there have been several mechanisms presented explaining this complex interaction between our environment, the lung, and diseases (Figure 1). For example, a link has been established between lipid peroxidation products and genetic (mutagenesis) [11-13] and epigenetic changes (e.g., changes in DNA conformation by histone modification) [5,14].
Figure 1. The lung as a target and source of systemic oxidative stress and inflammation.
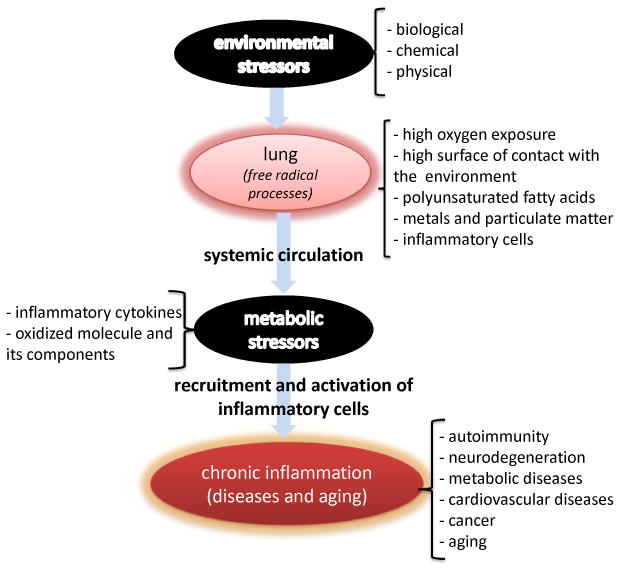
Inhaled biological, chemical, or physical environmental stressors produce irritation, recruitment, and activation of inflammatory cells in the airways and lung parenchyma. Reactive oxygen species produced by activation of these inflammatory cells damage proteins, lipids, and nucleic acids in the lung. Restoration mechanisms degrade modified macromolecules, which can reach the systemic circulation as single entities or as adducts (per example 4-HNE-modified proteins). Modified biomolecules, their degradation products and adducts of host proteins with oxidation products, inflammatory cytokines, bioactive lipids, and lipid peroxidation products can interact with cells in tissues away from the lung and alter cell physiology and metabolism depending on the presence of receptors and tissue accessibility. These modifications can elicit indirect oxidative damage and modification of tissue specific macromolecules, which can induce and or/exacerbate autoimmunity, neurodegenerative and other chronic inflammatory diseases, and aging.
In this review we discuss recent epidemiological and experimental evidence suggesting that systemic oxidative stress and inflammation are key hallmarks of inhalation exposure mediated by metabolic stressors (Figure 1). Metabolic stressors (bioactive mediators) induced by inhalation of environmental stressors may play a key pathogenic role by inducing or worsening chronic inflammatory diseases in genetically susceptible populations. Along with an increase in our understanding of the key role of lung-generated systemic inflammation and the ability to detect such a process earlier, we may be able to protect susceptible populations and find novel diagnostic strategies and therapies to prevent these diseases.
The lung as a target of inhaled environmental stressors
The lung has the largest surface area exposed to the environment in the human body. It is one of the most vascularized tissues in the body; it is a source of oxygen for loading hemoglobin and a way to eliminate the main product of metabolism (i.e., carbon dioxide) and other volatile metabolites in the exhaled breath [15]. Among the most common environmental stressors that reach our body through the lung (Table 1) are particulate matter comprising insect and microbial components, along with organic and inorganic chemicals usually associated with carbon particles, pet hair, and dust [7]. The most important vehicle is particulate matter (PM), through which microbial endotoxins, metals, and other chemicals gain access to our bodies by inhalation [7,16]. Particles smaller than 10 μm (PM10) have easy access to the airways, and if they are not removed by ciliary clearance, they may be deposited in the alveolar space and cause local inflammation in the lung.
Table 1. Environmental and metabolic stressors.
| Inhaled Environmental Stressors | |
|---|---|
| -Physical | |
| - particulate matter (carbon, silica, titanium dioxide, etc.) | |
| - diesel exhaust particles (DEP) | |
| - asbestos and silicates | |
| -Chemical | |
| - organic (agrochemicals, hydrocarbons) | |
| - inorganic (ozone, metals, N2O, N2O3, SO2, etc.) | |
| -Biological | |
| - animal (hair, feces, saliva, etc.) | |
| - insect (cockroach remains, chitin, mites, etc.) | |
| - microbial (parasites, bacteria, viruses, fungi and their components) | |
| Metabolic Stressors Produced in the lung | |
| -Oxidation products | |
| - proteins (carbonyl-, chloro-, nitro- and di-tyrosine, disulfide, methionine sulfoxide and degradation products) | |
| - lipids (malondialdehyde, F2α-isoprostanes, acrolein, saturated and unsaturated aldehydes, 4-hidroxy-2-alkenals, etc.) | |
| - sugar (glyoxal, methylglyoxal, pentocidine, acetone, etc.) | |
| - nucleic acids (uric acid, 8-oxo-dG, etc.) | |
| -Pro-inflammatory cytokines | |
| - tumor necrosis factor (TNF)-α | |
| - interleukin (IL)-1β | |
| - adipokines (leptin, bisfatin, resistin, etc.) | |
| - chemokines (IL-8, monocyte chemoattractant protein (MCP)-1, granulocyte-macrophage stimulating factor (GM-CSF)). | |
| - IL-6 | |
| -Bioactive lipids | |
| - prostaglandins | |
| - leukotrienes | |
| - thromboxanes | |
| -Acute phase proteins | |
| - C reactive protein | |
The airways and alveolar spaces are rich in redox-susceptible and redox-cycling components: polyunsaturated fatty acids (PUFAs), metals (e.g., iron), oxygen, and inflammatory cells. Inhaled environmental stressors induce redox changes that initiate a number of events leading to pulmonary inflammation. [See [17] for a recent comprehensive review about the oxidant and antioxidant profile of the lung in health and disease.] The key to explaining the role of the lung in systemic inflammation is understanding the mechanism of modification of the biomolecules produced by inhaled oxidants or generated by inflammatory cells in the upper and lower airways [18,19].
In the respiratory airways, inhaled environmental stressors usually produce irritation, a trigger of inflammation [20,21]. One of the most important aspects of irritation is the induction of the expression of chemokines such as IL-8, which attract inflammatory cells such as neutrophils and eosinophils to the airways [22-24]. Both neutrophils and macrophages are known to migrate in increasing numbers into a lung that has been exposed to particulate matter [25]. Other inhaled stressors also attract and activate neutrophils and other inflammatory cells in the lung's micro environment [22,26]; lung inflammation is mediated initially by neutrophils and later by macrophages [7,16].
Following the generation of mediators such as IL-8 by the airway and alveolar epithelia/endothelia, inflammatory cells are attracted to and home in on the pulmonary microcirculation. The inflammatory mechanism involves neutrophil adhesion to the endothelium and up-regulation of CD18 integrins, which are known to up-regulate NAPDH oxidase activity [26]. Cellular inflammation also involves the synthesis of inflammation mediators, cytokines, adhesion molecules, acute phase proteins, nucleic acid and protein oxidation products (nitrotyrosine, methionine sulfoxide, cystine, dityrosine, etc.), glycoxylation, bioactive lipids (prostaglandins, thromboxanes, leukotrienes, etc.) and lipid peroxidation products (Table 1) [24,27].
Activated inflammatory cells such as macrophages release a number of inflammatory mediators (e.g., cytokines and bioactive lipids) that can be sensed by cells in the surrounding tissue. Damage to airway epithelial/endothelial cells generates danger signals that activate the expression of genes coding for chemokines [28]. In addition, when stressors are in the airways, inflammatory cells may alter the signaling pathways and cellular responses to chemokines [22,24]. One of the most recent findings identifies adenosine triphosphate (ATP) as a danger signal that has an associated cell response. When ATP is released during airway injury, it activates dendritic cells by interacting with specific receptors (P2X and P2Y) on the cell surface, which in turn triggers purinergic signaling that is important in maintaining the asthmatic phenotype [29].
As macrophages and neutrophils in the lower respiratory tract are recruited and activated, the oxygen burden in the lungs may increase [18,26,30]. The cellular responses to such environmental stressors are controlled by reactive oxygen species (ROS), which include free radicals (superoxide radical anion, nitric oxide, radical hydroxyl, etc.) and non-free radical reactive species (H2O2, hypohalides, peroxynitrite, ozone, etc.) [31,32]. ROS can be generated directly by the stressor (for example ozone or nanoparticles) or produced by activation of the inflammatory cells in the lung [20]. The intracellular production of ROS may involve mitochondrial respiration, xanthine oxidase, and coupling to the membrane of the nicotine diamine phosphate reduced (NADPH) oxidase enzyme [27]. Activation of neutrophils and eosinophils generates superoxide radical anion (O2•−), which is rapidly converted to H2O2 by superoxide dismutase, and hydroxyl radicals are formed in the presence of Fe2+ by the Fenton reaction [17]. In neutrophils, myeloperoxidase (MPO) also catalyzes the peroxidation of chloride to form hypochlorous acid (HOCl), which requires H2O2 [33]. ROS are also released by activated lung epithelial cells and thus amplify lung inflammatory and oxidant events.
ROS are known as secondary messengers and are involved in the induction of NF-κB [34,35], the main transcriptional activator that controls the expression of a number of inflammatory genes for important mediators of inflammation such as chemokines, cytokines, and adhesion molecules [21,34-36]. Under physiological conditions, ROS molecules function as regulators of redox modulation [27], defined as a reversible change in a macromolecule necessary for the cell to adapt to a change in its environment. In these regulatory mechanisms, the ROS oxidize thiol groups at the active site of phosphatases, which inactivates their phosphatase activity [27]. Inactivation of the phosphatase activity is necessary for the accumulation of phosphorylated proteins, which are important as a cellular response to stress [27].
ROS are highly reactive and, when generated close to cell membranes, can induce lipid peroxidation and the accumulation of its products, including malondialdehyde (MDA), 4-hydroxy-2-alkenals, acrolein, and F2α-isoprostanes [19] (Table 1). Oxidized biomolecules are more susceptible to degradation, but they also can inhibit the removal of oxidatively modified proteins, such as the proteasome system [37]. When detected in elevated concentration in tissues and/or biological fluids, oxidatively modified biomolecules become important biomarkers of oxidative stress and inflammation [38].
The production of ROS in the lung almost invariably leads to depletion of antioxidants, followed by oxidation with biomolecule-centered radicals as intermediates [21,39]. Biomolecule-centered radicals decay very quickly by reacting with oxygen, by electron transfer, or by radical-radical reactions. These reactions produce oxidized species that can form adducts with other biomolecules, modifying their structure and thus their function (receptor, messenger, compartmentalization signal, enzyme, transporter, etc.) [27,40]. These changes are sensed by the cell leading to an inflammatory response and removal of the modified macromolecule by the proteasome, glycosylases, reductases, etc.) [27].
ROS can damage cellular proteins, lipids, and nucleic acids in the lung and in other inflamed tissue, such as the brain [31]. To defend against this damage, the lung and other inflamed tissues use up their stores of the key antioxidant glutathione (GSH). Failure to overcome the increased ROS production leads to production of further oxidative mediators (metabolic stressors) with activation of additional signaling pathways that regulate the expression of pro-inflammatory cytokine and chemokine genes [32]. These products are produced locally in the target tissue as well as systemically, which can lead to widespread pro-inflammatory effects remote from the lung [41].
Although a redox imbalance occurs in the lung as a natural protective response, an uncontrolled response may damage the lung and produce catastrophic systemic effects and even death [42,43]. Thus, the control of the inflammatory response, by preventing the redox process involved in the recruitment, hosting, and activation of inflammatory cells in the lung [23,24,35,44], may represent a therapeutic point to control the systemic effects of inhaled stressors.
In addition to their cytotoxic properties, ROS-modified biomolecules are increasingly recognized as being important in signal transduction for a number of important systemic diseases such as autoimmunity [45] and neurodegeneration [31,46]. Thus, when metabolic stressors are produced in the lung in response to environmental stressors, they may diffuse through the blood-brain barrier and produce neurotoxicity [31,46] or generate neoantigens [45], which can be involved in generating autoimmunity through a process known as epitope spreading [45,47].
The lung as a source of metabolic stressors
Oxidized biomolecules, their products of degradation, adducts with proteins, advanced glycoxidation end products (AGEs), and lipid peroxidation end products (ALEs) are generated [23] and eliminated in the breath [15] and/or transported to different tissues in the body [38,48-50] (Figure 1). When detected in the exhaled breath condensate [15] and/or in blood [38], these oxidation products are biomarkers of oxidative stress and inflammation. From an analytical point of view these products are used as indicators of the progress of systemic diseases (biomarkers) [15,38], but from a molecular point of view they are bioactive mediators because they can induce further stress signaling and systemic inflammatory responses [14,32,51,52]. Thus, they can be considered as metabolic stressors (Table 1), which include oxidized biomolecules, their products of degradation, adducts, and pro-inflammatory cytokines. Metabolic stressors can induce distinct responses by interacting with receptors whose distribution depends on tissues and whose affinity may depend on genetic polymorphisms [19]. Different organs vary in their distribution of receptors, and the receptors for metabolic stressors may be expressed differently depending on the endocrine profile and the cell type.
Pulmonary effects from environmental stressors include the triggering of inflammation in the airways and alveolar space, which can lead to induction or exacerbation of systemic chronic inflammatory diseases. Inflammation is the most important process occurring in the lung exposed to environmental stressors [53]. In the classical literature, inflammation is described as the principal response of the body to deal with injuries, the hallmarks of which include swelling, redness, pain, and fever. This often short-term adaptive response is a crucial component of tissue-repair remodeling and involves integration of many complex signals in distinct cells and tissues. However, the long-term consequences of prolonged inflammation are not beneficial and may be an important source of metabolic stressors (Table 1). For example, diabetic patients are more susceptible to the damaging effects of inhaled particulate matter than are healthy subjects [8], suggesting that during chronic inflammatory disease, the lung may be more susceptible to redox changes and subsequent inflammation induced by inhaled stressors.
Airway hyper-responsiveness (AHR) is one of the most significant indicators of airway inflammation induced by inhaled stressors [54], but the mechanism of the link between lung inflammation, respiratory physiology, and systemic effects of inhalation exposure to environmental stressors is unknown. Inhaled stressors, in particular ozone, chemicals associated with particles, cigarette smoke, and inflammatory cells, are the major source of ROS in the lung (Figure 1). Numerous cytokines (for example, IL-6, TNF-α, GM-CSF, and MCP-1) and acute phase proteins are involved in the activation of inflammatory cells, amplification of the oxidative damage, and the worsening of pre-existing inflammation [1,8], but the role of the oxidized products produced in the stressed lung has not been recognized. Recently this mechanism was considered following examination of data from the Framingham Heart Study [55]. Noteworthy, environmental stressors such as nanoparticles can reach the systemic circulation and induce injury in target organs [16]. Either directly (diffusion from the lung to systemic circulation) or indirectly (by metabolic stressors), tobacco smoke may produce pancreatic cancer [56] and arthritis [6] in genetically susceptible populations.
Clearly some materials deposited in the respiratory epithelia can be transported directly to other tissue and organs [16]. The transport of gases, molecules, allergens, and particles across the bronchial and alveolar epithelia/endothelia may have systemic effects in the heart and other organs [16]. Epidemiological studies continue to confirm the association between adverse health outcomes and inhalation of airborne particles and co-pollutants. For example, increased air pollution can increase heart rate variability as well as cardiac arrhythmias, especially in the elderly [1]. Moreover, experimental [41] and epidemiological studies suggest that inflammation in the lungs of patients suffering from chronic obstructive pulmonary disease, which are characterized by recruitment and activation of inflammatory cells in their airways, is associated with systemic inflammation and oxidative stress [53,55]. In addition, ultra-fine particles, especially those containing transition metals, may give rise to oxidative stress and, through low density lipoprotein oxidation, destabilize atherosclerotic plaques, leading to ischemic events [1,16,53].
Products of biomolecule oxidation are important in the propagation of lung inflammation to the systemic compartment. Such products are useful biomarkers of systemic oxidative stress and inflammation when evaluated in biological fluids. Reactive carbonyl compounds (RCCs) formed endogenously during lipid peroxidation and glycoxidation of carbohydrates are precursors of advanced glycoxidation end products (AGEs) and advanced lipid peroxidation end products (ALEs), which form cross-links on tissular proteins (carbonyl stress), and accumulate during aging and in chronic inflammatory diseases [10,50]. Oxidatively modified biomolecules and their adducts with proteins [48] can abrogate immuno-tolerance mechanisms and accumulate in the reticulo-endothelial system [46]. Oxidation of lipids induced by ROS generates a huge variety of lipid peroxidation products, including RCCs and more stable products such as ketones and alkanes [19]. RCCs such as aldehydes and dicarbonyls, including hydroxyalkenals, acrolein, malondialdehyde (MDA), glyoxal, and methylglyoxal, exhibit a large panel of biological properties that include modifications of tyrosine phosphorylation and of the cell cycle, cell development, and cell death [27,52]. These aldehydes react with cellular and tissular proteins to form adducts (ALEs) that induce protein dysfunction and alter cellular responses [57]. The signaling involved in these responses is a slow process countered by the rapid turnover of short-lived cellular proteins, whereas modified long-lived proteins accumulate and produce direct tissular damage and aging-related diseases [46].
The oxidation of polyunsaturated fatty acids (PUFAs) generates RCCs, including the highly reactive α,β-unsaturated hydroxyalkenals such as 4-hydroxynonenal (4HNE) and 4-hydroxyhexenal (4HHE) [19]. Oxidation of n-6 PUFAs (mainly linoleic and arachidonic acids) leads to the formation of 4HNE, whereas oxidation of n-3 PUFAs (decosahexanoic acid, eicosapentaenoic acid, and linolenic acid) leads to the formation of 4HHE. 4HNE can react with histidine, cysteine, or lysine residues in proteins, leading to the formation of stable Michael adducts with a hemi-acetyl structure [50,58]. The 1,2-Michael addition involves the reaction of the side chain epsilon amine group of lysine with the αβ-unsaturated carbonyl, resulting in the formation of a Schiff base at acidic pH. MDA and acrolein are formed during lipid peroxidation and bind to DNA and proteins to form adducts. MDA is one of the most abundant aldehydes resulting from peroxidation of arachidonic, eicosapentaenoic, and decodahexaenoic acid. MDA reacts with lysine residues to form Schiff bases, and plays a major role in modification of low density lipoproteins and their scavenging by macrophages, the hallmark of atherosclerosis [19].
ALE precursors play an active role in signal transduction by progressively altering the structure and function of circulating and tissular proteins, with consequences for inflammatory status and cell proliferation and viability [57]. The biological effects of ALE precursors are modulated by their local concentration, mechanism of uptake, and cellular detoxifying and metabolizing mechanisms [37,46]. For instance, ALEs are involved in inhibition of the proteasomal machinery in the cell [37], so oxidized products accumulate and cell stress occurs. Carbonyl stress and lipid peroxidation induce progressive protein modification, dysfunction, and formation of neoantigens [45]. As the lung may be an important source of oxidized and oxidant-modified biomolecules, inhibition of oxidative modifications of lung tissue exposed to inhaled stressors may prevent pathological consequences of carbonyl stress and may represent a new therapeutic strategy for patients suffering from chronic inflammatory diseases.
In human and animal studies, inhalation of particles elicits pro-inflammatory effects, cytokine production, and enhancement of allergic responses in the upper and lower airways [7,16]. PM exposure is likely to be linked to inflammation through the generation of ROS and oxidative modification of macromolecules [16]. Although there is still debate about which components of the inhaled air are responsible for producing ROS, there is accumulating evidence that pro-oxidative organic hydrocarbons, such as polycyclic aromatic hydrocarbons, quinones, and transition metals, play a role [16]. The PM provides the template for electron transfer to molecular oxygen in these redox cycling events [16]. In addition, target cells, such as airway epithelial cells and macrophages, generate ROS in response to particulate stressors by biologically active catalyzed redox reactions that occur in the cell membrane and mitochondria [7].
The role of nanoparticles in worsening cardiovascular events has been extensively investigated [7]. The precipitation of heart attacks is related to the level of PM in the air [1]. It is not only the particles that diffuse throughout the alveolar epithelium, but also the oxidized products of nanoparticle-induced activation of epithelial and endothelial cells [16]. Activation of lung epithelial cells induces the expression of adhesion molecules (e.g., intracellular adhesion molecule) and the synthesis of chemokines (e.g., IL-8, MCP-1) and pro-inflammatory cytokines (TNF-α, IL-1β) [22]. The expression of adhesion molecules, chemokines, and cytokines is controlled by transcription factors, the most important of which are NF-κB and activator protein-1 [21,35]. In addition, the role of toll-like receptors (TLR) in sensing environmental stressors such as silica particles, nanoparticles, and microbial components has been demonstrated [59].
Although oxidative stress and inflammation may explain certain aspects of systemic effects, other outcomes, such as sudden death, may result from altered autonomic regulation of the heart rate and changes in the coagulation system [1]. Although the cause of altered autonomic nervous activity is unknown, the systemic release of cytokines from the lung and vasculature may affect the production of clotting factors and anticoagulant enzymes in the liver [59]. This could lead to the formation of a dense clot on the top of a ruptured arteriosclerotic plaque, the pathological hallmark of fatal heart attacks [1]. The role of absorbed particles and chemicals in these systemic effects is uncertain. However, it is noteworthy that ultrafine particles and nanoparticles can gain access to systemic circulation by the increased cell permeability observed in the inflamed lung tissue [59,60].
Exposure of humans to bacterial lipopolysaccharide (LPS) and measurement of lung physiology, exhaled markers of inflammation such as nitric oxide, and systemic biomarkers of oxidative stress and inflammation provide a model for identification of individuals susceptible to airborne exposure. Studies suggest a link between inhalation of an environmental stressor and systemic effects [61]. Some individuals may be more prone to develop inflammation, asthma, cardiovascular effects, or cancer because of the accumulation of mutations in genes involved in the induction of antioxidant defenses. Other conditions that predispose individuals to environmental-stressor injury include old age and preexisting chronic inflammatory diseases, all of which are associated with redox changes and inflammation.
Genetic determinants of lung susceptibility to environmental and metabolic stressors
Humans do not generate adaptive immune responses to pollutants per se. Thus, the issue facing immunologists is how, when, and for whom environmental stressors can abrogate immunotolerance and produce autoimmunity [62]. It is known that inflammatory cells are attracted to a site of irritation to mount an immune response. Irritation, and the danger signals it produces, attracts cells from the adaptive and innate immune system. Activation of inflammatory cells can generate a number of ROS (see above) which can oxidize biomolecules and amplify the oxidative injury at the lung and at systemic levels. Controlled studies in humans have shown a great variability between humans in susceptibility to diesel exhaust particles (DEP), lipopolysaccharide (LPS), and even cigarette smoke exposure, suggesting that the existence of susceptible populations may be related to different polymorphisms.
Several lines of evidences have shown that both particulate and gaseous stressors (Table 1) can act on both the upper and lower airways to initiate and exacerbate irritation and inflammation. Increased numbers of neutrophils, B cells, and alveolar macrophages are found in the bronchoalveolar fluid (BALF) of both healthy and asthmatic patients exposed to DEP in chamber studies or in nasal washes after nasal provocation challenges [63,64]. Similar increases in inflammatory cells are found in BALF after exposure to ozone, sulfur dioxide (SO2), or NO2 [65,66]. Presumably as a consequence of this inflammation, altered lung function has been reported in humans, including increased nonspecific airway hyper-responsiveness (AHR) and increased airway resistance, most notably after ozone exposure [67].
A common finding in these exposure studies is an increase in inflammatory cells, chemokines, inflammatory cytokines, and adhesion molecules in the airway epithelial cells for hosting inflammatory cells [21]. The expression of adhesion molecules, granulocyte-macrophage colony stimulant factor (GM-CSF), and IL-8 is up-regulated by cytokines such as IL-1 and TNF-α; both DEP and DEP extract can induce in vitro production of these cytokines.
The key to protection from the harmful effects of pollutants is to mount an effective cytoprotective response. It follows that people with diminished ability to detoxify xenobiotics and metabolize ROS are at increased risk for adverse outcomes from inhalation of environmental stressors. The idea of a population susceptible to environmental stressors is not new [62]. Controlled human exposure to ozone, LPS, SO2, DEPs, and secondhand smoke have shown large inter-individual variation in response to pollutants; variable responses tend to be reproducible and intrinsic to each person. The idea of stressor susceptibility genes has gained prominence [62], with the identification of candidate genes now an area of intense research interest.
Two members of the glutathione – S-transferase superfamily of phase II xenobiotic metabolism enzymes, GSTP1 and GSTM1, are providing to be ideal candidate genes. Products encoded by these genes conjugate reactive intermediates with glutathione; they are present in the respiratory tract and they have common functional variant alleles. Epidemiological studies have shown that GSTP1 and GSTM1 variants that result in the absence of or a decrease in enzyme function are associated with airway hyperesponsiveness and asthma, as well as increased lung cancer risk in smokers [62].
A key enzyme in cigarette smoke-induced carcinogenesis is myeloperoxidase (MPO, 5% of the total protein in neutrophils) [68]. Based on the analysis of the Genetic Susceptibility to Environmental Carcinogens database, a polymorphism in the promoter region of the MPO gene (-463G→A) is inversely associated with lung cancer in both male and female smokers [69,70]. MPO is a phase I metabolism enzyme that can convert pro-carcinogens like benzo[α]pyrene to epoxy carcinogens [70]. Another important genotoxic species produced by MPO is the oxidant hypochlorous acid (HOCl); a substantial amount of evidence indicates that HOCl is an ROS induced by cigarette smoke in the airways [71], and its genotoxicity may be enhanced by nicotine, the main addictive alkaloid in cigarette smoke. Activated neutrophils produce a number of oxidants [72]; the best characterized, HOCl and taurine-chloramines, require active MPO, which is the only human enzyme known to produce HOCl under physiological conditions [73,74]. HOCl is a powerful oxidant that modifies lipids, proteins, sugars, and nucleic acids. The pKα of HOCl is 7.59; thus at physiological pH, a mixture of both HOCl and OCl– is present [75]. Therefore, HOCl has the potential to pass through the cell membranes of the target tissues and then chlorinate intracellular biomolecules [72]. Both exogenously and endogenously generated HOCl and oxidation products from biomolecules may be involved in the etiology of cigarette smoke-induced systemic inflammation [76], mutations, cell transformation, and lung cancer.
HOCl can be produced by activation of neutrophils. Neutrophil activation depends on the synthesis of superoxide radical anion by NADPH oxidase via a protein kinase C dependent (PKC) mechanism; the superoxide radical anion is then spontaneously or enzymatically (via superoxide dismutase, or SOD) dismutated to H2O2, which can then react with MPO and chloride anion [73,77,78]. Indeed, some 25% to 40% of the H2O2 produced by activated neutrophils may be converted to HOCl through reactions involving MPO, with subsequent DNA base modification, including formation of chlorinated bases [78]. The chlorination reactions of HOCl are enhanced by ternary amines, such as nicotine [79]. Chlorinated and oxidized biomolecules or their degradation products, generated in the airways, can reach the systemic circulation and accumulate in target organs producing tissue failures and inflammation.
Studies in humans examining variation in responsiveness to nasal provocation with DEPs have supported genetic variability in the response to this environmental stressor [80,81]. Additional proof for involvement of these genes comes from a randomized clinical trial of children who live in high-ozone areas in Mexico [82]. Asthmatic children with the GSTM1-null genotype (but not the functional variant) who received a placebo had greater ozone-related disorders in forced expiratory flow than children who received antioxidant supplementation with vitamins C and E. In mice, a quantitative trait locus analysis located susceptibility genes involved in AHR in response to ozone on chromosome 4 [83]. One of these genes is the toll-like receptor 4 (TLR-4) [84,85]. Mice deficient in TLR-4 are resistant to AHR induced by inhalation of LPS, and their response to ozone was completely abrogated. Epidemiological and controlled studies in humans are needed to establish the association between polymorphism in TLR-4 and airway response to environmental stressors.
Screening for genetic polymorphisms associated with an exaggerated inflammatory and oxidative response in the airways will help to identify susceptible patients who need to be more strictly protected from exposure. However, the development and testing of more potent and safer antioxidant inhalation therapies [86] to prevent the formation of oxidized biomolecules in the lungs and systemic inflammation in susceptible patients should not be too far in the future.
Inhalation of environmental stressors and autoimmunity
It has been suggested that immunopathologies per se are due to a combination of genetic and environmental factors. For example, DEPs can both induce and exacerbate in vivo allergic responses in the human upper respiratory tract [65]. In atopic patients, while allergen alone produces a two- to three-fold increase in allergen-specific IgE, a challenge of allergen combined with DEP enhances local allergen-specific IgE production 20- to 50-fold [62]. The study suggests that ROS and oxidatively modified bio-molecules might act as limiting factors in elucidating autoimmune responses by molecular mimicry and epitope spreading [47,87]. On the basis of this premise, it is argued that the amounts of metabolic stressors produced in the stressed lung and the adducts they form with proteins might play a pivotal role in inducing/accelerating autoimmune processes [88]. However, the manifestation of autoimmune diseases of exposed subjects depends on their genetic susceptibility.
Oxidative stress, as manifested by increased oxidation products from biomolecules (biomarkers), is a hallmark of many systemic autoimmune diseases, including rheumatic diseases such as arthritis and systemic erythematosus lupus (SEL) [89]. Inflammation, infection, drugs, ROS, and environmental factors such as tobacco smoke [6] induce formation of neo-antigens [90,91] and hence autoimmunity in genetically susceptible individuals [6,92]. High oxygen pressure (pO2), hemo-proteins, endotoxin, agrochemicals, metals associated with particles that reach the lung, and a high content of unsaturated fatty acids make the lung an important site for inflammation and oxidative stress when an irritant accesses the airways and alveolar air spaces (Figure 1). Oxidation of modified macromolecules or formation of adducts with the host proteins produces post-translational modification that can make self proteins be recognized as strange by the immune system; this effect in turn produces, in a genetically susceptible individual, autoimmunity [6,45]. Post-translational protein modifications are an important part of normal physiological processes (protein phosphorylation, for example) and have been associated with several autoimmune diseases [50,89].
The ability of the immune system to distinguish between self and non-self becomes largely incapacitated in autoimmunity. Studies have shown that even minor post-translational modifications, like the spontaneous conversion of an aspartic acid to isoaspartic acid, can cause self-antigens to become immunogenic [93]. Also, oxidatively induced modifications in a protein can affect its cell, tissue, or organ compartmentalization (immune privilege), which can be seen by the immune system to be in the wrong place or the wrong conformity and hence immunogenic. Our data with animal immunization using oxidatively modified Ro 60 autoantigen suggests that cryptic epitopes are revealed to the immune system after oxidative modification [94].
Autoimmune disorders may develop as organ-specific diseases (myasthenia gravis, type 1 diabetes, thyroiditis, primary biliary cirrhosis, Goodpasture's syndrome) or as systemic diseases (rheumatoid arthritis, systemic lupus erythematosus, progressive systemic sclerosis), and auto-antibodies occur in nearly all these diseases. Auto-antibodies were found to be present [95] many years before SLE diagnosis and serve as indicators of future disease [96]. Biomarkers of oxidative damage to proteins, lipids, and nucleic acids are usually detected in SLE and other autoimmune diseases, and serve as useful indicators for diagnosis, prognosis, and therapy in these patients. Oxidative damage has been detected in patients suffering autoimmune diseases [89]; however, the role of lung-generated oxidative products in inducing, promoting, or worsening autoimmunity has not received the attention that it deserves.
Because the development of autoimmunity has been linked to environmental contamination, we believe that inhaled air pollutants are important triggers of autoimmunity in genetically susceptible populations. Systemic lupus erythematosus (SLE) is a chronic, complex inflammatory disease whose etiology is multi-factorial, involving multiple systems. Auto-antibodies, directed against a variety of self-antigens, are characteristic features of the disease. These characteristics suggest the loss of immune privilege at the wrong time, during the immune development, and, of course, the environment-genetic interplay that encourages these individuals to develop SLE. The targets of these antibodies are found in the nucleus, cytoplasm, and/or cell membranes. SLE is thought to arise as a consequence of genetic and environmental factors or molecular mimicry [6,97,98]. Oxidative modification of bio-molecules may involve a change of the molecular conformation by formation of new bridges (e.g., dityrosine and cystine), modification of one or more residues (e.g., thiol oxidation, methionine sulfoxidation, and side-chain carbonylation), inclusion of a halide (chlorination, bromination, iodination, etc.), addition of a nitro group (nitration) or an addition of a hydroxyl group (hydroxylation), etc. (Table 1).
The diversification and amplification of autoimmunity in an individual could be explained by epitope spreading [47,99,100] (Figure 2). The concept of epitope spreading was first described in EAE, and has now been extended to other autoimmune diseases [47,100,101]. Epitope spreading is defined as the progression of an autoimmune response from initial activation to a chronic state, involving increased targeting of auto-antigens by T cells and antibodies (Figure 2).
Figure 2. 4HNE-modified autoantigens and epitope spreading in autoimmunity.
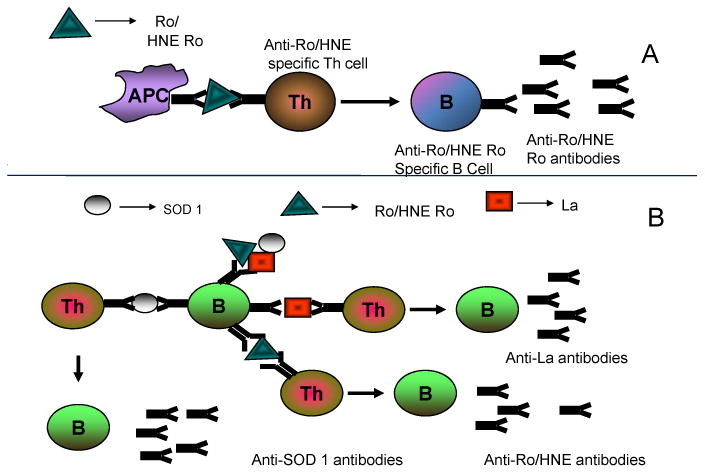
Experimental models of immunization with ALEs modified proteins have shown the induction of autoimmunity in mouse models [45]. A) antigen presenting cells (APC) such as macrophages or dendritic cells present self peptides from 60 kD Ro or 60 kD HNE-Ro adducts to T cells, which in turn provide help to autoreactive B cells. Clonal expansion of B cells capable of binding to 60 kD Ro or 60 kD HNE Ro occurs. B) APC present 4HNE-modified self 60 kD Ro to T-cells that then provide help to a diversified B-cell response. See text for more details.
Immune tolerance to self is maintained by the removal of self-reactive lymphocytes in the thymus during immune system development [102,103] and by making the T-lymphocytes that bind self-antigens anergic in the periphery [104,105]. Abrogation of self-tolerance results in the appearance of auto-reactive lymphocytes, leading to induction of autoimmunity. This autoimmune response is generally divided into three categories, namely B-cell dominant, T-cell dominant, and combinational types [106]. Autoimmune hemolytic anemia and myasthenia gravis are examples of B-cell dominant autoimmune diseases, while experimental autoimmune encephalomyelitis (EAE), insulin-dependent diabetes mellitus, and collagen-induced arthritis are T-cell dominant autoimmune diseases.
SLE is a complex disorder arising from the emergence of both auto-reactive T and B cells with an etiology that involves both genetic and environmental factors. Molecular mimicry of viral or bacterial antigens with self-determinants has been proposed as one of the pathogenic mechanisms in the appearance of auto-reactive cells [94,107].
A common target of auto-antibodies in SLE is the 60 kD Ro ribonucleoprotein. This structure is made up of a 60 kD protein non-covalently associated with at least one of four short uridine-rich RNAs (the hY RNAs). These hY RNAs are also associated with the 48 kDa La (or SSB) auto-antigen. Anti-Ro is found in 25-40% of patients with SLE, while anti-La is found in substantially fewer patients [105-107]. Scofield et al [94] showed that oxidative modification of Ro 60 auto-antigen occurred in a human liver during chronic inflammation. This lends credibility to the possibility that Ro 60 in SLE patients may be subject to modification, especially since increased oxidative damage has been reported in SLE [108,109]. Such a scenario proposes that the development of antibodies to 60 kDa Ro and thus autoimmunity to the entire Ro ribonucleoprotein particle starts after an initial immune response to oxidized 60 kD Ro. This effect has been seen under experimental conditions when rabbits were immunized with 4HNE-modified Ro 60 [94]. Distinct intra-molecular and inter-molecular epitope spreading effects were seen in these animals leading to accelerated autoimmunity.
In SLE and other autoimmune disorders, modification of macromolecules such as modification of self-antigens with 4HNE takes place, possibly originating in the environmentally stressed lung. Significantly higher 4HNE-modified protein levels occur in children with SLE [110]. Antibodies to oxLDL that are cross reactive with phospholipids are thought to be due to their binding to oxidized phospholipids [102]. Circulating oxLDL/ β-2-glycoprotein complexes and IgG immune complexes containing oxLDL/ β-2-glycoprotein occur in SLE and/or phospholipid syndrome [103].
Inhalation of environmental stressors and neuro-degeneration
If environmental toxins can induce neuro-degeneration, then it is likely that inhalation ought to be a significant contributing route of exposure to environmental neuro-toxicants. Surprisingly, the literature in this area is rather sparse. A search of the PubMed database (http://www.ncbi.nlm.nih.gov) using the query terms “neuro-degeneration” and “inhalation” yielded only 84 references; nonetheless, there are compelling reasons to explore this issue. Volatile organic hydrocarbons or hydrophobic reactive gases such as the anesthetic nitrous oxide (N2O) have long been documented to induce neurological problems including neuron loss in children and in patients suffering from certain compromised conditions such as chronic subclinical deficiencies of vitamin B12 (cyanocobalamin) [111-113]. Most of these anesthesia complications occur through direct reaction of a lipid-soluble, brain-accessible neuro-toxicant with target pathways. For instance, nitrous oxide elicits multiple molecular lesions including the loss of cobalt from methionine synthase, the crucial folate cycle enzyme that is dependent on vitamin B12, and from the lipid metabolic enzyme methylmalonyl CoA mutase [114].
Some circumstantial evidence suggests another more subtle mechanism of neurological injury that might arise from chronic inhalation of neurotoxins. The lung itself is sensitive to oxidative damage as a first target of exposure to these toxins. As discussed previously in this review and elsewhere, lipid oxidation products can be potently bioreactive and pro-inflammatory [115,116]. Thus it is conceivable that secondary toxins derived from neurotoxin activity in the lung might act as a diffusible, brain-accessible paracrine factor able to trigger neuro-inflammation or other deleterious processes in sensitive brain regions. This hypothesis has some support from work one of us (KH) performed in the 1990s using a gerbil model of hyperoxia-induced brain injury [117,118]. We found that in both young and aged gerbils, exposure to 90-100% isobaric hyperoxia for 24-48 hours induced brain synaptosomal protein oxidation similar to that seen in extreme aging or Alzheimer's disease. However, only the young animals were able to recover from and even reverse the indications of whole-body hyperoxia [117,118].
Hemoglobin is nearly saturated with oxygen at ambient (20%) levels, so it is very unlikely that isobaric hyperoxia could increase brain pO2 as a mechanism for synaptosomal protein alterations. We hypothesized that the brain protein oxidation occurring in the hyperoxia model might be a consequence of lung oxidation and release of diffusible neuro-effectors. To date, this hypothesis has not been proven or disproven.
Considering the plausibility that inhalation might cause or exacerbate neuro-degeneration, several studies have begun that will investigate whether suspect agents linked to common age-related disorders including Parkinson's disease might cause pathology when administered through the lung. Unfortunately these studies have proven largely unsuccessful at triggering a brain lesion. For instance, chronic nasal inoculations of the Parkinson's disease-relevant toxins rotenone and paraquat fail to induce Parkinson-like symptoms in rats or mice [119]. These toxins, however, are imperfect models and only induce Parkinson-like symptoms in certain strains and subpopulations of rodents when administered through injection [9]. Moreover, rotenone and paraquat are not highly volatile, so inhalation may not be a very important route of exposure for these agents unless the toxins are provided through a fine aerosol form. To our knowledge aerosol delivery has not been featured in published studies.
More recently, volatile trichloroethylene has been implicated as a Parkinson's disease risk factor amongst industrial workers. Oral administration of this agent does induce nigrostriatal degeneration in rodents [120]. Further studies need be done to refine and extend the possible inhalation route to neuro-degeneration for this and other agents. In particular, for each suspect agent, more work needs to be done to determine age, species, and strain specificity for inhalation sensitivity. Also, human exposure to agro-chemicals is likely to occur with simultaneous exposure to particulates and lung irritants (dust, allergens, etc), which to our knowledge have never been examined in co-exposed animal studies. Since conditions like Parkinson's disease are highly multi-factorial, it is likely that animal models will need to include the proper combination of genetic and environmental variables in order to produce robust models more relevant to human pathology.
Concluding remarks and future directions
As environmental health scientists, we work towards identifying mechanisms by which non-respiratory health effects occur and, by extension, facilitating the appropriate management of relationships between air quality and health [16]. Investigation of these mechanisms has spawned a new field of research: human exposure science, which studies human exposure to chemical, physical or biological agents occurring in our environment, and aims at advancing the knowledge of the mechanisms and dynamics of events causing adverse health effects [121]. We believe that this information not only will stimulate our thinking about the link between inhaled stressors and chronic inflammatory diseases, but will also provide an improved foundation for funding agencies and advisory groups to frame research strategies, programs, and priorities.
Inhalation of environmental stressors is an inevitable circumstance. Once a stressor reaches the alveolar epithelium a series of coordinated responses is initiated, beginning with irritation of the airways, the first step towards inflammation. Irritation attracts inflammatory cells, and the damage to the lung and oxidation products of lung tissue biomolecules induce their own regulatory and damaging effects. When these metabolic stressors are circulated through the system they may have damaging effects in organs far from the lungs, depending on receptors, signaling pathways, and genetics. The association between inhalation of environmental stressors and human disease is complex and deserving of in-depth and thorough analysis.
Increasing public health concerns about inhalation of environmental stressors and extra-pulmonary responses such as autoimmunity and neurodegeneration should help to drive future research. In order to develop preventive measures and reduce the negative health effects of inhalation of environmental stressors, we must understand the characteristics of the toxins and gain insights into how these characteristics are related to adverse health effects. As our understanding increases, we can use this knowledge to develop biomarkers that will in turn, identify susceptible individuals and hopefully reduce their exposure.
Acknowledgments
The project described was supported by Award Number R00ES015415 from the National Institute of Environmental Health Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health. DCR also acknowledge the start-up grant from the Presbyterian Health Foundation (PHF) to OMRF. We thank Drs Ann Motten for editing and Drs Luke Szweda and Maria S. Gimenez for critical review of this manuscript.
Abbreviations
- AGE
advanced glycoxidation end-product
- AHR
airway hyper-responsiveness
- ALE
advanced lipid peroxidation end-product
- BALF
bronchoalveolar fluid
- DEP
diesel exhaust particle
- DNA
deoxyribonucleic acid
- GSH
reduced glutathione
- 4HNE
4-hydroxy-2-nonenal
- IL
interleukin
- LDL
low density lipoprotein
- LPS
lipopolysaccharide
- MDA
malondialdehyde
- MPO
myeloperoxidase
- NF-κB
nuclear factor-κB
- NADPH
nicotinamide adenine dinucleotide reduced
- PM
particulate matter
- PUFA
polyunsaturated fatty acid
- RCC
reactive carbonyl compound
- ROS
reactive oxygen species
- SLE
systemic lupus erythematosus
- TLR
toll-like receptor
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaiser J. Mounting evidence indicts fine-particle pollution. Science. 2005;307:1858–1859. doi: 10.1126/science.307.5717.1858a. [DOI] [PubMed] [Google Scholar]
- 2.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 3.Walter R, Gottlieb DJ, O'Connor GT. Environmental and genetic risk factors and gene-environment interactions in the pathogenesis of chronic obstructive lung disease. Environ Health Perspect. 2000;108 4:733–742. doi: 10.1289/ehp.00108s4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aoshiba K, Nagai A. Chronic lung inflammation in aging mice. FEBS Lett. 2007;581:3512–3516. doi: 10.1016/j.febslet.2007.06.075. [DOI] [PubMed] [Google Scholar]
- 5.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harel-Meir M, Sherer Y, Schoenfeld Y. Tobacco smoking and autoimmune rheumatic diseases. Nat Clin Practice Rheumatology. 2007;3:707–715. doi: 10.1038/ncprheum0655. [DOI] [PubMed] [Google Scholar]
- 7.Nel A. Atmosphere. Air pollution-related illness: effects of particles. Science. 2005;308:804–806. doi: 10.1126/science.1108752. [DOI] [PubMed] [Google Scholar]
- 8.Liu L, Ruddy TD, Dalipaj M, Szyszkowicz M, You H, Poon R, Wheeler A, Dales R. Influence of personal exposure to particulate air pollution on cardiovascular physiology and biomarkers of inflammation and oxidative stress in subjects with diabetes. J Occup Environ Med. 2007;49:258–265. doi: 10.1097/JOM.0b013e31803220ef. [DOI] [PubMed] [Google Scholar]
- 9.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic pesticide exposure reproduces feature of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 10.Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston CJ, Driscoll KE, Finkelstein JN, Baggs R, O'Reilly MA, Carter J, Gelein R, Oberdörster G. Pulmonary chemokine and mutagenic responses in rats after subchronic inhalation of amorphous and crystalline silica. Toxicol Sci. 2000;56:405–413. doi: 10.1093/toxsci/56.2.405. [DOI] [PubMed] [Google Scholar]
- 12.Risom L, Dybdahl M, Moller P, Wallin H, Haug T, Vogel U, Klungland A, Loft S. Repeated inhalations of diesel exhaust particles and oxidatively damaged DNA in young oxoguanine DNA glycosylase (OGG1) deficient mice. Free Radic Res. 2007;41:172–181. doi: 10.1080/10715760601024122. [DOI] [PubMed] [Google Scholar]
- 13.Risom L, Moller P, Loft S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res d. 2005;592:119–137. doi: 10.1016/j.mrfmmm.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 15.Kharitonov SA, Barnes PJ. Exhaled biomarkers. Chest. 2006;130:1541–1546. doi: 10.1378/chest.130.5.1541. [DOI] [PubMed] [Google Scholar]
- 16.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 17.Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533:222–239. doi: 10.1016/j.ejphar.2005.12.087. [DOI] [PubMed] [Google Scholar]
- 18.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochem Soc Symp. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 19.Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153:6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol. 2001;429:195–207. doi: 10.1016/s0014-2999(01)01320-6. [DOI] [PubMed] [Google Scholar]
- 21.Gorbunov NV, Das DK, Goswami SK, Gurusamy N, Atkins JL. Spatial coordination of cell-adhesion molecules and redox cycling of iron in the microvascular inflammatory response to pulmonary injury. Antioxid Redox Signal. 2007;9:483–495. doi: 10.1089/ars.2006.1296. [DOI] [PubMed] [Google Scholar]
- 22.Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol. 2007;119:1055–1062. doi: 10.1016/j.jaci.2007.01.015. quiz 1063-1064. [DOI] [PubMed] [Google Scholar]
- 23.Thacker EL. Lung inflammatory responses. Vet Res. 2006;37:469–486. doi: 10.1051/vetres:2006011. [DOI] [PubMed] [Google Scholar]
- 24.Li XY, Gilmour PS, Donaldson K, MacNee W. Free radical activity and pro-inflammatory effects of particulate air pollution (PM10) in vivo and in vitro. Thorax. 1996;51:1216–1222. doi: 10.1136/thx.51.12.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alfaro-Moreno E, Nawrot TS, Nemmar A, Nemery B. Particulate matter in the environment: pulmonary and cardiovascular effects. Curr Opin Pulm Med. 2007;13:98–106. doi: 10.1097/MCP.0b013e328013f47e. [DOI] [PubMed] [Google Scholar]
- 26.Chignard M, Balloy V. Neutrophil recruitment and increased permeability during acute lung injury induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1083–1090. doi: 10.1152/ajplung.2000.279.6.L1083. [DOI] [PubMed] [Google Scholar]
- 27.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 28.Donnelly LE, Barnes PJ. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharmacol Sci. 2006;27:546–553. doi: 10.1016/j.tips.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Idzko M, Hammad H, Van Nimwegen M, Kool M, Willart MAM, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Vigilio F, Virchow CJ, Jr, Lambrecht BN. Extracellular ATP triggers and mantains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 30.Edgington SM. as we live and breathe: Free radicals and aging. Nat Biotechnol. 1994;12:37–40. doi: 10.1038/nbt0194-37. [DOI] [PubMed] [Google Scholar]
- 31.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 32.Uchida K. A Lipid-derived Endogenous Inducer of COX-2: A bridge between Inflammation and Oxidative Stress. Mol Cells. 2008;25:347–351. [PubMed] [Google Scholar]
- 33.Thomas EL. Myeloperoxidase-hydrogen peroxide-chloride antimicrobial system: effect of exogenous amines on antibacterial action against Escherichia coli. Infect Immun. 1979;25:110–116. doi: 10.1128/iai.25.1.110-116.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karin M, Greten FR. NF-kB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 35.Kutuk O, Basaga H. Inflammation meets oxidation: NF-kB as a mediator of initial lesion development in atherosclerosis. Trends Mol Med. 2003;9:549–557. doi: 10.1016/j.molmed.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Chung KF, Adcock IM. Signaling and transcriptional regulation in inflammatory and immune cells: importance in lung biology and disease. Eur Respir J. 2005;26:762–763. doi: 10.1183/09031936.05.00093305. [DOI] [PubMed] [Google Scholar]
- 37.Grune T, Davies KJ. The proteasomal system and HNE-modified proteins. Mol Aspects Med. 2003;24:195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 38.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 39.Sato K, Kadiiska M, Ghio A, Corbett J, Fann YC, Holland SM, Thurman RG, Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J. 2002;16:1713–1720. doi: 10.1096/fj.02-0331com. [DOI] [PubMed] [Google Scholar]
- 40.Negre-Salvayre A, Vieira O, Escargueil-Blanc I, Salvayre R. Oxidized LDL and 4-hydroxynonenal modulate tyrosine kinase receptor activity. Mol Aspects Med. 2003;24:251–261. doi: 10.1016/s0098-2997(03)00020-7. [DOI] [PubMed] [Google Scholar]
- 41.Niwa Y, Hiura Y, Sawamura H, Iwai N. Inhalation exposure to carbon black induces inflammatory response in rats. Circ J. 2008;72:144–149. doi: 10.1253/circj.72.144. [DOI] [PubMed] [Google Scholar]
- 42.Sin DD, Man SF. Systemic inflammation and mortality in chronic obstructive pulmonary disease. Can J Physiol Pharmacol. 2007;85:141–147. doi: 10.1139/y06-093. [DOI] [PubMed] [Google Scholar]
- 43.Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF. Systemic effects of smoking. Chest. 2007;131:1557–1566. doi: 10.1378/chest.06-2179. [DOI] [PubMed] [Google Scholar]
- 44.Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–1181. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 45.Kurien BT, Hensley K, Bachmann M, Scofield RH. Oxidatively modified autoantigens in autoimmune diseases. Free Radic Biol Med. 2006;41:549–556. doi: 10.1016/j.freeradbiomed.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 46.Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA. Age-Related Macular Degeneration and Retinal Protein Modification by 4-Hydroxy-2-nonenal. Invest Ophthalmol Vis Sci. 2007;48:3469–3479. doi: 10.1167/iovs.06-1058. [DOI] [PubMed] [Google Scholar]
- 47.Scofield RH, Kaufman KM, Baber U, James JA, Harley JB, Kurien BT. Immunization of mice with human 60-kd Ro peptides results in epitope spreading if the peptides are highly homologous between human and mouse. Arthritis Rheum. 1999;42:1017–1024. doi: 10.1002/1529-0131(199905)42:5<1017::AID-ANR22>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 48.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381–2386. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 50.Poli G, Biasi F, Leonarduzzi G. 4-hydroxynonenal-protein adducts: a reliable biomarker of lipid oxidation in liver diseases. Mol Asp Med. 2008;29:67–71. doi: 10.1016/j.mam.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 51.Uchida K. Future of toxicology--lipid peroxidation in the future: from biomarker to etiology. Chem Res Toxicol. 2007;20:3–5. doi: 10.1021/tx600304n. [DOI] [PubMed] [Google Scholar]
- 52.Uchida K. Lipid peroxidation and redox-sensitive signaling pathways. Curr Atheroscler Rep. 2007;9:216–221. doi: 10.1007/s11883-007-0022-7. [DOI] [PubMed] [Google Scholar]
- 53.Tetrault GA. Air pollution and lung function. N Engl J Med. 2004;351:2652–2653. author reply 2652-2653. [PubMed] [Google Scholar]
- 54.Fogarty AW, Jones S, Britton JR, Lewis SA, McKeever TM. Systemic inflammation and decline in lung function in a general population: a prospective study. Thorax. 2007;62:515–520. doi: 10.1136/thx.2006.066969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walter RE, Wilk JB, Larson MG, Vasan RS, Keaney JF, Jr, Lipinska I, O'Connor GT, Benjamin EJ. Systemic inflammation and COPD: the Framingham Heart Study. Chest. 2008;133:19–25. doi: 10.1378/chest.07-0058. [DOI] [PubMed] [Google Scholar]
- 56.Malfertheiner P, Schütte K. Smoking-A trigger for chronic inflammation and cancer development in the pancreas. Am J Gastroenterol. 2006;101:160–162. doi: 10.1111/j.1572-0241.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- 57.Uchida K. Cellular response to bioactive lipid peroxidation products. Free Radic Res. 2000;33:731–737. doi: 10.1080/10715760000301251. [DOI] [PubMed] [Google Scholar]
- 58.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 59.Borm PJ, Muller-Schulte D. Nanoparticles in drug delivery and environmental exposure: same size, same risks? Nanomed. 2006;1:235–249. doi: 10.2217/17435889.1.2.235. [DOI] [PubMed] [Google Scholar]
- 60.Nurkiewicz TR, Porter DW, Hubbs AF, Cumpston JL, Chen BT, Frazer DG, Castranova V. Nanoparticle inhalation augments particle-dependent systemic microvascular dysfunction. Part Fibre Toxicol. 2008;5:1. doi: 10.1186/1743-8977-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitz R, Rose MA, Placzek K, Schulze J, Zielen S, Schubert R. LPS inhalation challenge: a new tool to characterize the inflammatory response in humans. Med Microbiol Immunol. 2008;197:13–19. doi: 10.1007/s00430-007-0053-2. [DOI] [PubMed] [Google Scholar]
- 62.Saxon A, Diaz-Sanchez D. Air pollution and allergy: you are what you breathe. Nat Immunol. 2005;6:223–226. doi: 10.1038/ni0305-223. [DOI] [PubMed] [Google Scholar]
- 63.Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol. 1997;158:2406–2413. [PubMed] [Google Scholar]
- 64.Diaz-Sanchez D, Tsien A, Casillas A, Dotson AR, Saxon A. Enhanced nasal cytokine production in human beings after in vivo challenge with diesel exhaust particles. J Allergy Clin Immunol. 1996;98:114–123. doi: 10.1016/s0091-6749(96)70233-6. [DOI] [PubMed] [Google Scholar]
- 65.Diaz-Sanchez D. Pollution and the immune response: atopic diseases--are we too dirty or too clean? Immunology. 2000;101:11–18. doi: 10.1046/j.1365-2567.2000.00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bosson J, Pourazar J, Forsberg B, Adelroth E, Sandstrom T, Blomberg A. Ozone enhances the airway inflammation initiated by diesel exhaust. Respir Med. 2007;101:1140–1146. doi: 10.1016/j.rmed.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 67.Saxon A, Diaz-Sanchez D. Air pollution and allergy: you are what you breathe. Nat Immunol. 2005;6:223–226. doi: 10.1038/ni0305-223. [DOI] [PubMed] [Google Scholar]
- 68.Murao S, Stevens FJ, Ito A, Huberman E. Myeloperoxidase: a myeloid cell nuclear antigen with DNA-binding properties. Proc Natl Acad Sci U S A. 1988;85:1232–1236. doi: 10.1073/pnas.85.4.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chevrier I, Stucker I, Houllier AM, Cenee S, Beaune P, Laurent-Puig P, Loriot MA. Myeloperoxidase: new polymorphisms and relation with lung cancer risk. Pharmacogenetics. 2003;13:729–739. doi: 10.1097/01.fpc.0000054143.14659.ba. [DOI] [PubMed] [Google Scholar]
- 70.Van Schooten FJ, Boots AW, Knaapen AM, Godschalk RW, Maas LM, Borm PJ, Drent M, Jacobs JA. Myeloperoxidase (MPO) -463G->A reduces MPO activity and DNA adduct levels in bronchoalveolar lavages of smokers. Cancer Epidemiol Biomarkers Prev. 2004;13:828–833. [PubMed] [Google Scholar]
- 71.Knaapen AM, Gungor N, Schins RP, Borm PJ, Van Schooten FJ. Neutrophils and respiratory tract DNA damage and mutagenesis: a review. Mutagenesis. 2006;21:225–236. doi: 10.1093/mutage/gel032. [DOI] [PubMed] [Google Scholar]
- 72.Ramos DR, Garcia MV, Canle LM, Santaballa JA, Furtmuller PG, Obinger C. Myeloperoxidase-catalyzed chlorination: The quest for the active species. J Inorg Biochem. 2008;102:1300–1311. doi: 10.1016/j.jinorgbio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 73.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 74.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian Heme Peroxidases: From Molecular Mechanisms to Health Implications. Antioxid Redox Signal. 2008;7:1199–234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 75.Foote CS, Goyne TE, Lehrer RI. Assessment of chlorination by human neutrophils. Nature. 1983;301:715–716. doi: 10.1038/301715a0. [DOI] [PubMed] [Google Scholar]
- 76.Haegens A, Vernooy JH, Heeringa P, Mossman BT, Wouters EF. Myeloperoxidase modulates lung epithelial responses to pro-inflammatory agents. Eur Respir J. 2008;31:252–260. doi: 10.1183/09031936.00029307. [DOI] [PubMed] [Google Scholar]
- 77.Furtmuller PG, Obinger C, Hsuanyu Y, Dunford HB. Mechanism of reaction of myeloperoxidase with hydrogen peroxide and chloride ion. Eur J Biochem. 2000;267:5858–5864. doi: 10.1046/j.1432-1327.2000.01491.x. [DOI] [PubMed] [Google Scholar]
- 78.Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry. 2000;39:5474–5482. doi: 10.1021/bi992809y. [DOI] [PubMed] [Google Scholar]
- 79.Masuda M, Suzuki T, Friesen MD, Ravanat JL, Cadet J, Pignatelli B, Nishino H, Ohshima H. Chlorination of guanosine and other nucleosides by hypochlorous acid and myeloperoxidase of activated human neutrophils. Catalysis by nicotine and trimethylamine. J Biol Chem. 2001;276:40486–40496. doi: 10.1074/jbc.M102700200. [DOI] [PubMed] [Google Scholar]
- 80.Saxon A, Diaz-Sanchez D. Diesel exhaust as a model xenobiotic in allergic inflammation. Immunopharmacology. 2000;48:325–327. doi: 10.1016/s0162-3109(00)00234-4. [DOI] [PubMed] [Google Scholar]
- 81.Diaz-Sanchez D, Riedl M. Diesel effects on human health: a question of stress? Am J Physiol Lung Cell Mol Physiol. 2005;289:L722–723. doi: 10.1152/ajplung.00217.2005. [DOI] [PubMed] [Google Scholar]
- 82.Barraza-Villarreal A, Sunyer J, Hernandez-Cadena L, Escamilla-Nunez MC, Sienra-Monge JJ, Ramirez-Aguilar M, Cortez-Lugo M, Holguin F, Diaz-Sanchez D, Olin AC, Romieu I. Air pollution, airway inflammation, and lung function in a cohort study of Mexico City schoolchildren. Environ Health Perspect. 2008;116:832–838. doi: 10.1289/ehp.10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hollingsworth JW, Kleeberger SR, Foster WM. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 2007;4:240–246. doi: 10.1513/pats.200701-023AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hollingsworth JW, 2nd, Cook DN, Brass DM, Walker JK, Morgan DL, Foster WM, Schwartz DA. The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med. 2004;170:126–132. doi: 10.1164/rccm.200311-1499OC. [DOI] [PubMed] [Google Scholar]
- 85.Hollingsworth JW, Chen BJ, Brass DM, Berman K, Gunn MD, Cook DN, Schwartz DA. The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med. 2005;171:806–813. doi: 10.1164/rccm.200407-953OC. [DOI] [PubMed] [Google Scholar]
- 86.Hamahata A, Enkhbaatar P, Kraft ER, Lange M, Leonard SW, Traber MG, Cox RA, Schmalstieg FC, Hawkins HK, Whorton EB, Horvath EM, Szabo C, Traber LD, Herndon DN, Traber DL. gamma-Tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008;45:425–433. doi: 10.1016/j.freeradbiomed.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurien BT, Scofield RH. Free radical mediated peroxidative damage in systemic lupus erythematosus. Life Sci. 2003;73:1655–1666. doi: 10.1016/s0024-3205(03)00475-2. [DOI] [PubMed] [Google Scholar]
- 88.Kurien BT, Scofield RH. Lipid peroxidation in systemic lupus erythematosus. Indian J Exp Biol. 2006;44:349–356. [PubMed] [Google Scholar]
- 89.Seven A, Guzel S, Aslan M, Hamuryudan V. Lipid, protein, DNA oxidation and antioxidant status in rheumatoid arthritis. Clin Biochem. 2008;41:538–543. doi: 10.1016/j.clinbiochem.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 90.Ahsan H, Ali A, Ali R. Oxygen free radicals and systemic autoimmunity. Clin Exp Immunol. 2003;131:398–404. doi: 10.1046/j.1365-2249.2003.02104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eggleton P, Haigh R, Winyard PG. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology (Oxford) 2008;47:567–571. doi: 10.1093/rheumatology/ken014. [DOI] [PubMed] [Google Scholar]
- 92.Kannan S. Free radical theory of autoimmunity. Theor Biol Med Modeling. 2006;3:1–16. doi: 10.1186/1742-4682-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci. 2005;1050:1–9. doi: 10.1196/annals.1313.001. [DOI] [PubMed] [Google Scholar]
- 94.Scofield RH, Kurien BT, Ganick S, McClain MT, Pye Q, James JA, Schneider RI, Broyles RH, Bachmann M, Hensley K. Modification of lupus-associated 60-kDa Ro protein with the lipid oxidation product 4-hydroxy-2-nonenal increases antigenicity and facilitates epitope spreading. Free Radic Biol Med. 2005;38:719–728. doi: 10.1016/j.freeradbiomed.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 95.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 96.Scofield RH. Autoantibodies as predictors of disease. Lancet. 2004;363:1544–1546. doi: 10.1016/S0140-6736(04)16154-0. [DOI] [PubMed] [Google Scholar]
- 97.Boyd GW. An evolution-based hypothesis on the origin and mechanisms of autoimmune disease. Immunol Cell Biol. 1997;75:503–507. doi: 10.1038/icb.1997.78. [DOI] [PubMed] [Google Scholar]
- 98.Rose NR. Mechanisms of autoimmunity. Semin Liver Dis. 2002;22:387–394. doi: 10.1055/s-2002-35708. [DOI] [PubMed] [Google Scholar]
- 99.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 100.James JA, Gross T, Scofield RH, Harley JB. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B'-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. J Exp Med. 1995;181:453–461. doi: 10.1084/jem.181.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scofield RH, Henry WE, Kurien BT, James JA, Harley JB. Immunization with short peptides from the sequence of the systemic lupus erythematosus-associated 60-kDa Ro autoantigen results in anti-Ro ribonucleoprotein autoimmunity. J Immunol. 1996;156:4059–4066. [PubMed] [Google Scholar]
- 102.Vaarala O. Antibodies to oxidised LDL. Lupus. 2000;9:202–205. doi: 10.1191/096120300678828280. [DOI] [PubMed] [Google Scholar]
- 103.Matsuura E, Lopez LR. Are oxidized LDL/beta2-glycoprotein I complexes pathogenic antigens in autoimmune-mediated atherosclerosis? Clin Dev Immunol. 2004;11:103–111. doi: 10.1080/10446670410001722186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bashir S, Harris G, Denman MA, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52:659–666. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Farris AD, Gross JK, Hanas JS, Harley JB. Genes for murine Y1 and Y3 Ro RNAs have class 3 RNA polymerase III promoter structures and are unlinked on mouse chromosome 6. Gene. 1996;174:35–42. doi: 10.1016/0378-1119(96)00279-x. [DOI] [PubMed] [Google Scholar]
- 106.O'Brien CA, Wolin SL. A possible role for the 60-kD Ro autoantigen in a discard pathway for defective 5S rRNA precursors. Genes Dev. 1994;8:2891–2903. doi: 10.1101/gad.8.23.2891. [DOI] [PubMed] [Google Scholar]
- 107.Yamagata H, Harley JB, Reichlin M. Molecular properties of the Ro/SSA antigen and enzyme-linked immunosorbent assay for quantitation of antibody. J Clin Invest. 1984;74:625–633. doi: 10.1172/JCI111460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Serban MG, Tanaseanu S. Lipid peroxidation in autoimmune systemic vasculitides. Effect of corticoid treatment on lipid peroxidation. Antioxidant protection with vitamin E. Rom J Intern Med. 1994;32:137–142. [PubMed] [Google Scholar]
- 109.Grune T, Michel P, Sitte N, Eggert W, Albrecht-Nebe H, Esterbauer H, Siems WG. Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Radic Biol Med. 1997;23:357–360. doi: 10.1016/s0891-5849(96)00586-2. [DOI] [PubMed] [Google Scholar]
- 110.Michel P, Eggert W, Albrecht-nebe H, Grune T. Increased lipid peroxidation in children with autoimmune diseases. Acta Paediatr. 1997;86:609–612. doi: 10.1111/j.1651-2227.1997.tb08943.x. [DOI] [PubMed] [Google Scholar]
- 111.Flippo TS, Holder WD., Jr Neurological degeneration associated with nitrous oxide anesthesia in patients with vitamin B12 deficiency. Arch Surg. 1993;128:1395–1395. doi: 10.1001/archsurg.1993.01420240099018. [DOI] [PubMed] [Google Scholar]
- 112.Culley DJ, Roghavan SV, Waly M, Baxter MG, Yukhananov R, Deth RC, Crosby G. Nitrous oxide decreases cortical methionine synthase transiently but produces lasting memory impairment in aged rats. Anesth Analg. 2007;105:83–88. doi: 10.1213/01.ane.0000266491.53318.20. [DOI] [PubMed] [Google Scholar]
- 113.Selzer RR, Rosenblatt DS, Laxova R, Hogan K. Adverse effect of nitrous oxide in child with 5,10-methylenetetrahydrofolate reductase deficiency. N Engl J Med. 2003;349:45–50. doi: 10.1056/NEJMoa021867. [DOI] [PubMed] [Google Scholar]
- 114.Chanarin I. The effect of nitrous oxide on cobalamins, folates, and on related events. Crit Rev Toxicol. 1982;10:179–213. doi: 10.3109/10408448209037455. [DOI] [PubMed] [Google Scholar]
- 115.Bochkov VN. Inflammatory profile of oxidized phospholipids. Thromb Haemost. 2007;97:348–354. [PubMed] [Google Scholar]
- 116.Maier KL, Alessandrini F, Beck-Speier I, Hofer TP, Diabaté S, Bitterle E, Stöger T, Jakob T, Behrendt H, Horsch M, Beckers JF, Ziesenis A, Hültner L, Frankenberger M, Kraus-Etschmann S, Schulz H. Health effects of ambient particulate matter: Biological mechanisms and inflammatory responses to in vitro and in vivo particle exposure. Inhal Toxicol. 2008;20:319–337. doi: 10.1080/08958370701866313. [DOI] [PubMed] [Google Scholar]
- 117.Hensley K, Howard BJ, Carney JM, Butterfield AD. Membrane protein alterations in rodent erythrocytes and synaptosomes due to aging and hyperoxia. Biochim Biophys Acta. 1995;1270:203–206. doi: 10.1016/0925-4439(95)00043-4. [DOI] [PubMed] [Google Scholar]
- 118.Howard BJ, Yatin S, Hensley K, Allen KL, Kelly JP, Carney J, Butterfield AD. Prevention of hyperoxia-induced alterations in synaptosomal membrane-associated proteins by N-tert-butyl-alpha-phenylnitrone and 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (tempol) J Neurochem. 1996;67:2045–2050. doi: 10.1046/j.1471-4159.1996.67052045.x. [DOI] [PubMed] [Google Scholar]
- 119.Rojo AI, Cavada C, de Sagarra MR, Cuadrado A. Chronic inhalation of rotenone or paraquat does not induce Parkinson's disease symptoms in rats or mice. Exp Neurol. 2007;208:120–126. doi: 10.1016/j.expneurol.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 120.Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi DY, Hunter RL, Gerhardt GA, Smith CD, Slevin JT, Prince TS. Trichloroethylene: parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol. 2008;63:184–192. doi: 10.1002/ana.21288. [DOI] [PubMed] [Google Scholar]
- 121.Barr DB. Human exposure science: a field of growing importance. J Exp Sci Environ Epidemiol. 2006;16:473. [Google Scholar]