

Abstract
Objective
Brain metabolism, as studied by magnetic resonance spectroscopy (MRS), has been previously shown to be abnormal in Rett syndrome (RTT). However the relationship of MRS findings to age, disease severity and genotype is unknown. This study reports MRS findings in 40 RTT girls (1–14 years old) and 12 age-matched controls.
Methods
Single voxel, short echo time proton MRS of left frontal lobe white matter was performed. Levels of myo-inositol (mI), total choline (Cho), glutamate/glutamine (Glx), and N-acetyl aspartate (NAA) were expressed as ratios relative to creatine (Cr).
Results
NAA/Cr ratios decreased and mI/Cr ratios increased with age in RTT (both p<0.03) while these ratios were stable in controls. The mean Glx/Cr ratio was 36% higher in RTT than in controls (p=0.043). The mean NAA/Cr ratio was 12.6% lower in RTT patients with seizures compared to those without seizures (p=0.017). NAA/Cr ratios also decreased with increasing clinical severity score (p=0.031). Compared to patients with T158X, R255X, and R294X mutations, and C-terminal deletions, patients with the R168X mutation tended to have the highest severity score (0.01≤p≤0.11) and the lowest NAA/Cr ratio (0.029≤p<0.14).
Interpretation
Decreasing NAA/Cr and increasing mI/Cr with age are suggestive of progressive axonal damage and astrocytosis in RTT respectively, while increased Glx/Cr ratio may be secondary to increasing glutamate-glutamine cycling at the synaptic level. The relationships between NAA/Cr, presence or absence of seizures, and disease severity suggest that MRS provides a non-invasive measure of cerebral involvement in RTT, with greatest impairment in patients with the R168X mutation.
Introduction
Rett Syndrome (RTT) (MIM 312750) is an X-linked neurodevelopmental disorder primarily affecting girls from early childhood.1, 2 Clinical features of RTT include acquired microcephaly, loss of purposeful hand use, respiratory irregularities during wakefulness, seizures, failure of speech development, varying degrees of intellectual disability and behavioral changes.3, 4 Mutations in the methyl-CpG binding protein-2 (MeCP2) gene, located on chromosome Xq28 are identified in more than 80% of affected girls.5, 6 MeCP2 is a transcriptional regulator that appears to repress gene expression through binding to methylated DNA.5 Its dysfunction contributes to synaptic pathology, mainly by affecting activity-dependent plasticity.7 Multiple studies suggest that genotypic differences in RTT may account for differences in clinical profile and disease severity.8–13
Decline in brain development in RTT begins before the age of 1 year. Global reductions in gray and white matter volume, and regional variations in brain maturation lead to microcephaly.14–16 The frontal lobe appears to be particularly affected - by tissue reduction, 17, hypoperfusion 18–20 and, somewhat surprisingly, hypermetabolism. 21 In early RTT, proton magnetic resonance spectroscopy (MRS) detected lower concentration of N-acetyl aspartate (NAA, a surrogate neuronal marker 22) in both the frontal lobe gray and white matter.23 Neuroanatomical studies of cortical abnormalities in RTT have shown increased neuronal cell packing density, reduced size of neuronal bodies and dendrites and astrocytic reaction.17, 24, 25 Investigation of cortical white matter did not reveal any major qualitative changes in axons or myelin.26
In contrast to neuropathological studies, techniques of physiological MRI can probe axonal integrity and function over the entire brain. Due to non-invasive nature of these techniques, neuronal tissue can be followed sequentially over the entire life span. One of the advanced methods of physiological imaging is proton MRS, which can detect neurometabolites associated with neuronal integrity and function (NAA), cell membrane turnover (choline-containing compounds, Cho), glial component of the brain tissue (myo-inositol, mI), and a major neurotransmitter, glutamate (Glu).22 Assessment of these neurometabolites in RTT may potentially provide clinically valuable information. However, variability of technical approaches, differences in brain regions studied, and a relatively small number of RTT subjects in most studies have lead to variable results.27, 28 As the genetic abnormality was identified in late 1999, some observations reported in earlier studies may also not be related to the MeCP2 gene defect. NAA in RTT is decreased, both in the gray matter and the white matter;23, 27 however, the decrease was observed only in certain brain regions, 23, 28 including frontal lobes, parietal lobes, and the insula.23 One study measured lower glutamate concentrations compared to controls in both gray and white matter 27 but later, slightly elevated glutamate/NAA ratio in gray matter was detected.29 Mean Cho levels in RTT were reported to be slightly elevated, in particular, in patients with seizures.23 However, no significant increase in Cho concentration was documented in individual brain regions.23 Levels of the glial marker, mI, were examined only in one study, and normal mI/Cr ratios were found.30 To date, a detailed investigation of the relationship between the levels of MRS-detected compounds and genetic impairment has not been carried out.
It is puzzling that despite findings of no major qualitative changes in myelin in RTT,26 cortical white matter is affected by diffuse volume reduction and14 metabolic abnormalities23 indicative of impaired tissue integrity.31 As discussed earlier in this section, there is a specific involvement of frontal lobe in RTT.17–21 In agreement with these findings, frontal white matter impairment was detected in an earlier MRSI23 and a preliminary DTI study.31 To further evaluate white matter pathology in the frontal lobe, the goal of the current MRS study was to examine metabolic impairment in the frontal white matter. The following hypotheses were considered:
In RTT patients, white matter NAA levels will be decreased, consistent with axonal impairment; Cho and mI levels will be increased, suggesting presence of gliosis.
The concentration of the neuronal marker NAA will decrease with age, consistent with progression of symptoms.
Glutamate levels in RTT will be increased, particularly in younger subjects, in agreement with higher density of NMDA glutamate receptors demonstrated by postmortem autoradiography studies,32 and increased CSF glutamate levels.33
The degree of metabolic impairment will be associated with disease severity and will vary among MeCP2 mutation types.
Subjects and Methods
Study population
Forty girls diagnosed with RTT (mean age 6.1 years, range 1.1–14.1 years, standard deviation 3.1 years) and 12 healthy girls (mean age 8.9 years, range 3.6–14.7 years, standard deviation 3.5 years) were examined. Diagnosis of RTT was confirmed by identification of the MeCP2 mutation and presence of clinical features.34 Symptom severity was classified by severity scores.12 The list of mutations and corresponding age and severity scores ranges are presented in Table 1. The girls in the control group were either healthy siblings of the RTT patients or unrelated healthy volunteers. During the MRS examination all RTT patients were sedated with chloral hydrate; examination in all controls was performed without sedation. The study was approved by the local Institutional Review Board and all families provided a written informed consent.
Table 1.
MeCP2 gene mutations
| Mutation | N | Age range (years) | Severity score |
|---|---|---|---|
| P101S | 1 | 3.1 | 4 |
| R106Q | 1 | 8.4 | 8 |
| R106W | 1 | 3.2 | 5 |
| R133C | 1 | 4.3 | 5 |
| P152R | 1 | 5.1 | 6 |
| T158M | 9 | 2.5 – 13.1 | 4 – 12 |
| R168X | 5 | 1.1 – 8.5 | 5 – 13 |
| R255X | 4 | 1.1 – 7.7 | 3 – 7 |
| R270X | 3 | 6.6 – 14.1 | 6 – 15 |
| R294X | 5 | 4.3 – 10.4 | 4 – 12 |
| R305R | 1 | 6.6 | 6 |
| R306C | 1 | 5.0 | 8 |
| R306H | 1 | 9.3 | 8 |
| P322L | 1 | 5.7 | 9 |
| C-terminal deletions | 5 | 2.9 – 7.5 | 3 – 7 |
Proton Magnetic Resonance Spectroscopy
Single voxel proton MRS was performed with the Point Resolved Spectroscopy (PRESS) sequence (repetition time/echo time = 1500/30 ms, 2048 data points, 2000 Hz spectral width, 128 repetitions) at 1.5 T. Single voxel proton MRS was a component of an integrated MRI examination, including clinical MRI, volumetric MRI, and DTI. Due to time constrains in sedated patients, proton MRS examination was limited to one region of interest. The 2.0 × 2.0 × 2.0 cm3 voxel was positioned in the left forceps minor and contained predominantly white matter. In 26 RTT subjects, spectra without water suppression from the same voxel (acquired with 8 repetitions) were also obtained. LCModel (Version 6.1)35 was used for automatic analysis of the spectra. Individual metabolite ratios were obtained by the LCModel as ratios of N-acetyl aspartate (NAA), myo-inositol (mI), glutamate (Glu), glutamate and glutamine (Glx) and total choline (Cho) concentrations to the creatine (Cr) concentration. It should be noted that concentration ratios are not necessarily the same as peak area ratios, which are reported by other software packages. In 26 RTT patients apparent concentrations of individual metabolites (in arbitrary units, a.u.) were also estimated based on white matter water concentration as an internal reference. Due to incomplete resolution of the glutamate and glutamine (Gln) signals, the intensity of the complex Glu/Gln signal (Glx) was measured. Good quality spectra were obtained in all subjects. For a given metabolite, only data with the LCModel Cramer-Rao bounds ≤20% were included. In the Results section, the total number of subjects (N) is noted for analyses with missing data on metabolite ratios or metabolite concentrations.
Statistical Analysis
Normal distribution of all metabolite ratios was confirmed with the Kolmogorov-Smirnov test. General linear model (GLM) ANOVA was used to evaluate the differences in metabolite ratios between the RTT and control groups. Age and group (patients and controls) were the independent variables; an interaction term age × group was also used. Linear regression of metabolite ratios and metabolite concentrations in RTT patients on age was employed as a post-hoc test to examine age-related differences in RTT and control subjects. GLM ANOVA was also applied to examine the differences in metabolite ratios between patients with seizures and without seizures, among different types of MeCP2 gene mutations, and to assess the relationship between metabolite ratios and severity scores, controlling for age. Significance level was set to p<0.05. Data are presented as age-adjusted (marginal) means ± standard deviations.
Results
Figure 1 shows the region of interest in the left frontal forceps minor and examples of spectra in a RTT patient and a healthy control subject.
Figure 1.

Proton MR spectra of the left frontal white matter in a 10.4 year-old girl with Rett syndrome (RTT) and a 10 year-old healthy girl. The T2-weighted localizer images show the position of the region of interest. Microcephaly and prominent sulci are evident in the RTT patient. Lower NAA/Cr and higher mI/Cr and Glx/Cr ratios are apparent in the RTT patient.
The main GLM ANOVA of NAA/Cr ratios detected a significant interaction term group × age (p=0.027). In RTT patients, NAA/Cr ratios decreased with age (p=0.021) while stable NAA/Cr ratios with age were observed in controls (Figure 2). The differences in NAA/Cr ratios were more pronounced in older subjects; in RTT patients younger than 7 years, mean NAA/Cr ratio was similar to controls (group: p=0.77). The age-related decrease in NAA/Cr ratios in RTT was due to a decrease in NAA concentration (N=26; p=0.05); Cr concentration was stable (N=26; p=0.32) (Figure 3).
Figure 2.

Age-related differences in frontal white matter NAA/Cr, mI/Cr, Glx/Cr, and Cho/Cr ratios in RTT subjects and healthy controls. NAA/Cr decreased with age and mI increased in age in RTT (both p<0.03), while stable ratios were detected in controls. Glx/Cr was elevated in RTT subjects younger than 10 years (p=0.024). Cho/Cr ratio was stable across the examined age range in both groups.
Figure 3.

Age-related differences in frontal white matter NAA, mI, Glx, and Cr concentrations (in arbitrary units) in RTT patients. NAA concentration decreased (p=0.05) and mI concentration increased (p=0.016) with age. Stable Glx and Cr concentrations were detected.
The main GLM ANOVA of mI/Cr ratios detected a significant effect of group (p<0.0001) and age (p=0.003). In RTT patients, mI/Cr ratios increased with age (N=39, p=0.020) but were stable in controls (Figure 2). The age-related increase in mI/Cr ratios in RTT was due to an increase in mI concentration (N=25, p=0.016) (Figure 3).
The mean Glx/Cr ratio in RTT was 36% higher compared to controls (group: p=0.043) (Figure 2). Although no significant age-related differences in Glx/Cr ratios were detected the observed group differences were due to higher Glx/Cr ratios in patients younger than 10 years (p=0.024). No difference in Glx/Cr ratios was detected between patients older than 10 years (N=5) and controls. No group or age-differences in the Cho/Cr ratio were found (Figure 2).
RTT patients with seizures had a 12.6% lower mean NAA/Cr ratio compared to patients without seizures (p=0.017). NAA/Cr decreased linearly with increasing severity score (p=0.031) (Figure 4). Since both NAA/Cr and severity score were age-dependent, the effect of age was accounted for by using the residuals on the regression (of NAA/Cr and severity scores) with age. No relationship between any other ratio and presence of seizures or severity score was detected.
Figure 4.
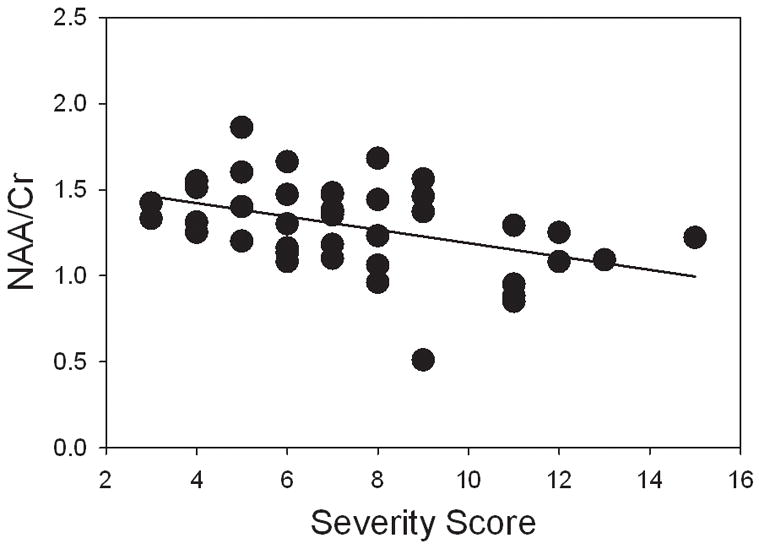
Relationship between NAA/Cr ratios in RTT and severity scores. After controlling for age, NAA/Cr linearly decreased with increasing severity score (p=0.035).
The most frequent mutations in the examined RTT group were T158M, R168X, R255X, R294X, and C-terminal deletions (Table 1). The R168X mutation tended to be associated with the highest mean severity score (0.01≤p≤0.11) and the lowest NAA/Cr ratio (0.029≤p<0.14) (Figure 5). No significant difference in mean NAA/Cr, mI/Cr or Glx/Cr ratio between the R168X and T158M mutations was detected (all p≥0.14). Patients with C-terminal deletions tended to have the highest mean Glx/Cr ratio compared to patients with the T158M, R168X, and R294X mutations (0.054≤p<0.15).
Figure 5.

Age-adjusted (marginal) means of severity scores, NAA/Cr, Glx/Cr, and mI/Cr ratios in the most frequent mutations in the RTT group: T158M (N=9), R168X (N=5), R255X (N=4), R294X (5), and deletions (N=5). The patients with the R168X mutation tended to have the highest severity score (0.010<p<0.11) and the lowest NAA/Cr ratio (0.029<p<0.14). A tendency to a high mean Glx/Cr ratio was observed in patients with C-terminal deletions compared to patients with the R168X, T158M, and R294X mutations (0.054<p<0.15).
Discussion
The main findings of this study were detection of elevated levels of mI, considered to be a glial marker, and low levels of NAA, a neuronal marker, in white matter in RTT. Compared to healthy children, the mean NAA/Cr ratio was 5% lower and mI/Cr ratio was 54% higher in RTT. The MRS results are therefore suggestive of mild white matter pathology, in agreement with the neuropathological findings that RTT is not associated with a recognizable degenerative, demyelinating or gross malformative process.36 Age-related decreases in NAA are consistent with clinical deterioration typically occurring in older subjects.37 NAA/Cr decreased with increasing severity scores, was also low in patients with seizures, and tended to be low in patients with the R168X mutation, which was associated with the highest mean severity score. Our results therefore suggest that NAA/Cr may be a valuable quantitative marker of clinical status in RTT.
Elevated white matter mI concentrations, which progressively increased with age, have not been previously reported in RTT. One earlier MRS study found normal mean white matter mI/Cr in 7 girls with RTT, including four girls aged 4 to 10 years who had decreased NAA/Cr ratio.30 The discrepancy may be due to differences in patient populations as the diagnosis of RTT in the previous study was based on clinical criteria alone. Low frontal white matter NAA concentrations and normal Cho concentrations in RTT reported in the current study are in agreement with a previous proton MR spectroscopic imaging (MRSI) study.23 Although a 12% increase in mean Cho concentration was observed, no significant differences were detected in individual regions.23 In this study,23 a MRSI technique with higher spatial resolution was performed but due to the use of a long echo time mI was not detected. Increased mI concentration, normal Cho levels, and decreased NAA concentration in the frontal white matter may be interpreted as astrocytic reaction in the context of mild axonal disruption (or dysfunction). Astroglial proliferation appears as a less likely correlate, due to lack of concurrent increase in Cho and mI.38 Also, as glial cells have relatively high concentrations of mI and Cr,39 in gliosis, elevation of Cr may be present. However, in the current study, Cr concentration in control subjects was not measured, so this cannot be confirmed. Although an 8% higher mean Cr concentration was previously found in the frontal white matter in the RTT group, the difference was not significant, due to a high variability in Cr levels in both RTT and control groups.23 Involvement of glial metabolism was also suggested in an in vivo MRS study of MeCP2-null male mice, though deficit in MeCP2 in these mice was associated with decreased mI, increased Cho, and decreased NAA concentration.40 Comparison with the results of this study suggests a different etiology for glial pathology in the MeCP2-null mouse than in human RTT females. Further experiments need to be performed to explain the difference. One of the limitations of the MeCP2-null male mouse model studied, besides difference in magnitude of MeCP2 gene involvement compared to human RTT subjects, is their severely restricted life span. MeCP2-null mice die at 10 weeks postnatal age, which precludes studies of later developmental stages. Neuropathological studies of RTT brain tissue reported high levels of GFAP immunoreactivity and prominent astrocytes;25, 41, 42 however, gene expression profiles characterized by upgregulation of astrocytic genes (e.g., GFAP, alpha B crystalline, glial EAAT1) suggest that the neuropathological abnormalities may not be typical astrocytic proliferation.41
Stable frontal white matter NAA concentrations in younger RTT patients were detected both in a previous study 23 and in this work. However, NAA levels were lower and decreased with age in older patients while no age-related differences were detected in controls. One recent study including older patients also noted lower NAA/Cr ratio in frontal white matter compared to controls.30 However, no age-related differences were detected, probably due to a limited sample size (total of 6 subjects, 3 – 21 years old).30 Lower NAA concentration in older RTT patients compared to younger patients was reported in one of the first MRS studies of RTT, which included 9 patients aged 2.3 to 21.3 years old. 27 It was noted that the decrease in NAA was less pronounced in the white matter than in the gray matter.27 To date, there are no MRS data from longitudinal studies, which could elucidate the time course of NAA concentration changes in individual patients.
The age-related differences in Glx/Cr found in the current study are similar to the pattern of developmental differences between RTT patients and healthy children in glutamate receptor binding in the frontal cortex.32 A higher Glx/Cr ratio was found in RTT patients younger than 10 years compared to controls but there was no difference in Glx/Cr between older patients and controls. In the glutamate receptor binding study, the density of NMDA receptors was higher in the frontal cortex of patients younger than 10 years but lower in the older RTT group.32 Similar, but not significant, age-dependent differences in AMPA, metabotropic, and GABA receptors were detected in the basal ganglia.43 The findings of increased NMDA receptor density, increased glucose utilization in the frontal lobe 21 and increased glutamate levels in the cerebrospinal fluid 33 are suggestive of enhanced excitatory neurotransmission in younger patients with RTT. A 4.1 Tesla MR spectroscopic imaging study of 6 girls with early RTT (4.5–6 years old) reported elevated Glu/NAA ratio in the gray matter and normal Glu/NAA in the white matter.29 However, that study reported mean white matter Glu/NAA ratio in a slice at the level of the cingulate gyrus 29 while our study examined only one specific region. Age-related differences for both Glx/Cr and Glu/Cr ratios observed in the current study were similar (Glu/Cr results are not reported here); however, statistical significance was reached only for the Glx/Cr ratio because because of the difficulty in separating Glu and Gln at 1.5 Tesla. Increased accuracy in assessment of Glu may be achieved at higher magnetic fields, where the separation between Glu and Gln resonances is improved.
Mutations in the MeCP2 gene are associated with a wide range of severity.4, 44 The common mutations in RTT are the R106W, R133C, T158M, R168X, R255X, R270X, R294X, R306C, and small insertions/deletions towards the 3′ end of the MeCP2 gene, leading to C-terminal truncations.8, 9, 44 In our RTT group, the R168X mutation tended to have the highest mean severity scores and lowest mean NAA/Cr ratio. Subjects with C-terminal deletions had relatively higher mean Glx/Cr ratios. Despite its milder overall severity, this genotype has been linked to a distinctive clinical course characterized by early onset of dystonia.45 Although our statistical analysis detected trend to significant differences in metabolites between common mutations, our results (obtained with a small number of patients in individual mutation groups) warrant studies in a larger series, which could permit more detailed understanding of association between genotype, phenotype, and concentration of neurochemicals detected with MR spectroscopy.
Patients with the same mutation may manifest variable phenotypes likely due to differences in pattern of X-chromosome inactivation.13, 46, 47 Due to the small number of cases, we could not evaluate the effect of X-chromosome inactivation on metabolite levels in individual mutations in this study.
In conclusion, proton MRS revealed presence of mild white matter pathologic processes, which appear to be complex and progressive in nature. Therefore, future physiological MRI studies in RTT should examine interactions between the glial and neuronal components to evaluate the relationship between metabolic, structural, clinical, and genetic impairment.
Acknowledgments
Supported by P01 HD 24448 NICHD, and UL1RR025005 Institute for Clinical and Translational Research grants.
We would like to thank Terri Brawner, Kathleen Kahl, and Carolyn Gillen for their assistance with patients and the MRI examination.
Footnotes
All authors: no conflict of interest
References
- 1.Einspieler C, Kerr AM, Prechtl HF. Is the early development of girls with Rett disorder really normal? Pediatr Res. 2005;57:696–700. doi: 10.1203/01.PDR.0000155945.94249.0A. [DOI] [PubMed] [Google Scholar]
- 2.Naidu S. Rett syndrome: a disorder affecting early brain growth. Ann Neurol. 1997;42:3–10. doi: 10.1002/ana.410420104. [DOI] [PubMed] [Google Scholar]
- 3.Jellinger KA. Rett Syndrome -- an update. J Neural Transm. 2003;110:681–701. doi: 10.1007/s00702-003-0822-z. [DOI] [PubMed] [Google Scholar]
- 4.Naidu S, Bibat G, Kratz L, et al. Clinical variability in Rett syndrome. J Child Neurol. 2003;18:662–668. doi: 10.1177/08830738030180100801. [DOI] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Sirianni N, Naidu S, Pereira J, et al. Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet. 1998;63:1552–1558. doi: 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer A, Qayumi J, Ronnett G. MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol Cell Neurosci. 2008;37:794–807. doi: 10.1016/j.mcn.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70:868–875. doi: 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- 9.Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leonard H, Moore H, Carey M, et al. Genotype and early development in Rett syndrome: the value of international data. Brain Dev. 2005;27(Suppl 1):S59–S68. doi: 10.1016/j.braindev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Colvin L, Leonard H, de Klerk N, et al. Refining the phenotype of common mutations in Rett syndrome. J Med Genet. 2004;41:25–30. doi: 10.1136/jmg.2003.011130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffbuhr K, Devaney JM, LaFleur B, et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology. 2001;56:1486–1495. doi: 10.1212/wnl.56.11.1486. [DOI] [PubMed] [Google Scholar]
- 13.Hoffbuhr KC, Moses LM, Jerdonek MA, et al. Associations between MeCP2 mutations, X-chromosome inactivation, and phenotype. Ment Retard Dev Disabil Res Rev. 2002;8:99–105. doi: 10.1002/mrdd.10026. [DOI] [PubMed] [Google Scholar]
- 14.Carter JC, Lanham DC, Pham D, et al. Selective cerebral volume reduction in Rett syndrome: a multiple-approach MR imaging study. AJNR Am J Neuroradiol. 2008;29:436–441. doi: 10.3174/ajnr.A0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramaniam B, Naidu S, Reiss AL. Neuroanatomy in Rett syndrome: cerebral cortex and posterior fossa. Neurology. 1997;48:399–407. doi: 10.1212/wnl.48.2.399. [DOI] [PubMed] [Google Scholar]
- 16.Reiss AL, Faruque F, Naidu S, et al. Neuroanatomy of Rett syndrome: a volumetric imaging study. Ann Neurol. 1993;34:227–234. doi: 10.1002/ana.410340220. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong DD. Review of Rett syndrome. J Neuropathol Exp Neurol. 1997;56:843–849. doi: 10.1097/00005072-199708000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Naidu S, Kaufmann WE, Abrams MT, et al. Neuroimaging studies in Rett syndrome. Brain Dev. 2001;23(Suppl 1):S62–71. doi: 10.1016/s0387-7604(01)00381-3. [DOI] [PubMed] [Google Scholar]
- 19.Burroni L, Aucone AM, Volterrani D, et al. Brain perfusion abnormalities in Rett syndrome: a qualitative and quantitative SPET study with 99Tc(m)-ECD. Nucl Med Commun. 1997;18:527–534. doi: 10.1097/00006231-199706000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Lappalainen R, Liewendahl K, Sainio K, et al. Brain perfusion SPECT and EEG findings in Rett syndrome. Acta Neurol Scand. 1997;95:44–50. doi: 10.1111/j.1600-0404.1997.tb00067.x. [DOI] [PubMed] [Google Scholar]
- 21.Villemagne PM, Naidu S, Villemagne VL, et al. Brain glucose metabolism in Rett Syndrome. Pediatr Neurol. 2002;27:117–122. doi: 10.1016/s0887-8994(02)00399-5. [DOI] [PubMed] [Google Scholar]
- 22.Barker PB, Lin DDM. In vivo proton MR spectroscopy of the human brain. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;49:99. [Google Scholar]
- 23.Horska A, Naidu S, Herskovits EH, et al. Quantitative 1H MR spectroscopic imaging in early Rett syndrome. Neurology. 2000;54:715–722. doi: 10.1212/wnl.54.3.715. [DOI] [PubMed] [Google Scholar]
- 24.Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology. 1995;45:1581–1586. doi: 10.1212/wnl.45.8.1581. [DOI] [PubMed] [Google Scholar]
- 25.Bauman ML, Kemper TL, Arin DM. Microscopic observations of the brain in Rett syndrome. Neuropediatrics. 1995;26:105–108. doi: 10.1055/s-2007-979737. [DOI] [PubMed] [Google Scholar]
- 26.Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol. 1988;76:142–158. doi: 10.1007/BF00688098. [DOI] [PubMed] [Google Scholar]
- 27.Hanefeld F, Christen HJ, Holzbach U, et al. Cerebral proton magnetic resonance spectroscopy in Rett syndrome. Neuropediatrics. 1995;26:126–127. doi: 10.1055/s-2007-979742. [DOI] [PubMed] [Google Scholar]
- 28.Khong PL, Lam CW, Ooi CG, et al. Magnetic resonance spectroscopy and analysis of MECP2 in Rett syndrome. Pediatr Neurol. 2002;26:205–209. doi: 10.1016/s0887-8994(01)00385-x. [DOI] [PubMed] [Google Scholar]
- 29.Pan JW, Lane JB, Hetherington H, Percy AK. Rett syndrome: 1H spectroscopic imaging at 4.1 Tesla. J Child Neurol. 1999;14:524–528. doi: 10.1177/088307389901400808. [DOI] [PubMed] [Google Scholar]
- 30.Gokcay A, Kitis O, Ekmekci O, et al. Proton MR spectroscopy in Rett syndrome. Comput Med Imaging Graph. 2002;26:271–275. doi: 10.1016/s0895-6111(02)00016-2. [DOI] [PubMed] [Google Scholar]
- 31.Izbudak I, Farage L, Bonekamp D, et al. Diffusion tensor imaging findings in Rett syndrome patients. International Society for Magnetic Resonance in Medicine, 14th Scientific Meeting; Seattle, WA, USA. 2006. p. 1557. [Google Scholar]
- 32.Blue ME, Naidu S, Johnston MV. Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol. 1999;45:541–545. doi: 10.1002/1531-8249(199904)45:4<541::aid-ana21>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Lappalainen R, Riikonen RS. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996;15:213–216. doi: 10.1016/s0887-8994(96)00218-4. [DOI] [PubMed] [Google Scholar]
- 34.Trevathan E, Naidu S. The clinical recognition and differential diagnosis of Rett syndrome. J Child Neurol. 1988;3(Suppl):S6–16. doi: 10.1177/0883073888003001s03. [DOI] [PubMed] [Google Scholar]
- 35.Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001;14:260–264. doi: 10.1002/nbm.698. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 37.Hagberg B. Rett syndrome: long-term clinical follow-up experiences over four decades. J Child Neurol. 2005;20:722–727. doi: 10.1177/08830738050200090401. [DOI] [PubMed] [Google Scholar]
- 38.Kim JP, Lentz MR, Westmoreland SV, et al. Relationships between astrogliosis and 1H MR spectroscopic measures of brain choline/creatine and myo-inositol/creatine in a primate model. AJNR Am J Neuroradiol. 2005;26:752–759. [PMC free article] [PubMed] [Google Scholar]
- 39.Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci. 1993;15:289–298. doi: 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- 40.Saywell V, Viola A, Confort-Gouny S, et al. Brain magnetic resonance study of Mecp2 deletion effects on anatomy and metabolism. Biochem Biophys Res Commun. 2006;340:776–783. doi: 10.1016/j.bbrc.2005.12.080. [DOI] [PubMed] [Google Scholar]
- 41.Colantuoni C, Jeon OH, Hyder K, et al. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann WE, Naidu S, Budden S. Abnormal expression of microtubule-associated protein 2 (MAP-2) in neocortex in Rett syndrome. Neuropediatrics. 1995;26:109–113. doi: 10.1055/s-2007-979738. [DOI] [PubMed] [Google Scholar]
- 43.Blue ME, Naidu S, Johnston MV. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 1999;156:345–352. doi: 10.1006/exnr.1999.7030. [DOI] [PubMed] [Google Scholar]
- 44.Philippe C, Villard L, De Roux N, et al. Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: a molecular update. Eur J Med Genet. 2006;49:9–18. doi: 10.1016/j.ejmg.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Smeets E, Terhal P, Casaer P, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet A. 2005;132A:117–120. doi: 10.1002/ajmg.a.30410. [DOI] [PubMed] [Google Scholar]
- 46.Huppke P, Maier E, Warnke A, et al. Very mild cases of Rett syndrome with skewed X inactivation. J Med Genet. 2006;43:814–816. doi: 10.1136/jmg.2006.042077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chae JH, Hwang H, Hwang YS, et al. Influence of MECP2 gene mutation and X-chromosome inactivation on the Rett syndrome phenotype. J Child Neurol. 2004;19:503–508. doi: 10.1177/08830738040190070501. [DOI] [PubMed] [Google Scholar]