

Abstract
Light chain amyloidoses arise from the overproduction and abnormal deposition of immunoglobulin light chain in various organs. LEN is the variable domain of an immunoglobulin light chain originally isolated from the urine of a patient suffering from multiple myeloma, with no sign of renal dysfunction or amyloidosis. LEN was shown to form fibrils in vitro under mildly destabilizing conditions. In this work we investigated the changes induced by methionine oxidation in structural properties, conformational stability, and aggregation behavior of the immunoglobulin light chain domain LEN. We established that LEN was well-protected from the oxidation in its native state, but successful oxidation was achieved in the presence of 4M GuHCl. Oxidation induced noticeable structural changes in LEN and destabilized this protein. The methionine-oxidized LEN preferred to form amorphous aggregates instead of fibrils. The results indicated that the LEN oxidation may play an important role in amorphous deposition of the protein, but not in its fibrillation.
Keywords: Amyloidosis, immunoglobulin light chain, protein aggregation, conformational stability, partially folded intermediate, conformational disease
The amyloidoses are lethal diseases involving the extracellular deposition of amyloid fibrils and plaques (1, 2). All amyloidoses arise from the conversion of specific proteins from their soluble functional states into stable, highly-ordered, filamentous protein aggregates, known as amyloid fibrils, and from the deposition of these aggregated material in a variety of organs and tissues (3, 4). Although the amyloid fibrils from different diseases are structurally and morphologically similar to each other, the amyloidogenic polypeptides causing diseases are extremely diverse and, prior to fibrillation, may be rich in β-sheet, α-helix, β-helix, or be intrinsically disordered (5). Aggregation and subsequent development of protein deposition diseases originate from conformational changes in corresponding amyloidogenic proteins. In vitro studies show that the fibrils are produced from partially folded intermediates following either the partial destabilization of physiologically folded proteins, in the case of globular proteins (3-13), or the partial folding, in the case of natively unfolded (or intrinsically disordered) proteins (5, 14-16). Presumably, such a partially unfolded conformation enables specific intermolecular interactions, including electrostatic attraction, hydrogen bonding, and hydrophobic contacts, which are necessary for oligomerization and fibrillation.
There are four common light chain-related deposition diseases: AL amyloidosis, light chain deposition disease (LCDD), myeloma cast nephropathy, and acquired Fanconi’s syndrome, all arising from pathological deposition of light chains of monoclonal antibodies (17-20). Light chain amyloidosis originates from the overproduction and abnormal accumulation of light chain protein aggregates in various organs, with the most common organs affected being the kidney, heart, liver, and autonomic or peripheral nerves (21). AL amyloidosis is a rare disease, as only 1200 to 3200 new cases are reported each year in the United States, which is usually associated with myeloma. Amyloid deposits in organs affected by AL amyloidosis can cause shortness of breath, fatigue, edema (swelling of ankles and legs), dizziness upon standing, a feeling of fullness in the stomach (especially after eating), diarrhea, weight loss, enlarged tongue, numbness of the legs and arms, and protein in the urine.
Contrarily to AL amyloidoses, LCDD is characterized by the deposition of amorphous, noncongophilic light chains in multiple organs. These deposits do not exhibit a fibrillar structure when examined ultrastructurally. As such, the morphology of the deposited light chain aggregates in AL amyloidoses and LCDD is clearly different. Patients typically exhibit only one form of light chain deposition. However, there is at least one report of a patient exhibiting both AL amyloidosis and LCDD involving the same light chain (22).
Myeloma kidney or myeloma cast nephropathy occurs in the presence of excess free light chains in the plasma and urine. Although the mechanism by which urinary light chains lead to renal failure is not understood completely, it is believed that the pathology of this disease is associated with the precipitation of light chains in the tubules, leading to obstructing, dense, intratubular casts in the distal and collecting tubules that may initiate a giant cell reaction (23). Finally, acquired Fanconi’s syndrome represents a complex of defects in the functioning of renal tubules associated with multiple myeloma.
Human antibody light chains originate from 30-40 κ and λ variable domain gene segments, each of which serves as the progenitor of a family of related light chains. Remarkable diversity of light chains is determined by the accumulation of somatic mutations. Therefore, the primary structures of amyloidogenic and nonamyloidogenic light chains differ at numerous sites, with no single amino acid substitution or combination thereof that would be responsible for the doubtless determination of a pathological propensity (24). Most variable domains form dimers at moderately high concentrations (a few milligrams/ml), and the dimerization constants and the geometry of the interface depend on the specific residues at the interface (25).
LEN is a 114-residues-long variable domain of the immunoglobulin light chain (kIV Bence Jones protein). Earlier, based on the analysis of the amino acid sequences, LCs were classified in several groups and several hypervariable or complementarity-determining regions (CDRs) and framework regions were defined according to the sequence variability (26). According to the nomenclature of Kabat et al. (26), extra residues in CDR1, after residue 27, are labeled 27a to 27f. There are six such extra residues in LEN (SVLYSS) and in other Bence Jones proteins from the kIV subgroup. Therefore, the numbering of their sequences traditionally goes from 1 (D) to 27 (Q), then 27a-27f, then 28 (S) to 108 (R). Crystal structure of LEN solved at 1.95 Å resolution revealed that this domain is a prototypical β-domain that consists of two β-sheets organized in a specific fold β—sandwich fold (27). The LEN X-ray crystal structure further indicated that the hypervariable or complementarity-determining region 1 (CDR1) formed a large loop that extends out from the domain (27).
LEN was originally isolated from the urine of a patient suffering from multiple myeloma, with no sign of renal dysfunction or amyloidoses (28). However, LEN was shown to form fibrils in vitro under mildly destabilizing conditions (low pH or certain amount of denaturant such as urea and GuHCl) (29-31). Benign LEN is very similar to amyloidogenic light chain domains, SMA and REC, which were initially extracted from the AL amyloidosis patients (28). In fact, amino acid sequences of SMA and LEN are different only in eight positions, whereas REC is different from LEN at 14 positions (28). Interestingly, amyloidogenic LCs were shown to be significantly less stable than LEN in vitro (24). Furthermore, a series REC- or SMA-mimic mutations of LEN affected both conformational stability and aggregation propensity of this protein (24). The systematic analysis of LEN structural properties and aggregation revealed the accumulation of two partially folded intermediates: a relatively native-like intermediate was shown to accumulate at pH values between 4 and 6, and a relatively unfolded, but compact, intermediate was formed at pH below 3. Unfolded intermediate was shown to readily form amyloid fibrils, whereas native-like intermediate preferentially assembled into the amorphous aggregates (32).
Protein oxidation is associated with numerous conditions characterized by oxidative stress, as well as a number of human diseases including Alzheimer’s disease, Parkinson’s disease, diabetes, rheumatoid arthritis, muscular dystrophy, cataractogenesis, induction of renal tumors, bronchopulmonary dysplasia, amyloidosis, chronic ethanol ingestion, acute carbon tetrachloride toxicity, amyotrophic lateral sclerosis, and the progeria (33). All amino acid residues are potential targets for oxidation, which can be induced, for example, by HO• generated upon exposure to ionizing radiation or in the presence of high concentrations of hydrogen peroxide and Cu2+ or Fe2+. Cysteine and methionine are by far the most sensitive to oxidation by almost all kinds of reactive oxygen species (ROS) (34). Methionine is easily oxidized to methionine sulfoxide by 2-electron oxidation induced by H2O2, among many other biological oxidants (35). The methionine oxidation to methionine sulfoxide can be reversed by the methionine sulfoxide reductase (MSR) (36, 37). Based on this consideration it was proposed that the methionine oxidation in proteins might serve as a first line of antioxidant defense against ROS damage (37).
In this work, we investigated the conformational changes of the light chain domain LEN induced by methionine oxidation, and analyzed the effect of oxidation on conformational stability and aggregation of this protein. We found that the LEN was well-protected against oxidation in its native state. The methionine-oxidized (MetO) protein preferred to form amorphous aggregates instead of fibrils. The results indicate that oxidation of LEN could not play a role in its fibrillation.
MATERIALS AND METHODS
Chemicals
Guanidine hydrochloride (GuHCl) with the purity of 99.5% was purchased from EM Science. Peptone and yeast extract used in the medium were purchased from Difco. All other chemicals were purchased from Fisher or Sigma and were of the highest grade available. The water was doubly deionized.
Predictions of intrinsic disorder
Disorder predictions for LEN were made using PONDR® VSL2B (38) and FoldIndex™ (39) algorithms. PONDR® VSL2 (version 2 Predictor Of Natural Disordered Regions, Various Short-Length) is an ensemble of logistic regression models that predicts per-residue order-disorder (38). Two models predict either long or short disordered regions - greater or less than 30 residues — based on features similar to those used by PONDR® VLXT (40). The algorithm calculates a weighted average of these predictions, where the weights are determined by a meta-predictor that approximates the likelihood of a long disordered region within its 61-residue window (38). FoldIndex™, a freely available graphic web server (http://bioportal.weizmann.ac.il/fldbin/findex), predicts if a given protein sequence is intrinsically unfolded implementing the algorithm of Uversky and co-workers (41), which is based on the average residue hydrophobicity and net charge of the sequence (39).
Expression and purification of LEN
A frozen stock of Escherichia coli JM83 cloned with the plasmid pkIVlen004, the expression system constructed by Dr. F. Stevens (28), was grown in a rich medium and LEN was expressed as described earlier (29, 30). The over-expressed protein LEN was purified using the procedure of Stevens et al. (28): Protein dialyzed against a 10 mM Tris buffer at pH 7.4 after sucrose and water extracts were pooled together, and then eluted through a HiTrip Q FF column (Pharmacia) to remove some protein impurities. After dialysis against a 10mM acetate buffer at pH 4.0, the flow through from the HiTrip Q FF column was loaded onto Hi Trip SP FF column (Pharmacia) and protein was eluted using a NaCl gradient from 0 to 200mM over 30 min. The fractions were assayed by SDS gel-electrophoresis and the fractions containing the recombinant protein were pooled, concentrated by ultra filtration using a membrane with a 3,000 KD cutoff (Amicon). Resulting protein solution was load to a size-exclusion column (Sephacryl 200 (Pharmacia)) for further purification. Protein concentrations were measured via optical density at 280 nm using the extinction coefficient of E0.1% = 1.71 calculated from the amino acid sequence. The concentration of the LEN stock solution was ∼10 mg/ml. The purified LEN was stored in glass vials in 10 mM phosphate butter pH 7.4 at 4 °C. The purity of the protein was assessed by SDS gel-electrophoresis and by electro-spray mass spectrometry.
Oxidation of LEN by hydrogen peroxide
The oxidation of LEN was induced by the incubation of protein solution in the presence of 4 M GuHCl and 4% H2O2 at room temperature for 20 min. GuHCl and non-reacted peroxide were removed from the protein solution by dialysis against 10 mM phosphate buffer solution at pH 7.4 (changing the buffer 3 times over 24 hours). The extent of LEN oxidation was confirmed by mass spectrometry analysis (MicroMass Quattro II).
In vitro fibril formation assays
Amyloid fibrils and/or aggregates were grown from purified protein solutions in 10 mM phosphate buffer containing 100 mM NaCl. The proteins underwent an ultra-centrifugation at 178,000 g on an Airfuge (Beckman) for 30 minutes to eliminate the pre-existed aggregates before incubation under the desired conditions. Protein solutions with concentration of 0.5 mg/ml (10 mM HCl and 100 mM NaCl) were stirred in 2 ml HPLC glass vials (Fisher) containing micro-stirring bars (8.5mm, Fisher). Volumes ranged from 500 to 1000 μl. The stirring rate was at ∼600 rpm. LEN fibril formation was monitored using a fluorescence assay based on the enhanced fluorescence of the dye Thioflavin T (ThT) on binding to amyloid fibrils (42). Rayleigh light scattering and fluorescence spectra were collected using a SPEX/Jobin-Yvon Fluoromax-2 spectrofluorometer.
Circular dichroism measurements
Far-UV circular dichroism (CD) spectra were measured on an AVIV model 62DC spectropolarimeter between 250 and 200 nm with a step size of 1.0 nm, averaging time of 3 second and collecting 5 repeat scans. Cell with the path-length of 0.01 cm was used. Near-UV CD spectra were recorded from 320 to 250 nm using a cuvette with a path-length of 1 cm. Protein concentration was kept at 0.5-1 mg/ml throughout these experiments. The background was subtracted from all the spectra and the data were converted into mean residue ellipticities.
Protein stability toward GuHCl-induced unfolding
The conformational stability of LEN at pH 7.4 (10 mM phosphate buffer) was assessed by measuring intrinsic fluorescence using a FluoroMax-2 fluorescence spectrometer (Jobin Yvon-Spex) at 25 °C with protein concentration of 0.1 mg/ml. Intrinsic fluorescence measurements were performed by recording the emission spectra between 305 and 450 nm, with an excitation wavelength of 295 nm. The samples were incubated in 10 mM phosphate buffer (pH 7.4) and varying amounts of GuHCl (0-5 M) for 1 hour to make sure that the unfolding reaction reaches equilibrium.
Fluorescence intensity and wavelength of emission maximum values were analyzed by nonlinear least-squares that fit to two-state unfolding model. The fraction unfolded (Fu) was calculated by using the equation:
where yf and yu represent the intrinsic fluorescence of the folded and unfolded states and y represents the observed fluorescence at the given GuHCl concentration.
Thioflavin T and ANS Fluorescence
Fluorescence measurements were performed using a FluoroMax-2 fluorescence spectrophotometer (Jobin Yvon-Spex, Edison, NJ). ANS fluorescence was excited at 380 nm and the emission spectra were recorded from 420 to 600 nm at 25°C. In these experiments, the final concentration of ANS and protein were 10 μM and 0.1 mg/ml, respectively. ThT fluorescence was excited at 450 nm and the emission spectra were recorded from 465 to 560 nm. In these experiments, 10 μl protein solutions were added to the 1.0 ml of ThT solution (10 μM) before the measurements.
Thin-film attenuated total-reflectance Fourier transform infrared spectroscopy (ATR-FTIR)
FTIR spectra of LEN were recorded on a ThermoNicolet Nexus 670 FTIR spectrophotometer from 4000 to 400 cm-1 using a 2 cm-1 resolution and an accumulation of 64 scans. 10 μl of protein solutions of non-oxidized and MetO-LEN were applied evenly on the surface of a germanium crystal and dried to form a hydrated thin film by flowing a nitrogen stream. Background and water vapor subtractions were performed. Curve fitting of the amide I regions (raw spectra) were performed using Gaussian/Lorentzan functions (Grams). Second derivative and Fourier self-deconvoluted spectra were used as a peak position guide for the curve fitting procedure.
Transmission electron microscopy
LEN aggregates were analyzed by EM. The stirred protein solutions were sampled at pre-determined time points during the kinetic experiments by dropping 10 μl protein samples on Formarvar-coated copper grids. After draining the excess sample with filter paper, the grids were rinsed 3 times with water and air-dried. A freshly prepared 2% (w/v) uranyl acetate solution was then applied on the grids for 5 minutes. After the removal of the excess negative staining solution, the grids were air-dried again. Electron micrographs were collected using a JEOL JEM-100B microscope with a voltage accelerator of 80 kV. A magnification of 75,000 was used.
RESULTS
In vitro oxidation of LEN
There is one methionine (Met4), two cystinine (Cys23, Cys88), two tryptophan (Trp35 and Trp50) and nine tyrosine residues (Tyr31, Tyr38, Tyr42, Tyr55, Tyr92, Tyr93, Tyr97, Tyr98, and Tyr102) in LEN, all of which are readily oxidized under the appropriate conditions. Among these residues, the methionine is the most easily oxidized to methionine sulfoxide, by 2-electron oxidations induced by H2O2, among many other biological oxidants (35). Unexpectedly, the LEN was well-protected from oxidative modification, even being incubated in the presence of 4% H2O2 for 2 hours at room temperature (data not shown). In the presence of 4 M GuHCl, where LEN was mostly unfolded, the methionine residue was oxidized to methionine sulfoxide (MetO) with 4% H2O2 after the incubation at room temperature for 20 min. The oxidation was confirmed by the increase in the molecular mass from 12,636 to 12,652 Da detected by mass spectrometric analysis. This 16 Da increase in the LEN mass was attributed to the methionine oxidation (adding one oxygen atom).
Changes in secondary structure of LEN induced by methionine oxidation
LEN is a β-rich protein with a classical β-sandwich immunoglobulin fold (27). As shown in Figure 1, the far-UV CD spectrum of non-oxidized LEN under native conditions (10 mM phosphate buffer, pH 7.4) was characterized by two minima (235 nm and 219 nm). The minimum at 219 nm is the characteristic of β-structure, whereas 235 nm minimum is attributed to the contribution of aromatic residues (tyrosine clusters) to the far-UV CD region (43, 44). Methionine oxidation resulted in a significant increase in the degree of disorder in comparison with the non-oxidized protein. This was manifested by a noticeable shift of the 219 nm minimum to 214 nm accompanied by a dramatic increase in the negative ellipticity (Figure 1A). In addition, a substantial decrease in the negative ellipticity at 235 nm suggested that the environment of the aromatic residues in MetO-LEN is more symmetric than that in the non-oxidized protein.
Figure 1.
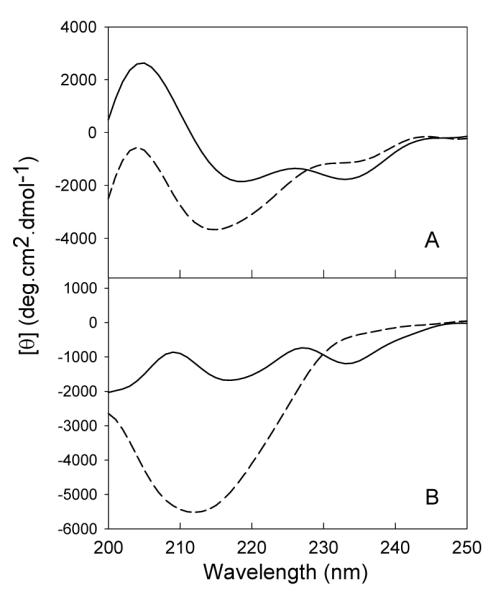
Comparison of the secondary structures of methionine oxidized LEN and native LEN at neutral and acidic pH monitored by far-UV CD. Panel A: Far-UV CD spectra at pH 7.4 measured with protein concentration of 1.0 mg/ml, 0.1 mm of path length, in 10 mM phosphate buffer at 25 °C. Panel B: Far-UV CD spectra at pH 2.0, measured with protein concentration of 1.0 mg/ml, 0.1 mm of path length, in 10 mM phosphate buffer at 25 °C. The solid and dashes lines correspond to non-oxidized and MetO proteins.
Substantial evidence supports the hypothesis that “pathological” or abnormal aggregation of LEN arises from a key partially-folded intermediate precursor which is formed under mildly destabilizing conditions (pH 2 or pH 7 with some amounts of detergents) (29). To characterize conformational changes induced in LEN by methionine oxidation, far-UV CD spectra of both non-oxidized and MetO-LEN were collected at pH 2.0; i.e., under conditions favoring the fibril formation (32). As shown in Figure 1B, the far-UV CD spectrum of the non-oxidized protein measured at pH 2.0 was rather similar to that measured at pH 7.4, with a small decrease in the negative ellipticity at 235 nm. This reflected a slightly increased degree of disorder in the environment of the aromatic residues (tyrosine clusters), likely due to the partial unfolding of LEN at low pH. In the MetO-LEN, a significant increase in the ellipticity at 212 nm, together with the complete disappearance of the 235 nm minimum were observed, reflecting noticeable rearrangement of β-structure and/or aromatic cluster.
The secondary structures of non-oxidized and MetO-LEN in both neutral and acidic environments were further analyzed by hydrated thin-film ATR-FTIR. Figure 2 shows that the FTIR amide I spectrum of native LEN (10 mM phosphate buffer, pH 7.4) possesses a main peak at 1637 cm-1, reflecting the predominantly β-sheet structure. No dramatic changes of the FTIR spectra in amide I region were observed at acidic pH (see Figure 2 and Table 1), which is consistent with the far-UV CD results. The detailed analysis of the secondary derivative spectra revealed that the β-structure content increased slightly from 73.9% at pH 7.4 to 77.7% at pH 2, indicating a small structural reorganization (Table 1). Figure 2C and Table 1 show that the methionine oxidation induced a small decrease in the LEN β-structure content (from 73.9% in non-oxidized LEN to 65.1% in MetO-LEN). When pH was decreased to 2, the β-structure content in the MetO-LEN increased slightly compared to that at neutral pH (Table 1). However, the close similarity of both spectra indicated the absence of the major changes in the secondary structure (cf. Figures 2C and 2D). These observations suggested that the significant changes induced by the methionine oxidation in the far-UV spectra of LEN should be attributed to the rearrangement of the tyrosine cluster and its environment, rather than to the secondary structure changes.
Figure 2.

Amide I region of the FTIR spectra of methionine oxidized and non-oxidized LEN at pH 7.4 and pH 2.0 (10 mM phosphate buffer) (A: LEN at pH 7.4; B: MetO LEN at pH 7.4; C: LEN at pH 2.0; and D: MetO LEN at pH 2.0). The solid lines represent the raw ATR-FTIR spectra and the dash lines represent the curve-fitted components used for secondary structure analysis.
Table 1.
Secondary Structure Content of LENa
| Peak position (cm-1) |
Secondary structure assignment |
Area under the curve (%) |
|
|---|---|---|---|
| LΕΝ at pH 7.4 | 1688 | turns/loops | 2.56 |
| 1682 | β-sheet | 2.53 | |
| 1675 | β-sheet | 9.08 | |
| 1663 | turns/loops | 17.20 | |
| 1653 | turns/loops | 6.30 | |
| 1637 | β-sheet | 54.83 | |
| 1617 | β-sheet | 7.50 | |
| Met. Oxidized LEN at pH 7.4 | 1689 | turns/loops | 0.07 |
| 1683 | β-sheet | 0.34 | |
| 1677 | β-sheet | 6.50 | |
| 1667 | turns/loops | 16.33 | |
| 1654 | turns/loops | 18.65 | |
| 1638 | β-sheet | 55.79 | |
| 1615 | β-sheet | 2.33 | |
| LΕΝ at pH 2.0 | 1688 | turns/loops | 1.47 |
| 1680 | β-sheet | 3.88 | |
| 1671 | β-sheet | 6.57 | |
| 1662 | turns/loops | 14.34 | |
| 1654 | turns/loops | 6.56 | |
| 1637 | β-sheet | 58.59 | |
| 1617 | β-sheet | 8.59 | |
| Met. Oxidized LEN at pH 2.0 | 1691 | turns/loops | 0.81 |
| 1682 | β-sheet | 7.33 | |
| 1675 | β-sheet | 4.8 | |
| 1667 | turns/loops | 12.58 | |
| 1656 | turns/loops | 14.36 | |
| 1639 | β-sheet | 56.35 | |
| 1618 | β-sheet | 4.50 |
From hydrated thin-film ATR-FTIR spectra at 25 °C.
Changes in tertiary structures of LEN induced by methionine oxidation
The near-UV CD spectra of non-oxidized and MetO-LEN were collected at pH 7.4 and pH 2.0 at 0.5mg/ml (Figure 3). In near-UV CD spectrum of native LEN, three maxima at 297, 289, and 280 nm were observed. The methionine oxidation of the protein resulted in a significant decrease in the intensities of these peaks. However, they were still observed at pH 7.4. This indicated that although the oxidized protein lost the majority of its tertiary structure, some residual tertiary structure might be present in MetO-LEN. At pH 2.0, i.e., under conditions promoting LEN fibrillation, non-oxidized LEN was characterized by the pronounced near-UV CD spectrum, which was rather similar to that measured at pH 7.4 (Figure 3B). This indicated that the tertiary structure of LEN, being slightly modified, was still maintained at these conditions. However, the near-UV CD spectrum of MetO-LEN was featureless, suggesting the complete loss of the tertiary structure at pH 2.0.
Figure 3.

Methionine oxidation induced change of tertiary structures monitored by near-UV CD spectra. Panel A: Near-UV CD spectra at pH 7.4, measured with protein concentration of 1.0 mg/ml, 1.0 cm of path length, in 10 mM phosphate buffer at 25 °C. Panel B: Near-UV CD spectra at pH 2.0, measured with protein concentration of 1.0 mg/ml, 1.0 cm of path length, in 10 mM phosphate buffer at 25 °C. The solid and dashes lines correspond to non-oxidized and MetO proteins.
Analysis of the near-UV CD, far-UV CD, and FTIR spectra revealed that the methionine oxidation induced significant structural alterations in LEN. In fact, MetO-LEN was characterized by a well-developed native-like secondary structure, whereas its tertiary structure was almost completely diminished even at neutral pH. These differences between the non-oxidized and oxidized LEN became even more evident at pH 2.0, where non-oxidized protein preserved the majority of its native structure, whereas MetO-LEN completely lost its tertiary structure while still preserving native-like secondary structure. This strongly suggested that methionine oxidation induced transformation of LEN into a molten-globule-like conformation, which is a compact denatured state characterized by a native like-secondary structure and a high affinity to a hydrophobic fluorescent probe ANS (45, 46).
Changes in LEN interaction with ANS induced by methionine oxidation
ANS fluorescence is frequently used to probe protein conformational reorganizations associated with the changes in the solvent exposure of hydrophobic regions (47, 48), usually observed upon partial protein unfolding (48). Therefore, this technique is frequently used to detect the formation of molten globule-like species (48). Both an increase in the fluorescence intensity and a decrease in the λmax values (blue shift) are usually observed upon ANS binding to exposed hydrophobic clusters. As shown in Figure 4, the ANS spectrum in the presence of non-oxidized LEN at neutral pH was characterized by λmax at ∼524 nm, indicating the absence of ANS binding to the protein under these conditions. ANS fluorescence in the presence of the methionine oxidized protein, however, possessed λmax at around 495 nm and enhanced intensity, indicating noticeable ANS binding to the protein. This gave further support to the hypothesis that the methionine oxidation of LEN induced the structural rearrangement of the protein into the molten globule-like conformation, where the native-like secondary structure was preserved, whereas the tertiary structure was diminished and the hydrophobic core became exposed to the solvent.
Figure 4.

Comparison of ANS binding capabilities of non-oxidized and methionine oxidized LEN at neutral and acidic pH. ANS fluorescence measurements were performed using an excitation wavelength of 380 nm and recording the emission spectra from 420 to 600 nm at 25 °C by adding 10 μM of ANS to 1.0 ml of protein solutions of 0.1 mg/ml at pH 7.4 (panel A) and pH 2.0 (panel B). The solid and dashes lines correspond to the non-oxidized and MetO proteins.
At pH 2.0, some enhancement in the ANS binding to the non-oxidized LEN was observed as indicated by the λmax blue shift from 520 nm to 500 nm, accompanied by a slight increase in ANS fluorescence intensity. This suggested that the hydrophobic clusters were still mostly buried in the core of LEN, though a small exposure took place. However, the methionine oxidized LEN at low pH showed very high affinity to ANS, as indicated by the λmax at 475 nm and significantly enhanced fluorescence intensity. In combination with other spectroscopic data, this indicated that the decrease in pH completed the structural transformation of MetO-LEN into the molten globule.
Changes in the Trp environment induced by methionine oxidation
The tryptophan fluorescence is known to be very sensitive to subtle structural reorganizations in the vicinity of a chromophore. The intrinsic fluorescence of LEN is determined by the presence of two tryptophan residues, Trp35 and Trp50. In the native state, Trp35 is buried in the hydrophobic core of the protein and is completely quenched by the spatial proximity of the disulfide bridge Cys23-Cys88, whereas Trp50 is significantly solvent-exposed (27) (see Figure 6A, below). Therefore, the intrinsic fluorescence of native LEN is mostly determined by the solvent-exposed Trp50, which is indicated in the λmax = 352.5 nm at pH 7.4 in the absence of GuHCl (Figure 5). Intrinsic fluorescence of MetO-LEN was characterized by λmax = 349 nm at pH 7.4 in the absence of GuHCl. This suggested that either Trp50 was more buried in MetO-LEN than in the native protein, or the spatial positions of Trp35 and the disulfide bridge Cys23-Cys88 was changed, leading to less effective quenching. The fact that the Trp fluorescence intensity of MetO-LEN was almost two-fold higher than that of the non-oxidized protein supported the hypothesis that changes in intrinsic fluorescence were due to the changes in Trp35 environment. This hypothesis was further supported by the almost complete elimination of the characteristic features in the LEN near-UV CD spectrum induced by the methionine oxidation (see Figure 3).
Figure 6.


Spatial pattern of LEN. (A) The surface of the protein. Met4 is shown in blue. (B) Cartoon diagram of the protein. Localization of Met4 (blue), Trp35 (green), Trp50 (light green), and disulfide bridge made between Cys23 and Cys88 (red) are shown as spheres. The figure is constructed on the basis of LEN structure given in the file 1LVE.pdb (27). The drawing was generated by the graphic program VMD (83).
Figure 5.

The change in conformational stability of LEN induced by methionine oxidation measured by intrinsic fluorescence in presence of various concentrations of Guanidine HCl (GuHCl). Panel A (non-oxidized LEN) and panel B (MetO LEN) show the GuHCL unfolding curves monitored by Trp fluorescence intensity (▯) and λmax (▯). Panel C: Comparison of fraction unfolded curves for the non-oxidized (▯) and the MetO-LEN (▯) derived from the Trp fluorescence λmax values in the presence of various concentrations of GuHCl.
Changes in conformational stability induced by methionine oxidation
The GuHCl-induced unfolding of LEN and LEN-MetO at pH 7.4 was studied by characteristic changes in the intrinsic fluorescence. Figure 5A shows that GuHCl-induced unfolding of native LEN was accompanied by a significant increase in the Trp fluorescence intensity, and by the shift of the λmax from 352.5 to 356 nm. Changes of these two parameters occurred almost simultaneously, being characterized by C1/2 ∼ 1.8 M GuHCl. These spectral perturbations indicated that Trp35, buried in the hydrophobic core of the native protein, went away from the disulfide bridge Cys23-Cys88 and became solvent-exposed. In MetO-LEN, the increase in the GuHCl concentration was accompanied by a red shift from 349 to 358 nm (C1/2 ∼ 1.2 M GuHCl) and a set of complex changes in the fluorescence intensity, which first sharply increased almost two-fold (C1/2 ∼ 0.2 M GuHCl) and then showed sigmoid changes with the C1/2 ∼ 2.0 M GuHCl (Figure 5B). Finally, Figure 5C shows that methionine oxidation resulted in a noticeable destabilization of a protein.
Analysis of the Met4 local environment
Analysis of the local environment of the single methinione residue of LEN (Met4) revealed that although it is located in the periphery of the hydrophobic core of the macromolecule (Figure 6B), the density of its microenvironment is rather high. In fact, it contains 10 side chains (including Val3, Thr5, Cys23, Lys24, Ser25, Leu33, Cys88, Gln90, Ser97, and Gly99) and 22 atoms (within a 4 Å sphere). Furthermore, Met4 is rather close to the disulfide bond (5.25 Å), being 15 Å apart from Trp35 (Figure 6A). Therefore, folded structure of this LC includes the snug-fitting environment for the buried Met side chain and hence protects it from H2O2 (despite the small size of the OH radical). However, the addition of the oxygen to the Met4 sulfur presumably radically breaks the snugness of this burial, significantly loosening the structure of entire protein. In fact, in the close proximity to Met4 there are residues from both N- and C-termini as well as some residues from the middle part of LEN. In agreement with this hypothesis, our experimental data suggest that Trp35 and disulfide bond move apart in response to Met4 oxidation.
Interestingly, BLASTing (49) LEN against Swiss-Prot database revealed that Met4 is rather conserved (Figure 7, central plot). In fact, this residue possesses the conservation degree of 65%, being conservatively substituted by either Leu (33%) or Ile (2%). Earlier, the Met4 to Leu (M4L) substitution in the core of LEN was shown to result in a noticeable stabilization of this domain (by 1.0 kcal/mol) (25). Similar stabilizing effect was reported for the M4L mutation in a murine Fv fragment (50). It has been proposed that the leucine residue, being completely buried, occupies the same space as the methionine in the wild-type LEN (25). In the interior of a protein, a Met-to-Leu substitution is expected to stabilize the protein by ∼1.4 kcal/mol if it does not introduce strain in the structure (51). The fact that the M4L substitution in LEN produced comparable stabilization (1.0 kcal/mol) suggested that it did not introduce noticeable alteration into the packing of the hydrophobic core.
Figure 7.

Computational analysis of the LEN sequence. Middle plot illustrates the conservation of the LEN sequence based on the BLAST analysis of Swiss-Prot database. Top and bottom panels represent the results of the LEN analysis by the PONDR® VSL2 and FoldIndex™ algorithm, respectively. Dashed vertical line illustrates the Met4 position. Bars on the top of figure represent the localization of four framework regions (FR1, FR2, FR3 and FR4) as well as the localization of three complementarity determining regions (CDR1, CDR2, and CDR3).
Predicted flexibility of the Met4-containing N-terminal fragment
Additional clues on the conformational behavior of non-oxidized and MetO-LEN can be derived from the analysis of its intrinsic disorder propensities. Recently, protein flexibility and especially its extreme case, intrinsic protein disorder, attracted considerable attention of the researchers. Recent reviews (41, 52-68) clearly reflect the growing interest in the intrinsically disordered proteins. A special database, DisProt, has been created to collect structural and functional information on these proteins (69). In part this stably increasing interested is due to the fact that these flexible regions are crucial for many non-enzymatic protein functions (70-72). Statistical analyses of amino acid composition, flexibility, hydropathy, charge, coordination number and several other factors have revealed that sequences of intrinsically disordered (and thus highly flexible) proteins, or regions of proteins, are significantly different from intrinsically ordered (regions of) proteins (55, 73, 74). An intrinsically disordered region is characterized by low sequence complexity coupled with a compositional bias; a low content of order-promoting residues, such as bulky hydrophobic amino acids (V, L, I, M, F, W and Y) normally forming the core of a folded globular protein, and cysteine; and a high proportion of polar, charged and structure-braking amino acids (Q, S, P, E, K, and less often, G and A) promoting disorder (40, 75). These differences were utilized to develop a number of disorder predictors, many of which can be accessed at the DisProt database (69). The results of application of two of these, PONDR® VSL2 (38) and FoldIndex™ (39), to the LEN sequence are shown in the top and the bottom panels of Figure 7, respectively. High PONDR® VSL2 scores for the N-terminal fragment of LEN indicate that this protein segment is predisposed to be flexible. On the other hand, FoldIndex™ predicts this fragment to be ordered due to the relatively high content of the hydrophobic residues (Figure 7). Taken together, these results suggest that ∼20 N-terminal residues of LEN are likely in the metastable folded state, which easily can be affected by some environmental factors, e.g., by the Met4 oxidation, which make this residue more polar.
Effect of methionine oxidation on LEN aggregation behavior
Changes in the thioflovin T (ThT) fluorescence were used to monitor the kinetics of fibril formation, whereas Rayleigh light scattering was utilized to follow the formation of large aggregates. As shown in Figure 8A, incubation of the non-oxidized LEN produced a sigmoid increase in the ThT fluorescence intensity with a lag-time of 10 hours. The slight increase in the signal during the lag-time reflected the formation of oligomeric species with some affinity to ThT (29-31). The large-scale changes in ThT fluorescence occurred simultaneously with the increase in the Rayleigh light scattering (cf. Figures 8A and 8B), indicating the formation of large fibrillar aggregates. The fibrils formed after the exponential growth phase (after 50 hours of incubation) were visualized by the negatively stained transmission electron microscopy (Figure 9C).
Figure 8.

Kinetics of the aggregation of non-oxidized (▯) and MetO-LEN (▯) monitored by thiflovin T (ThT) (Panel A) and light scattering (Panel B). The protein solutions were stirred at 0.5 mg/ml, 37 °C, and 600 rpm in phosphate buffer with 100 mM NaCl.
Figure 9.
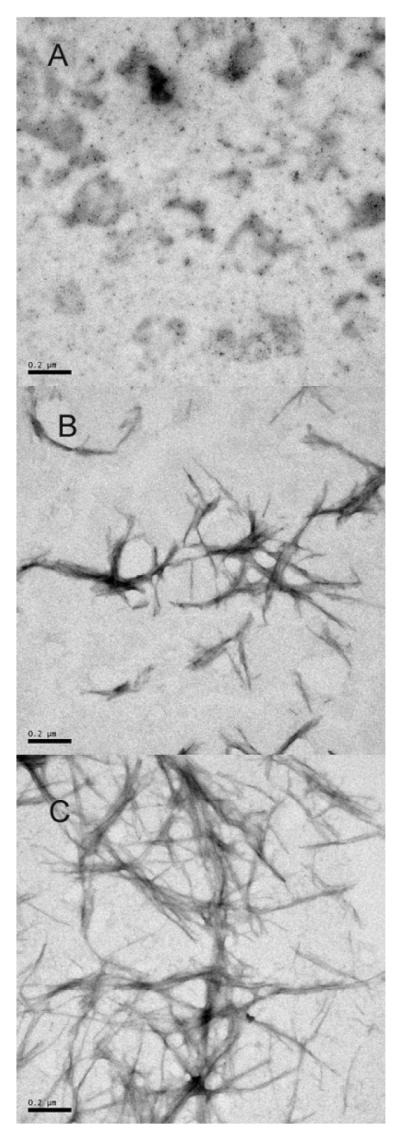
Negatively stained transmission electron micrographs of aggregates of representative nonoxidized and MetO-LEN. A: MetO-LEN after incubation for 5 hours. B: MetO-LEN after incubation for 50 hours. C: Non-oxidized LEN after incubation for 50 hours.
In the case of MetO-LEN, the fast formation of aggregates was observed at the beginning of the incubation period (in 5 hours) as indicated by the increase in both the ThT fluorescence (Figure 8A) and the Rayleigh light scattering (Figure 8B). The formation of amorphous aggregates was also confirmed by EM (Figure 9A). Although the lag-time was longer than that of the non-oxidized protein, the formation of fibrils after the prolonged incubation of MetO-LEN was detected by the ThT fluorescence (Figure 8A) and EM image (Figure 9B). The significantly lower intensity of the ThT fluorescence indicated that a lesser amount of fibrils was formed in the methionine-oxidized sample. This conclusion was also confirmed by the EM analysis (cf. Figure 9B and 9C).
Changes in the secondary structure of non-oxidized and MetO-LEN during incubation
Far-UV CD spectra of both non-oxidized and MetO-LEN were recorded during the early stages of aggregation induced by incubation with stirring at pH 2.0. In the first 10 hours of incubation, which was the lag time of fibrillation, a slight increase in the ellipticity at 217 nm was observed, indicating the existence of a slight conformational adjustment of LEN during the nucleation period. Simultaneously with the significant increase in the ThT signal, the ellipticity of the non-oxidized protein dramatically increased, and the shape of the far-UV CD spectrum changed reflecting the conformational reorganization to a typical β-sheet-enriched structure during the fibril elongation (Figures 10A and 10B).
Figure 10.

Comparison of kinetics of the secondary structure changes of non-oxidized and MetO-LEN during incubation monitored by far-UV circular dichroism. The protein solutions were stirring at 600 rpm at 37 °C in 10 mM phosphate buffer in the presence of 100 mM NaCl. A, spectra of non-oxidized LEN at the time points of incubation. B, analysis of transitions of both the ellipticity and the positions of minimum derived from panel A. C, spectra of methionine-oxidized LEN at the time points of incubation, and D, transitions of both the ellipticity and the positions of minimum derived from panel C.
At the beginning of incubation, the MetO-LEN showed immediate changes in both the spectral intensity and the minimum position. The increase in ellipticity and shift of the minimum position from 212 to 210 nm occurred in first 6 hours of incubation. However, no evidence of fibril formation was found during this time period, suggesting that the observed spectral changes reflected the conformational reorganization during oligomerization and amorphous aggregate formation (Figures 10C and 10D). Interestingly, both ellipticity and minimum position in the far-UV CD spectra of the MetO-LEN were not changed in the period between 6 to 20 hours of incubation, followed by another phase in which the continuous increase in negative ellipticity was accompanied by a shift in minimum position from 210 nm to 217 nm, indicating the formation of fibrils.
The secondary structure of LEN and MetO-LEN during aggregation was further analyzed by hydrated thin-film ATR-FTIR (Figure 11). Initially, the major peak in both LEN and MetO-LEN FTIR amide I spectra was observed at 1638 cm-1, which is typical for β-proteins. In the first 12 hours of incubation, which was within the lag-time of LEN fibrillation, there was only a slight change in the spectrum of the non-oxidized LEN, reflecting the lack of dramatic structural reorganization (Figure 11A). After 34 hours of stirring, a significant shift of the peak maximum toward the lower wave numbers (1630 cm-1) was observed concomitantly with a significant decrease in the intensity in the 1660∼1640 cm-1 region. These spectral changes reflected the structural reorganization of LEN to more ordered β-structure (i.e., to so-called aggregation β-structure), indicating the formation of fibrils.
Figure 11.

Amide I region of the FTIR spectra of LEN (A) and methionine-oxidized LEN (B) solutions at pH 2 (20 mM HCl and 100 mM NaCl) as a function of stirring time: 0 h (solid), 4 h (long dash), 6 h (short dash), 12 h (dotted), and 34 h (dash and dotted). LEN solutions were stirred at 0.5 mg/ml, 37 °C, and 600 rpm.
In contrast to the non-oxidized LEN, structural reorganization of the MetO-LEN occurred in the first 2 hours of incubation. This was indicated by the peak maximum shift from 1638 cm-1 to 1630 cm-1 (Figure 11B). This structural reorganization was accompanied by the oligomer and amorphous aggregate formation, as indicated by ThT/light scattering assay (Figure 8). The continuous right-shift of the peak maximum (toward lower wave numbers) was observed during the 12 hours of incubation with stirring, likely reflecting further protein association. The FTIR amide I spectrum of MetO-LEN after 34 hours of incubation showed a peak maximum at 1630 cm-1 with decreased intensity in the vicinity of 1660∼1640 cm-1, indicating the formation of fibrils. This was also confirmed by ThT assay (Figure 8) and electron microscopy (Figure 9).
ANS fluorescence was also used to probe protein conformational changes during the association/aggregation processes of the non-oxidized and MetO-LEN (see Figure 12). In this study, 10 μM ANS was used, as at this concentration no binding of ANS to wild type LEN was detected, but there was a significant ANS binding to MetO-LEN as shown in Figure 4. At the beginning of the non-oxidized LEN incubation, both an increase in the ANS fluorescence intensity and a blue shift of its λmax were observed. Importantly, the changes in the ANS λmax took place prior to the major changes in the ThT fluorescence. This indicated a structural transformation into a conformation with the solvent-exposed hydrophobic clusters. In the case of MetO-LEN, no changes in the λmax were detected, but the immediate increase in the ANS fluorescence intensity at the beginning of incubation was found, suggesting that the aggregates of MetO-LEN had a more solvent-exposed hydrophobic area accessible to ANS (Figure 12).
Figure 12.

Comparison of the conformational changes in the non-oxidized and MetO-LEN during incubation monitored by ANS fluorescence. The protein solutions were stirring at 600 rpm at 37 °C in 10 mM phosphate buffer in the presence of 100 mM NaCl. The transition of ANF fluorescence intensity and maximum emission wavelength for non-oxidized (Panel A) and MetO-LEN (Panel B) were plotted against incubation time.
DISSCUSSION
Pathological outcomes of aggregation, fibril formation and/or aggregate/fibril deposition are different for different diseases, ranging from the targeted neuron death in the highly localized brain regions in neurodegenerative diseases and ending with the gross tissue disruption by large masses of fibrils in classical peripheral amyloidoses. However, a common molecular mechanism is believed to underlie the fibril formation process associated with various amyloidosis diseases. The hypothesis that fibril formation arises from partially folded intermediates has been usually favored (5, 7, 12, 76). This hypothesis is supported by the following facts:
The fibrils from different pathologies are rather similar and possess a number of common structural features including a twisted, rope-like structure and a cross-β-sheet structure, where continuous β-sheets are formed with β-strands running perpendicular to the long axis of the fibrils (77).
Despite similarities characteristic of amyloid fibrils, more than 20 proteins known to be involved in deposition diseases are structurally unrelated (7). Prior to fibrillation, amyloidogenic proteins may be rich in β-sheet, α-helix, β-helix, or contain both α-helices and β-structure; they may be well-folded globular proteins with unique 3D-structures or be intrinsically unstructured (5). This suggests that there might be a unifying mechanism of protein fibrillation that involves structural transformation of a polypeptide chain into a partially folded conformation, which originates due to the partial destabilization of physiologically folded globular proteins (3-13), or because of the partial folding of natively unfolded (or intrinsically disordered) proteins (5, 14-16, 76). Presumably, such a partially unfolded conformation enables specific intermolecular interactions, including electrostatic attraction, hydrogen bonding, and hydrophobic contacts, which are necessary for oligomerization and fibrillation.
Furthermore, fibril formation has been hypothesized to occur stepwise, a slow phase of nuclei formation preceding a relatively fast elongation/growth phase (14, 78). However, both the triggering factors and the mechanisms involved in the abnormal formation of in vivo insoluble fibrillar aggregates from soluble proteins are poorly understood (79).
Given the fact that the benign light chain domain LEN with no sign of renal dysfunction or amyloidosis in a patient suffering from multiple myeloma may still form fibrils under the destabilizing conditions, and the fact that its molecular analogue SMA, being only 8 residues different from LEN, is amyloidogenic protein, suggest that some environmental factors could play a role in its fibrillation.
LEN Oxidation
Oxidative damage, probably through the intervention of various ROS, is one of the contributing factors in amyloid disease pathogenesis. The only methionine, which is located at the N-terminal part of LEN amino acid sequence, is the most readily oxidized side-chain. Given that mildly destabilizing conditions can lead to light chain aggregation, we oxidized LEN by adding hydrogen peroxide and studied the effect of methionine oxidation on protein structural properties, conformational stability, and aggregation. Surprisingly, the protein was well-protected from oxidation by H2O2 in its native state under physiological conditions. Two mechanisms can contribute to the observed high resistance toward the methionine oxidation: compactness/rigidity of a native structure and LEN dimerization. The analysis of the crystal structure (Brookhaven Protein Data Bank 1lve) revealed that the methionine residue is not located at the dimer interface, being deeply buried in the hydrophobic core of LEN. Thus, it is likely that this core location of methionine protects it from being oxidized.
However, in the presence of 4 M GuHCl, 4% of hydrogen peroxide was able to add one oxygen atom to the protein molecule, which was confirmed by mass spectrometry. The addition of 4 M GuHCl resulted in complete LEN unfolding and disassociation of its dimers. We believe that the mentioned oxygen addition was due to the oxidation of the methionine residue.
Conformational modification induced by methionine oxidation
It is recognized that mildly destabilizing conditions such as 4 M Urea, 2 M GuHCl, or pH 2.0 are necessary for LEN to form fibrils in vitro in a reasonable time-scale. However, it is unknown what triggers the light chain fibrillation in vivo under physiological conditions. Though in light chain amyloidosis, the most affected organ is the kidneys, where the pH is lower than the physiological pH, it is still not low enough to induce fibril formation found in in vitro experiments (32). Furthermore, the fact that several organs other than the kidneys are involved in the light chain amyloidosis also indicates that the acidic pH may not be the triggering factor to initiate the light chain fibrillation in vivo. On the other hand, some intra-molecular modifications, such as point mutations or oxidative modifications, can decrease the conformational stability of a light chain, and therefore promote its aggregation and fibrillation.
Data presented in this paper shows that methionine oxidation of LEN has significant effect on its structural properties, conformational stability, and propensity to aggregate. Although no noticeable changes in the secondary structure were detected in MetO-LEN in comparison with the non-oxidized protein at both physiological and acidic conditions, the far-UV and near-UV CD spectra indicated that a significant alteration of the tertiary structure took place upon oxidation. Overall, spectroscopic analysis revealed that MetO-LEN possessed many features of the molten globule, including native-like secondary structure, lack of tertiary structure, and high affinity to ANS. In addition, oxidation induced a decrease in the stability against GuHCl unfolding. All these factors can facilitate the MetO-LEN aggregation.
Oxidized and non-oxidized LEN aggregation and light chain deposition diseases
Overall, the results of our analysis show that the non-oxidized and MetO-LEN have different aggregation pathways. Non-oxidized LEN forms soluble oligomers without significant structural reorganization. Earlier, it has been established that these rapidly formed soluble oligomers are off the LEN fibrillation pathway (31). Furthermore, although most of the protein was present in these off-pathway intermediates at early aggregation stages, eventually all the protein forms fibrils. A structural rearrangement from the non fibril-prone off-pathway oligomers to a more fibril-prone species was analyzed by a variety of techniques (31). These analyses revealed the existence of dramatic structural changes within the soluble oligomers and suggested that fibrils were formed from the oligomers containing a less stable conformation of LEN, either directly or via dissociation (31).
In the case of MetO-LEN, the aggregation started with a significant structural rearrangement, leading to the formation of insoluble amorphous aggregates, which inhibited the MetO-LEN fibrillation. This difference in aggregation behavior is determined by the structural alterations induced by methionine oxidation. In fact, MetO-LEN was in a molten globule-like conformation even at neutral pH, and was further destabilized at pH 2.0. Although the oxidized protein aggregated essentially faster than the non-oxidized LEN, the molten globule-like conformation of MetO-LEN described in this paper seems to represent a species located off the fibrillation pathway.
It was observed that the morphology of light chain deposits varies in various light chain deposition diseases. In AL amyloidosis, which is the most common form of systemic amyloidosis, the amyloid fibrils derived from light chain is the dominant proteinaceous deposits. LCDD, however, is the light chain deposition disease, where the amorphous deposits are found associated with the basement membranes (17, 80). Occasionally, patients may present both AL and LCDD pathologies originated from the fibrillar and amorphous deposition of the same variable domain of the light chain (VL), sometimes in the same tissue (81). Some other deposit morphologies are present in other light chain deposition diseases. For example, amorphous cast composed of light chain in myeloma cast nephropathy and light chain crystals in Fanconi’s syndrome. As reported earlier (30, 31), in vitro studies of the LEN and SMA aggregation revealed that the light chains tend to form fibrils from their partially unfolded states. Whereas at the mild denaturing conditions, where the light chain is in its native-like intermediate, the protein readily forms amorphous aggregates (82). In present study we showed that the methionine oxidation of LEN induced the amorphous aggregation, and partially inhibited protein fibrillation. During the early stage of aggregation, the rapid formation of molten globule-like oligomers, which are different from the pro-fibrillation species formed during the early stage of non-oxidized protein fibrillation, blocks the fibrillation pathway favoring amorphous aggregate formation. Therefore, these in vitro observations, taken together with the data on morphologically different proteinaceous deposition in various light chain deposition diseases, suggest that the multiple morphologies of light chain deposition could result from multiple factors, both intramolecular and external.
Acknowledgements
We express our deepest gratitude to Alexey Uversky for carefully reading and editing the manuscript.
ABBREVIATIONS
- AL
amyloidosis, immunoglobulin light-chain amyloidosis
- LCDD
light-chain deposition disease
- ROS
reactive oxygen species
- MSR
methionine sulfoxide reductase
- MetO
methionine-oxidized
- GuHCl
guanidinim hydrochloride
- ThT
Thioflavin T
- CD
circular dichroism
- ATR-FTIR
attenuated total reflectance Fourier transform infrared spectroscopy
- EM
electron microscopy
Footnotes
This research was supported in part by grants R01 NS39985 (to D.H., Z.Q. and A.L.F.), R01 LM007688-01A1 (V.N.U.) and GM071714-01A2 (V.N.U.) from the National Institutes of Health. We gratefully acknowledge the support of the IUPUI Signature Centers Initiative.
References
- 1.Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts) N Engl J Med. 1980;302:1283–92. doi: 10.1056/NEJM198006053022305. [DOI] [PubMed] [Google Scholar]
- 2.Sipe JD. Amyloidosis. Annu Rev Biochem. 1992;61:947–75. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 3.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein deposits as the molecular basis of amyloidosis. I. Systemic amyloidoses. Med Sci Monitor. 1999;5 [Google Scholar]
- 4.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein Deposits As the Molecular Basis of Amyloidosis. II. Localized Amyloidosis and Neurodegenerative Disorders. Med Sci Monitor. 1999;5:1238–1254. [Google Scholar]
- 5.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–53. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–6. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 7.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 8.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–91. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rochet JC, Lansbury PT., Jr. Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–8. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 10.Lansbury PT., Jr. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci U S A. 1999;96:3342–4. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 12.Dobson CM. The structural basis of protein folding and its links with human disease. Philos Trans R Soc Lond B Biol Sci. 2001;356:133–45. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zerovnik E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur J Biochem. 2002;269:3362–71. doi: 10.1046/j.1432-1033.2002.03024.x. [DOI] [PubMed] [Google Scholar]
- 14.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr. Assembly of A beta amyloid protofibrils: an in vitro model for a possible early event in Alzheimer’s disease. Biochemistry. 1999;38:8972–80. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 15.Uversky VN, Lee HJ, Li J, Fink AL, Lee SJ. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem. 2001;276:43495–8. doi: 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- 16.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–44. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 17.Gallo G, Picken M, Buxbaum J, Frangione B. The spectrum of monoclonal immunoglobulin deposition disease associated with immunocytic dyscrasias. Semin Hematol. 1989;26:234–45. [PubMed] [Google Scholar]
- 18.Buxbaum J. Mechanisms of disease: monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol Oncol Clin North Am. 1992;6:323–46. [PubMed] [Google Scholar]
- 19.Solomon A, Weiss DT, Pepys MB. Induction in mice of human light-chain-associated amyloidosis. Am J Pathol. 1992;140:629–37. [PMC free article] [PubMed] [Google Scholar]
- 20.Solomon A. Clinical implications of monoclonal light chains. Semin Oncol. 1986;13:341–9. [PubMed] [Google Scholar]
- 21.Solomon A. Light chains of human immunoglobulins. Methods Enzymol. 1985;116:101–21. doi: 10.1016/s0076-6879(85)16008-8. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan B, Vidal R, Kumar A, Ghiso J, Frangione B, Gallo G. Amino-terminal identity of co-existent amyloid and non-amyloid immunoglobulin kappa light chain deposits. A human disease to study alterations of protein conformation. Clin Exp Immunol. 1997;110:472–8. doi: 10.1046/j.1365-2249.1997.4421454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanders PW, Booker BB. Pathobiology of cast nephropathy from human Bence Jones proteins. J Clin Invest. 1992;89:630–9. doi: 10.1172/JCI115629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffen R, Dieckman LJ, Szpunar M, Wunschl C, Pokkuluri PR, Dave P, Stevens P. Wilkins, Cai X, Schiffer M, Stevens FJ. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci. 1999;8:509–17. doi: 10.1110/ps.8.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pokkuluri PR, Raffen R, Dieckman L, Boogaard C, Stevens FJ, Schiffer M. Increasing protein stability by polar surface residues: domain-wide consequences of interactions within a loop. Biophys J. 2002;82:391–8. doi: 10.1016/S0006-3495(02)75403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. U.S. Department of Health and Human Services U.S. Government Printing Office; Washington, DC: Sequences of Proteins of Immunological Interest. (5th ed) 1991 NIH publication 91-3242.
- 27.Huang DB, Chang CH, Ainsworth C, Johnson G, Solomon A, Stevens FJ, Schiffer M. Variable domain structure of kappaIV human light chain Len: high homology to the murine light chain McPC603. Mol Immunol. 1997;34:1291–301. doi: 10.1016/s0161-5890(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 28.Stevens PW, Raffen R, Hanson DK, Deng YL, Berrios-Hammond M, Westholm FA, Murphy C, Eulitz M, Wetzel R, Solomon A, et al. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Protein Sci. 1995;4:421–32. doi: 10.1002/pro.5560040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souillac PO, Uversky VN, Millett IS, Khurana R, Doniach S, Fink AL. Effect of association state and conformational stability on the kinetics of immunoglobulin light chain amyloid fibril formation at physiological pH. J Biol Chem. 2002;277:12657–65. doi: 10.1074/jbc.M109230200. [DOI] [PubMed] [Google Scholar]
- 30.Souillac PO, Uversky VN, Millett IS, Khurana R, Doniach S, Fink AL. Elucidation of the molecular mechanism during the early events in immunoglobulin light chain amyloid fibrillation. Evidence for an off-pathway oligomer at acidic pH. J Biol Chem. 2002;277:12666–79. doi: 10.1074/jbc.M109229200. [DOI] [PubMed] [Google Scholar]
- 31.Souillac PO, Uversky VN, Fink AL. Structural transformations of oligomeric intermediates in the fibrillation of the immunoglobulin light chain LEN. Biochemistry. 2003;42:8094–104. doi: 10.1021/bi034652m. [DOI] [PubMed] [Google Scholar]
- 32.Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, Millett I, Fink AL. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry. 2001;40:3525–35. doi: 10.1021/bi001782b. [DOI] [PubMed] [Google Scholar]
- 33.Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 34.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 35.Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 36.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A. 2001;98:12920–5. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50:301–7. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 38.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–8. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 40.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 42.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–9. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 43.Sreerama N, Manning MC, Powers ME, Zhang JX, Goldenberg DP, Woody RW. Tyrosine, phenylalanine, and disulfide contributions to the circular dichroism of proteins: circular dichroism spectra of wild-type and mutant bovine pancreatic trypsin inhibitor. Biochemistry. 1999;38:10814–22. doi: 10.1021/bi990516z. [DOI] [PubMed] [Google Scholar]
- 44.Strickland EH, Mercola D. Near-ultraviolet tyrosyl circular dichroism of pig insulin monomers, dimers, and hexamers. Dipole-dipole coupling calculations in the monopole approximation. Biochemistry. 1976;15:3875–84. doi: 10.1021/bi00662a035. [DOI] [PubMed] [Google Scholar]
- 45.Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 46.Ptitsyn OB, Bychkova VE, Uversky VN. Kinetic and equilibrium folding intermediates. Philos Trans R Soc Lond B Biol Sci. 1995;348:35–41. doi: 10.1098/rstb.1995.0043. [DOI] [PubMed] [Google Scholar]
- 47.Stryer L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J Mol Biol. 1965;13:482–95. doi: 10.1016/s0022-2836(65)80111-5. [DOI] [PubMed] [Google Scholar]
- 48.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–28. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 49.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benhar I, Pastan I. Identification of residues that stabilize the single-chain Fv of monoclonal antibodies B3. J Biol Chem. 1995;270:23373–80. doi: 10.1074/jbc.270.40.23373. [DOI] [PubMed] [Google Scholar]
- 51.Gassner NC, Baase WA, Matthews BW. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc Natl Acad Sci U S A. 1996;93:12155–8. doi: 10.1073/pnas.93.22.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 53.Namba K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells. 2001;6:1–12. doi: 10.1046/j.1365-2443.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 54.Demchenko AP. Recognition between flexible protein molecules: induced and assisted folding. J Mol Recognit. 2001;14:42–61. doi: 10.1002/1099-1352(200101/02)14:1<42::AID-JMR518>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 56.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 57.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–56. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 59.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–71. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–5. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 61.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 62.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–84. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 63.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–56. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 65.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 66.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 67.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–33. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 68.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35:D786–93. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–32. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–98. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romero P, Obradovic Z, Dunker AK. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Informatics. 1997;8:110–124. [PubMed] [Google Scholar]
- 74.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–84. [PubMed] [Google Scholar]
- 75.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52:573–84. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 76.Uversky VN. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res. 2008;5:260–87. doi: 10.2174/156720508784533312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sunde M, Blake C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv Protein Chem. 1997;50:123–59. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 78.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–72. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 79.Kisilevsky R. Review: amyloidogenesis-unquestioned answers and unanswered questions. J Struct Biol. 2000;130:99–108. doi: 10.1006/jsbi.2000.4222. [DOI] [PubMed] [Google Scholar]
- 80.Gallo G, Goni F, Boctor F, Vidal R, Kumar A, Stevens FJ, Frangione B, Ghiso J. Light chain cardiomyopathy. Structural analysis of the light chain tissue deposits. Am J Pathol. 1996;148:1397–406. [PMC free article] [PubMed] [Google Scholar]
- 81.Stokes MB, Jagirdar J, Burchstin O, Kornacki S, Kumar A, Gallo G. Nodular pulmonary immunoglobulin light chain deposits with coexistent amyloid and nonamyloid features in an HIV-infected patient. Mod Pathol. 1997;10:1059–65. [PubMed] [Google Scholar]
- 82.Qin Z, Hu D, Zhu M, Fink AL. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry. 2007;46:3521–31. doi: 10.1021/bi061716v. [DOI] [PubMed] [Google Scholar]
- 83.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]