

Abstract
It has been suggested that sensorimotor gating deficits as indexed by prepulse inhibition (PPI) of the acoustic startle reflex represent an endophenotypic marker of psychotic conditions such as schizophrenia (SCZ). This hypothesis has been questioned as several studies have found that PPI levels change with improvement in symptoms and are responsive to medications. We tested PPI in a sample of acutely decompensated schizophrenia patients who were re-tested after 2 weeks of hospital treatment. PPI was assessed at three interstimulus intervals (30, 60, and 120 ms) in 23 SCID-diagnosed SCZ patients shortly after admission to an inpatient psychiatric hospital. Eight of these patients were initially tested in a medication-free state, and all were re-tested approximately 2 weeks later after initiation or increase/change of antipsychotic medications. Symptom ratings were collected at both sessions. 20 nonpatient comparison subjects (NCS) were also tested at a 2-week interval. While SCZ patients showed lower PPI at the first session than NCS, after 2 weeks of treatment their PPI increased to levels not different than those of NCS. In contrast, the PPI of NCS remained consistent over a 2-week period. For the SCZ patients, increase in PPI was correlated with a decrease in symptom scores. Our results suggest that PPI can be improved by short-term treatment, and that improvement in sensorimotor gating is associated with treatment-related improvement of symptoms of schizophrenia.
Keywords: Sensorimotor gating, Prepulse inhibition, Schizophrenia, Acoustic startle, Antipsychotic medication, Symptoms
1. Introduction
Prepulse inhibition (PPI) is the reduction of the startle reflex by the presence of a non-startling stimulus and is an operational measure of sensorimotor gating (Braff and Geyer, 1990). This reflex, present in all mammals, is mediated by inhibitory cortical–striatal neural circuits (Braff et al., 2001; Swerdlow and Koob, 1987) and hypothesized to be critical for inhibiting or screening out internal and external stimuli (Braff et al., 1978, 1995; Perry and Braff, 1994). Accordingly, individuals with neuro-psychiatric disorders characterized by inhibitory deficits of sensory, motor and cognitive functions have impaired PPI, namely patients with schizophrenia (Braff et al., 1978; Perry et al., 2002), as well as obsessive-compulsive disorder (Swerdlow et al., 1993), Huntington’s (Swerdlow et al., 1995), Tourette’s syndrome (Swerdlow et al., 2001), bipolar disorder (Perry et al., 2001), and adult autistic disorder (McAlonan et al., 2002; Perry et al., 2006). PPI has been found to be highly stable and reliable in non-psychiatrically ill humans (Schwarzkopf et al., 1993). PPI can be studied across species and schizophrenia-like PPI deficits in animals resulting from genetic, environmental, and pharmacological manipulations have become useful animal models for studying the mechanisms of psychiatric illness and the efficacy of antipsychotic compounds (Geyer et al., 2001; Swerdlow et al., 2000).
Some evidence suggests that PPI deficits improve in schizophrenia patients treated with atypical antipsychotic medication (Kumari et al., 1999; Leumann et al., 2002; Oranje et al., 2002; Weike et al., 2000). In particular, several authors (Kumari et al., 1999; Oranje et al., 2002) have reported that clozapine was superior to other antipsychotics in normalizing PPI. These cross-sectional studies are promising but do not address whether PPI improvement is mediated by antipsychotic medication or antipsychotic-related symptom reduction.
Several longitudinal studies have addressed the effect of medication status on PPI among schizophrenia patients. Duncan and colleagues (2003) tested unmedicated, de-compensated schizophrenia patients and retested them 3 months after antipsychotic treatment (Duncan et al., 2003). They found continued impaired PPI in the face of symptomatic improvement; however, this study did not include a non-schizophrenia comparison group, limiting the ability to draw definitive conclusions. Mackeprang et al. studied PPI in a drug-naïve, first-episode schizophrenia cohort and retested them 3 months later following treatment with risperidone or a typical antipsychotic (Mackeprang et al., 2002). At baseline the patients had lower PPI than comparison subjects which did not significantly improve with treatment. These findings in addition to others’ work showing PPI deficits in first-degree relatives of schizophrenia patients (Cadenhead et al., 2000), have led some to suggest that PPI deficits may be a stable endophenotypic marker of schizophrenia. There have been, however, other longitudinal studies that have reported PPI improvement following treatment. Meincke and his colleagues (Meincke et al., 2004) tested medicated schizophrenia patients and a nonpatient group on two occasions 2–3 weeks apart. The patients were tested following psychiatric admission and after the onset of treatment. They found that the patients had impaired PPI at baseline but were not different from the nonpatients at the second session, when patients’ symptoms had improved. These authors also reported a significant relationship between PPI deficits and positive symptoms. Recently, Quednow et al. (2006) tested hospitalized schizophrenia patients once prior to treatment with either olanzapine or amisulpride, and again after 4 and 8 weeks of treatment, and compared them to a group of nonpatients tested at the same time intervals. Schizophrenia patients showed PPI deficits at the first session, but the groups were not significantly different at the two subsequent test sessions.
These (Meincke et al., 2004; Quednow et al., 2006) studies suggest that, rather than being a fixed characteristic of schizophrenia, PPI deficits improve with treatment. Still, it remains unclear whether improvement in PPI is a direct function of the effects of antipsychotic medications, or is related to improvement in symptoms. In the present study we sought to: 1) examine PPI in schizophrenia patients who are hospitalized for decompensation and are treated with antipsychotic medications for 2 weeks, and test non-patient comparison subjects over the same time course, and 2) determine the relationship between PPI and changes in symptoms over the course of treatment. Additionally, by comparing PPI changes in schizophrenia patients who were initially unmedicated to patients initially medicated, we sought to address whether treatment with antipsychotic medications is directly related to an amelioration of sensorimotor gating deficits.
2. Method
2.1. Participants
Twenty-three schizophrenia (SCZ) participants (16 M, 7 F) (mean age=35.3, S.D.=10.8) diagnosed with the Structured Clinical Interview for DSM-IV Disorders (SCID) were tested within 72 h of admission to the psychiatric service at UCSD-Medical Center and retested approximately 2 weeks after their first session. Twenty (10 M, 10 F) nonpatient comparison subjects (NCS) (mean age=31.9, S.D.=9.2) were tested twice, 2 weeks apart. Potential participants from both groups were excluded for: Axis I and II disorders as assessed by the SCID (apart from SCZ for the SCZ group), neurological illness or head trauma, unstable medical illness, drug abuse or dependence within the past 6 months, treatment with electroconvulsive therapy, or a positive result on a toxicology screen. The data from the first test session of 17 SCZ pts and 4 NCS have been previously reported in Perry et al. (2002).
The SCZ patients were divided into groups based upon medication status at the time of the first testing session (Session 1). Eight SCZ patients were identified as being medication free for at least 1 week prior to admission (see Perry et al., 2002). These “unmedicated” participants were tested prior to initiating antipsychotic medication treatment. Patients’ medication regimens were not altered in any way because of participation in this research. Some patients refused medications and others were in a brief period of observation prior to selection of an appropriate medication as per normal clinical practice. Fifteen SCZ patients were treated with antipsychotic medications for at least 1 week prior to admission (“medicated”) and continued treatment at the time of the testing. In most cases (approximately 80%), medication status was verified by our research staff with the patient’s caregivers. Medicated and unmedicated SCZ patients were not significantly different in terms of age, years of education, or WAIS-R Vocabulary scaled scores. By the second testing session, almost all of the SCZ patients in both groups had completed 2 weeks of antipsychotic treatment (Table 1).
Table 1.
Antipsychotic medication regimen for initially unmedicated (n=8) and initially medicated (n=15) SCZ patients
| Session 1 | Session 2 | |
|---|---|---|
| Unmedicated SCZ: | ||
| #1 | None | Risperidone 4 mg (266.7) |
| #2 | None | Refusing medications |
| #3 | None | Risperidone 4 mg (266.7) |
| #4 | None | Risperidone 2 mg (133.3) |
| #5 | None | Haloperidol 10 mg (500) |
| #6 | None | Refusing medications |
| #7 | None | Olanzapine 10 mg (250) |
| #8 | None | Risperidone 4 mg (266.7) |
| Medicated SCZ: | ||
| #1 | Quetiapine 800 mg, | Quetiapine 800 mg, |
| Risperidone 2 mg(933.3) | Risperidone 2 mg(933.3) | |
| #2 | Olanzapine 20 mg (500) | Olanzapine 20 mg (500) |
| #3 | Olanzapine 20 mg (500) | Refusing medications (500) |
| #4 | Quetiapine 400 mg (400) | Quietiapine 300 mg (300) |
| #5 | Haloperidol 10 mg (500) | Loxapine 50 mg (500) |
| #6 | Olanzapine 20 mg (500) | Haloperidol 15 mg (750) |
| #7 | Olanzapine 20 mg (500) | Olanzapine 20 mg (500) |
| #8 | Risperidone 6 mg (400) | Risperidone 6 mg (400) |
| #9 | Risperidone 6 mg (400) | Risperidone 6 mg (400) |
| #10 | Olanzapine 10 mg (250) | Olanzapine 10 mg (250) |
| #11 | Risperidone 4 mg (266.7) | Risperidone 5 mg (333.3) |
| #12 | Risperidone 4 mg (266.7) | Risperidone 4 mg (266.7) |
| #13 | Risperidone 2 mg (133.3) | Risperidone 6 mg (400) |
| #14 | Fluphenazine 10 mg (500) | Fluphenazine decanoate 25 mg (250) |
| #15 | Risperidone 6 mg (400) | Risperidone 6 mg (400) |
Chlorpromazine equivalent in parentheses ( ).
2.2. Procedure
After participants received a complete description of the study, written informed consent was obtained. SCZ patients were assessed using the Positive and Negative Syndrome Scale (PANSS) (Kay et al., 1987). The WAIS-R Vocabulary subtest was administered to obtain an estimate of Verbal IQ. Participants refrained from nicotine use 30 min prior to startle testing. All participants underwent a brief hearing screening using an audiometer. Participants were seated comfortably in a reclining chair. Acoustic startle and prepulse stimuli were presented binaurally through headphones. The eyeblink component of the auditory startle reflex was measured using electromyography (EMG) of the orbicularis oculi muscle and EMG activity was recorded and filtered per our established methods (Braff et al., 1992; Perry et al., 2002, 2001).
The startle session was consistent with previous methodology (Braff et al., 1992; Perry et al., 2002, 2001), beginning with a 5-min acclimation period of 70 db white noise followed by four blocks. The first and last block consisted of five pulse-alone trials of 40 ms 115 dB startle stimuli. Blocks two and three consisted of pulse-alone and prepulse–pulse trials presented in pseudorandom order. The 20 ms prepulse stimuli preceded the startle stimulus by 30, 60 or 120 ms and were 15 dB above the 70 dB background noise. The inter-trial interval averaged 15 s with a range of 8 to 22 s.
2.3. Data processing and statistical analyses
The startle measures were: 1) amplitude of the startle response to pulse alone trials as measured in digital units. 2) Habituation of the startle response was measured by assessing the percentage decrement in the amplitude of the startle response to pulse alone trials (Block 1 and Block 4). 3) Prepulse inhibition (PPI), calculated as the percent decrement in startle amplitude in the presence of the prepulse compared to the amplitude without the prepulse [100− (prepulse amplitude/pulse amplitude)×100]. The data from subjects who showed extreme inconsistency in PPI within the same session (≥ 1.5 standard deviations from mean PPI change from Block 2 to Block 3 in at least one interstimulus interval) were not included in the analyses. Average PPI was calculated over Blocks 2 and 3 (the two blocks containing prepulse trials). Data were inspected for normality and homogeneity. All statistical analyses were performed with SPSS 11.5. Significance level for the a priori analyses was set at p<.05, and for post-hoc analyses it was conservatively set at p≤.025. Effect sizes were calculated using Cohen’s d.
3. Results
3.1. Startle amplitude, habituation and PPI between SCZ and NCS groups
NCS and SCZ patients did not significantly differ in age [t (41)=1.1, ns] or gender (χ2 =1.7, ns). A Repeated Measures ANOVA revealed that SCZ patients had higher startle amplitude than NCS overall [F (1, 41)=8.5, p=.01]. Post-hoc t-tests indicated that this significant difference was at both Session 1 [t (41)=2.8, p=.01] and 2 [t (41)=2.6, p=.01]. There was no main effect of session [F (1, 41)=.01, ns] nor a session-by-group interaction [F (1, 41)=.14, ns], suggesting that startle amplitude for both groups was stable across sessions. For percent habituation, there was no session main effect [F (1, 41)=.11, ns], no group main effect [F (1, 41)=.31, ns], and no session-by-group interaction [F (1, 41)=.65, ns].
For PPI, a 3×2×2 mixed ANOVA was conducted with interstimulus interval (30, 60, 120 ms) and session (1, 2) as the repeated measures and group (SCZ, NCS) as the between-subjects measure. There was a significant main effect of session [F (1, 41)=5.5, p=.02] such that PPI improved over time. However, the significant session-by-group interaction [F (1, 41)=4.5, p=.04] (Fig. 1) indicated that this improvement is largely accounted for by the SCZ patients. There was a significant main effect of interstimulus interval [F (2, 40)=89.9, p<.001] such that PPI was greater at the longer intervals, and a significant session-by-interstimulus interval interaction [F (2, 40)= 3.4, p=.04] (Fig. 2). There was no significant main effect of group [F (1, 41)=3.4, ns], no significant group-by-interstimulus interval interaction [F (2, 40)=.98, ns], nor a significant group-by-session-by-interstimulus interval interaction [F (2, 40)=.64, ns].
Fig. 1.
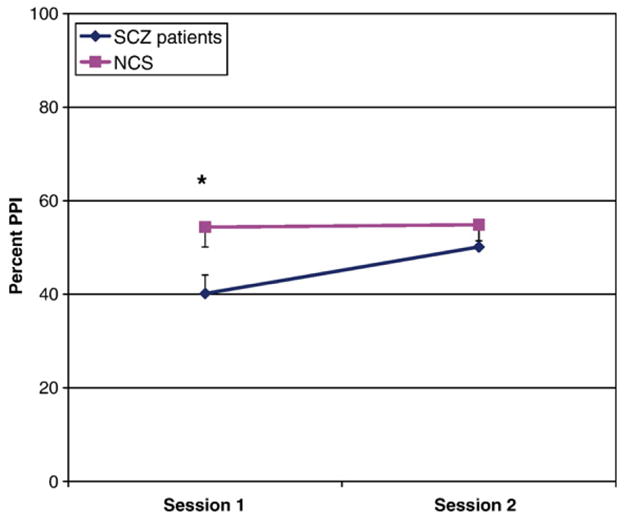
PPI averaged across 30 ms, 60 ms, and 120 ms interstimulus intervals across sessions for SCZ patients (n=23) and NCS (n=20). * Significant difference between groups, p<.05.
Fig. 2.
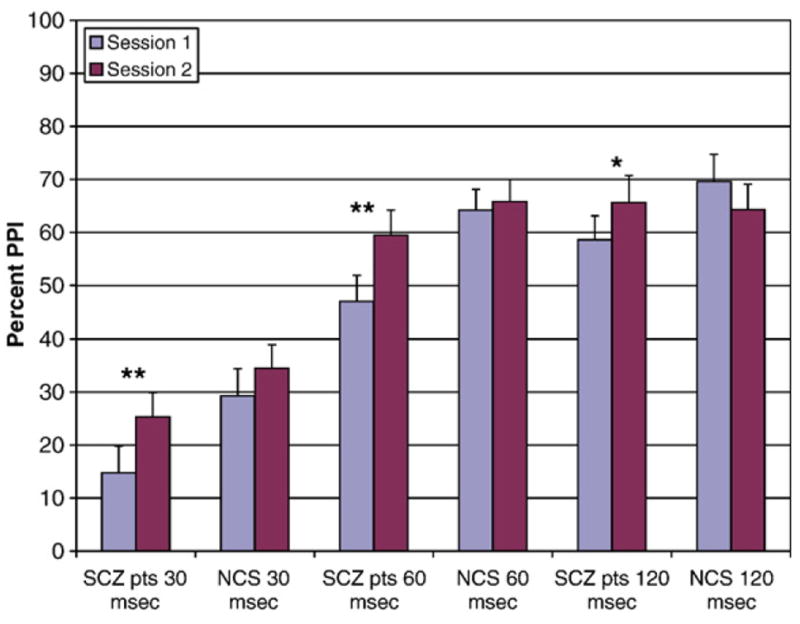
PPI for 30 ms, 60 ms, and 120 ms interstimulus intervals across sessions for SCZ patients (n=23) and NCS (n=20). ** Significant difference between Session 1 and Session 2, p<.01. * Significant difference between Session 1 and Session 2, p<.05.
To interpret the session-by-group interaction, post-hoc t-tests were conducted on average PPI across all three interstimulus intervals. SCZ patients had significantly lower PPI than NCS at Session 1 [t (41)=2.4, p=.02], but there was no group difference in PPI at Session 2 [t (41)=.89, ns]. Paired-samples t-tests conducted separately for SCZ and NCS indicated that, while SCZ patients’ PPI significantly increased from Session 1 to Session 2 [t (22)=3.2, p=.004], NCS’ PPI remained relatively consistent [t (19)=.16, ns]. Effect sizes for the difference in PPI from Session 1 to Session 2 for the SCZ patients were as follows: 30 ms d=.46, 60 ms d=.55, 120 ms d=.39.
Because SCZ patients had larger startle amplitude compared to the NCS, we assessed whether amplitude differences accounted for PPI differences. A mixed 3×2×2 ANOVA on PPI was conducted on a subset of the two groups that were matched on amplitude (SCZ n=20, NCS n=17). There was a significant session-by-group interaction [F (1, 35)=6.1, p =.02], and no group-by-session-by-interstimulus interval interaction [F (2, 34)=.99, ns].
3.2. Relationship between PPI and symptom status
Pearson R correlations were calculated between PPI at Session 1 and PANSS scores at Session 1, as well as between change in PPI from Session 1 to Session 2 and change in PANSS scores from Sessions 1 to 2. Analyses were conducted using PPI averaged across all inter-stimulus intervals. We also conducted separate analyses for the interstimulus intervals based upon our and others’ findings showing significant relationships between symptoms and PPI at the longer intervals (Perry and Braff, 1994; Perry et al., 1999; Weike et al., 2000). There was a trend for negative symptom scores at Session 1 to be inversely correlated with PPI at the 30 ms condition. Improved PPI over the two sessions was related to a decrease in all PANSS subscales, most notably in the 60 ms condition and with the negative symptom subscale (Table 2).
Table 2.
Pearson R correlations between PPI and Positive and Negative Syndrome Scale (PANSS) scores for SCZ patients (n=23)
| Overall PPI Session 1 | 30 ms PPI Session 1 | 60 ms PPI Session 1 | 120 ms PPI Session 1 | |
|---|---|---|---|---|
| Positive scale Session 1 | R=−.12 | R=−.09 | R=−.20 | R=.00 |
| Negative scale Session 1 | R=−.20 | R=−.41 | R=−.06 | R=.00 |
| General Scale Session 1 | R=−.07 | R=−.14 | R=−.16 | R=.14 |
| Total PANSS score Session 1 | R=−.18 | R=−.33 | R=−.17 | R=.08 |
| Overall PPI change | 30 ms PPI change | 60 ms PPI change | 120 ms PPI change | |
| Positive scale change | R=−.30 | R=−.02 | R=−.52** | R=−.11 |
| Negative scale change | R=−.55** | R=−.54** | R=−.47* | R=−.27 |
| General scale change | R=−.29 | R=−.18 | R=−.41 | R=−.03 |
| Total PANSS score change | R=−.48* | R=−.35 | R=−.57** | R=−.17 |
“Change” for both PPI and PANSS scores refers to Session 2 value minus Session 1 value.
p≤.05.
p<.01.
3.3. Medication status
SCZ patients who were initially unmedicated and medicated were compared on startle and PPI at Sessions 1 and 2. There was no main effect of session on startle amplitude [F (1, 21)=.11, ns], no main effect of group [F (1, 21)=1.04, ns], and no session-by-group interaction [F (1, 21)=.35, ns]. A 3×2×2 mixed ANOVA with medication status (medicated, unmedicated) as the between-subjects measure revealed no main effect of group [F (1, 21) =.68, ns], no group-by-session interaction [F (1, 21)=.77, ns], no group-by-interstimulus interval interaction [F (2, 20)=.39, ns], and no group-by-session-by-interstimulus interval interaction [F (2, 20)=.53, ns].
4. Discussion
In the present study we again found that at cross-section, decompensated schizophrenia patients show PPI deficits when compared to nonpatients (Braff et al., 1978, 2001; Perry and Braff, 1994; Perry et al., 1999). We further found that the PPI levels of schizophrenia patients improved after 2 weeks of psychiatric treatment, with a medium effect size. In contrast, PPI levels of comparison subjects remained relatively unchanged across the same two time points. The present findings support those of Meincke et al. (2004) and Quednow et al. (2006) who found PPI deficits in schizophrenia that dissipated after a period of treatment. Furthermore, change in PPI among the schizophrenia patients was inversely correlated with change in symptom scores. Change in positive symptoms was correlated with change in PPI in one of the three interstimulus interval conditions, and change in negative symptoms was correlated to change in overall PPI. Thus, improvement in PPI over this relatively short treatment period did appear to be related to symptom improvement.
In contrast to our results, Duncan et al. (2003) and Mackeprang et al. (2002) reported that that treatment or symptom reduction was unrelated to PPI levels in schizophrenia patients. However Duncan and colleagues also pointed out that restriction of range may have prevented the reliable detection of improvement in PPI, and as they did not have a comparison group it remains difficult to assess whether schizophrenia patients had noticeable PPI deficits at their first test session. Mackeprang et al. noted that they tested a small group of patients treated with an atypical antipsychotic (n=13). Therefore, further studies are required to resolve these conflicting findings. Findings from first-degree family members of schizophrenia patients indicate that PPI deficits may be a trait characteristic of schizophrenia. If subsequent research confirms our findings that PPI varies within individuals across symptom states, this will suggest that PPI in schizophrenia patients reflects both state and trait features relevant to the disorder.
The significant correlations between improvement in PPI and improvement in symptoms are consistent with our (Perry et al., 1999) as well as others’ (Meincke et al., 2004) results that sensorimotor gating deficits and psychotic symptomatology are related, and further highlight that these two areas of impairment improve in relation to one another. However, to our knowledge this is the first report showing not only a correlation in PPI and symptoms cross-sectionally, but a systematic relationship between improvement in symptoms and improvement in PPI. Interestingly, this improvement is most evident for negative symptoms, which has not been widely reported in the literature (but see (Braff et al., 1999)). This finding is noteworthy given that there are reports that atypical antipsychotics appear to be more efficacious than typicals at improving PPI (Kumari et al., 1999; Leumann et al., 2002; Oranje et al., 2002; Weike et al., 2000) and also negative symptoms (Moller, 2003). Increased dopamine in the prefrontal cortex associated with atypical antipsychotic medications (Horacek et al., 2006) has been implicated as a basis for amelioration of negative symptoms. It is possible therefore that this mechanism also underlies the apparent improvement in PPI.
Quednow and colleagues (Quednow et al., 2006) recently reported that treatment with both a selective dopamine (D2/D3) antagonist and a serotonin-2/D2 antagonist ameliorated PPI deficits in schizophrenia patients. Accordingly, they suggest that dopamine regulation is the crucial link in PPI and suggest a putative relationship between dopamine blockade and restoring PPI. We had previously reported no differences in PPI between unmedicated and medicated schizophrenia patients when assessed cross-sectionally (Perry et al., 2002). In the present study, we found medication status at baseline did not differentially influence PPI following 2 weeks of treatment. These findings suggest that PPI may not be solely mediated by dopamine blockade and underscore the complex interplay between dopamine and symptomatology in schizophrenia. Because of a small sample size and the naturalistic nature of this study where medication status was not controlled or randomly assigned, our results are not conclusive. Therefore, it remains unclear what direct impact antipsychotic medications have on PPI.
In conclusion, the current results suggest that PPI levels in acutely decompensated schizophrenia patients are impaired at baseline and improve following 2 weeks of treatment, and that improvement in PPI was related to a decrease in symptoms of the disease. More rigorously controlled studies are needed using a consistent dose of an active antipsychotic compound versus placebo, or testing the effects of antipsychotic medications on non-schizophrenia populations who have PPI deficits.
Acknowledgments
This study was supported by a NARSAD Young Investigator Award (WP) and R01-MH071916-01. AM was supported in part by NIMH Training Grant T32-MH18399.
References
- Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47(2):181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15(4):339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49(3):206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Braff DL, Swerdlow NR, Geyer MA. Gating and habituation deficits in the schizophrenia disorders. Clin Neurosci. 1995;3(2):131–139. [PubMed] [Google Scholar]
- Braff DL, Swerdlow NR, Geyer MA. Symptom correlates of prepulse inhibition deficits in male schizophrenic patients. Am J Psychiatry. 1999;156(4):596–602. doi: 10.1176/ajp.156.4.596. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156(2–3):234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Cadenhead KS, Swerdlow NR, Shafer KM, Diaz M, Braff DL. Modulation of the startle response and startle laterality in relatives of schizophrenic patients and in subjects with schizotypal personality disorder: evidence of inhibitory deficits. Am J Psychiatry. 2000;157(10):1660–1668. doi: 10.1176/appi.ajp.157.10.1660. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Szilagyi S, Efferen TR, Schwartz MP, Parwani A, Chakravorty S, Madonick SH, Kunzova A, Harmon JW, Angrist B, et al. Effect of treatment status on prepulse inhibition of acoustic startle in schizophrenia. Psychopharmacology (Berl) 2003;167(1):63–71. doi: 10.1007/s00213-002-1372-z. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156(2–3):117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Horacek J, Bubenikova-Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, Hoschl C. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs. 2006;20(5):389–409. doi: 10.2165/00023210-200620050-00004. [DOI] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- Kumari V, Soni W, Sharma T. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999;156(7):1046–1051. doi: 10.1176/ajp.156.7.1046. [DOI] [PubMed] [Google Scholar]
- Leumann L, Feldon J, Vollenweider FX, Ludewig K. Effects of typical and atypical antipsychotics on prepulse inhibition and latent inhibition in chronic schizophrenia. Biol Psychiatry. 2002;52(7):729–739. doi: 10.1016/s0006-3223(02)01344-6. [DOI] [PubMed] [Google Scholar]
- Mackeprang T, Kristiansen KT, Glenthoj BY. Effects of antipsychotics on prepulse inhibition of the startle response in drug-naive schizophrenic patients. Biol Psychiatry. 2002;52(9):863–873. doi: 10.1016/s0006-3223(02)01409-9. [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, et al. Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain. 2002;125(Pt 7):1594–1606. doi: 10.1093/brain/awf150. [DOI] [PubMed] [Google Scholar]
- Meincke U, Morth D, Voss T, Thelen B, Geyer MA, Gouzoulis-Mayfrank E. Prepulse inhibition of the acoustically evoked startle reflex in patients with an acute schizophrenic psychosis—a longitudinal study. Eur Arch Psychiatry Clin Neurosci. 2004;254(6):415–421. doi: 10.1007/s00406-004-0523-0. [DOI] [PubMed] [Google Scholar]
- Moller HJ. Management of the negative symptoms of schizophrenia: new treatment options. CNS Drugs. 2003;17(11):793–823. doi: 10.2165/00023210-200317110-00003. [DOI] [PubMed] [Google Scholar]
- Oranje B, Van Oel CJ, Gispen-De Wied CC, Verbaten MN, Kahn RS. Effects of typical and atypical antipsychotics on the prepulse inhibition of the startle reflex in patients with schizophrenia. J Clin Psychopharmacol. 2002;22(4):359–365. doi: 10.1097/00004714-200208000-00005. [DOI] [PubMed] [Google Scholar]
- Perry W, Braff DL. Information-processing deficits and thought disorder in schizophrenia. Am J Psychiatry. 1994;151(3):363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- Perry W, Geyer MA, Braff DL. Sensorimotor gating and thought disturbance measured in close temporal proximity in schizophrenic patients. Arch Gen Psychiatry. 1999;56(3):277–281. doi: 10.1001/archpsyc.56.3.277. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Feifel D, Braff DL. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry. 2001;50(6):418–424. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]
- Perry W, Feifel D, Minassian A, Bhattacharjie I, Braff DL. Information processing deficits in acutely psychotic schizophrenia patients medicated and unmedicated at the time of admission. Am J Psychiatry. 2002;159(8):1375–1381. doi: 10.1176/appi.ajp.159.8.1375. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2005.09.025. (E-pub ahead of print) [DOI] [PubMed] [Google Scholar]
- Quednow BB, Wagner M, Westheide J, Beckmann K, Bliesener N, Maier W, Kuhn KU. Sensorimotor gating and habituation of the startle response in schizophrenic patients randomly treated with amisulpride or olanzapine. Biol Psychiatry. 2006;59(6):536–545. doi: 10.1016/j.biopsych.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Schwarzkopf SB, McCoy L, Smith DA, Boutros NN. Test–retest reliability of prepulse inhibition of the acoustic startle response. Biol Psychiatry. 1993;34(12):896–900. doi: 10.1016/0006-3223(93)90059-m. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Koob GF. Dopamine, schizophrenia, mania, and depression: toward a unified hypothesis of cortico-stratio-pallido-thalamic function. Behav Brain Sci. 1987;10:197–245. [Google Scholar]
- Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biol Psychiatry. 1993;33(4):298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1995;58(2):192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol. 2000;11(3–4):185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Karban B, Ploum Y, Sharp R, Geyer MA, Eastvold A. Tactile prepuff inhibition of startle in children with Tourette’s syndrome: in search of an “fMRI-friendly” startle paradigm. Biol Psychiatry. 2001;50(8):578–585. doi: 10.1016/s0006-3223(01)01164-7. [DOI] [PubMed] [Google Scholar]
- Weike AI, Bauer U, Hamm AO. Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biol Psychiatry. 2000;47(1):61–70. doi: 10.1016/s0006-3223(99)00229-2. [DOI] [PubMed] [Google Scholar]