

Abstract
Laulimalide is a potent microtubule stabilizing agent and a promising anticancer therapeutic lead. The identification of stable, efficacious and accessible analogues is critical to clinically exploiting this novel lead. To determine which structural features of laulimalide are required for beneficial function and thus for accessing superior clinical candidates, a series of side chain analogues were prepared through a last step cross metathesis diversification strategy and their biological activities were evaluated. Five analogues, differing in potency from 233 nM to 7.9 μM, effectively inhibit cancer cell proliferation. Like laulimalide, they retain activity against multidrug resistant cells, stabilize microtubules and cause the formation of aberrant mitotic spindles, mitotic accumulation, Bcl-2 phosphorylation and initiation of apoptosis. Structural modifications in the C23–C27 dihydropyran side chain can be made without changing the overall mechanism of action, but it is clear that this subunit has more than a bystander role.
Keywords: Laulimalide, laulimalide analogues, metathesis, antimitotics, microtubule stabilizers, synthetic chemistry
Introduction
Tubulin-binding agents are among the most important drugs used for the treatment of a wide variety of cancers.1 The microtubule-stabilizing taxanes paclitaxel and docetaxel are used to treat breast, ovarian, prostate and non-small cell lung cancer. The important clinical activities of the taxanes coupled with the desire to improve upon these agents has stimulated the search for new microtubule-stabilizing compounds that would offer therapeutic advantages including improved ADME-toxicology, selectivity, and activity against resistant disease. Several chemically diverse classes of microtubule-stabilizing compounds have been identified, including the laulimalides that have novel if not unique tubulin targeting features.2
Laulimalide (also known as fijianolide) is a structurally complex macrolide originally isolated from the sponge Cacospongia mycofijiensis.3,4 Laulimalide potently inhibits cancer cell proliferation and it has advantages over the taxanes in that it can circumvent multidrug resistance arising from enhanced expression of P-glycoprotein (Pgp) and mutations in the paclitaxel binding site. The development of multidrug resistance is the major cause of clinical failure of many drugs. Overcoming multidrug resistance imparted by Pgp expression represents a significant clinical challenge, because Pgp expression in several tumor types is linked with poor prognosis. Laulimalide is highly effective against Pgp-expressing multidrug resistant cell lines.2,5 Laulimalide and another sponge-derived macrolide, peloruside A, do not bind to the taxane binding site and they appear to share the same tubulin binding site,5,6 allowing them to circumvent drug resistance mediated by tubulin mutations within the paclitaxel-binding site.5,7
Apart from distinct advantages against resistant cancer, the cellular effects of laulimalide are mechanistically almost identical to the effects of other microtubule stabilizers. Laulimalide increases the density of interphase microtubules, and it causes the formation of thick, short microtubule bundles in the cytoplasm of interphase cells. Abnormal circular mitotic spindles are formed in cells treated with laulimalide.2 Laulimalide-induced aberrant mitotic spindles cause mitotic arrest and subsequent initiation of apoptosis. In tubulin polymerization assays laulimalide promotes the assembly of purified tubulin in a manner nearly identical to paclitaxel, showing a direct interaction with tubulin, albeit at nonoverlapping binding sites.2,5 Significantly and consistent with these data, laulimalide and paclitaxel synergistically promote purified tubulin assembly.6,8 In cells, laulimalide and paclitaxel also act synergistically to inhibit cell proliferation and cause mitotic arrest.9 However, laulimalide is also synergistic with the microtubule depolymerizing agent 2-methoxyestradiol, suggesting that the specific antimitotic actions of these compounds and not overall changes in tubulin polymerization are responsible for the synergistic antiproliferative and antimitotic actions. Interestingly, laulimalide and docetaxel also provide synergistic activity against VEGF-induced endothelial cell migration and tubule formation, effects that predict antiangiogenic actions.10 The synergistic actions appear to be due to the ability of these two microtubule stabilizers to have slightly different effects on integrin signaling pathways.10 Despite the exceptional in vitro activity of laulimalide, a recent report has highlighted the need for synthetically accessible analogues that improve on its stability as well as its in vivo efficacy and toxicity profile.11
We have previously reported the synthesis of laulimalide analogues, with the ultimate goal of identifying simpler and therefore more synthetically accessible as well as more stable leads that could offer superior clinical performance.12,13 The function-oriented synthesis approach has proven exceptionally effective in the design and synthesis of analogues superior to otherwise difficult to access and therapeutically unoptimized natural leads.14 Our laulimalide analogues were initially designed to address the acid-promoted decomposition of laulimalide to isolaulimalide involving nucleophilic attack of the C20 hydroxyl on the C16–C17 epoxide,12 and to provide structurally simplified analogues (e.g., 11-desmethyl derivatives) that could be accessed through a more step-economical synthetic route.15 Complementing these studies directed at stability, we also directed attention at the functional role of other subunits in the biological activities of laulimalide. Toward this end, a route to analogues that differ from the natural product in only the structure of the C22 “side chain” substituent was recently disclosed.16 One focus of this effort has been to explore whether the C23 dihydropyran unit present in the natural product could be replaced with a more readily accessible group that would retain or improve upon the activity of the lead compound. Our initial study established that one side chain analogue of laulimalide, the C22-cyclohexane analogue LA14, could be accessed in several steps through a diversification strategy utilizing a cross metathesis reaction, between a C20-vinyl macrolactone and an olefin, as the key diversification step.16 An important finding resulting from that initial study was that the side chain has a dramatic influence on potency. Whereas laulimalide has an IC50 of 5.7 nM against the MDA-MB-435 cell line, the corresponding cyclohexyl analogue LA14 is roughly 200-fold less potent (IC50 = 1170 nM). An important and more recent report describes several new natural products of the laulimalide (fijianolide) class whose activities are also significantly influenced by side chain variations.17 Given the emerging importance of this subunit in activity, we devised a new and more step economical diversification strategy based on a cross metathesis reaction that allows for the single step diversification of the core structure to directly produce new analogues differing systematically only in the side chain structure. This last step diversification allowed us to interrogate the biological consequences of specific structural modifications incorporating changes only to the C22 side chain. We report herein the first laulimalide analogues derived from this new strategy and elucidation of their antiproliferative potencies and their ability to retain microtubule stabilizing activities. Detailed mechanistic consequences of side chain modifications have not been previously identified.
Experimental Section
Chemistry
Commercially available reagents were used as supplied from the manufacturer unless otherwise noted. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian INOVA 600 (1H at 600 MHz, 13C at 150 MHz) or Varian INOVA 500 (1H at 500 MHz, 13C at 125 MHz) NMR spectrometer. Chemical shifts are reported in parts per million relative to tetramethylsilane using the solvent resonance as an internal standard. Infrared spectra were recorded on a Perkin-Elmer 1600 series FTIR and are reported in wavenumbers per centimeter. Optical rotations were measured using a JASCO DIP-360 digital polarimeter and are reported as specific rotation (in degrees) followed by the concentration of the analogue (in g/100 mL) and the solvent used for the measurement. High-resolution mass spectra (HRMS) were recorded at the Vincent Coates Foundation mass spectrometry laboratory at Stanford University. Reported mass values are within error limits of 5 ppm. Analogues LA13 and LA14 were synthesized using the methods described in our previous publication, and LA15 was isolated in 19% yield (1.5:1 mixture of epoxide diastereomers) as a byproduct in the final step of the synthesis of LA14.16
Representative Procedure for Cross Metathesis
A solution of cyclohexane side chain analogue LA14 (2.0 mg, 4 μmol, 1 equiv) in dichloromethane (1 mL) was degassed using the freeze–pump–thaw method. To this solution was added 3-methylstyrene (27 μL, 0.2 mmol, 50 equiv) followed by Grubbs second generation metathesis catalyst A (0.7 mg, 0.8 μmol, 0.2 equiv). The resulting solution was allowed to stir at room temperature for 18 h. The solvent was evaporated and the residue was purified by flash chromatography (silica gel, EtOAc:pentane 1:2) to provide side chain analogue LA16 (1.0 mg, 50%) as a clear oil.
LA16
Rf: 0.48 (EtOAc:pentane 1:1). [α]D: −83.9° (c = 0.2, CDCl3). 1H NMR: (600 MHz, CDCl3) δ 7.23−7.24 (m, 3H), 7.10−7.06 (m, 1H), 6.64 (d, J = 16.2 Hz, 1H), 6.46−6.40 (m, 1H), 6.15 (dd, J = 16.2, 6 Hz, 1H), 5.94−5.90 (m, 1H), 5.86−5.82 (m, 1H), 5.71−5.66 (m, 1H), 5.26−5.22 (m, 1H), 4.86 (s, 1H), 4.85 (s, 1H), 4.38−4.34 (m, 1H), 4.32−4.27 (m, 1H), 4.11−4.06 (m, 1H), 3.79−3.69 (m, 2G), 3.11−3.07 (m, 1H), 2.92−2.90 (m, 1H), 3.79−3.69 (m, 2H), 3.11−3.07 (m, 1H), 2.29−2.10 (m, 3H), 2.07−1.97 (m, 3H), 1.96−1.88 (m, 2H), 1.82−1.75 (m, 1H), 1.49−1.41 (m, 1H), 1.37−1.26 (m, 2H), 0.83 (d, J = 6.6 Hz, 3H) ppm. 13C NMR: (125 MHz, CDCl3) δ 166.1, 150.5, 144.8, 138.2, 136.0, 132.9, 128.8, 128.5, 127.3, 126.9, 125.2, 123.8, 120.4, 112.5, 74.3, 73.1, 72.5, 67.9, 66.4, 60.6, 52.0, 45.5, 43.4, 37.0, 33.7, 31.7, 29.7, 29.6, 21.4, 20.7 ppm. IR (thin film) ν = 3429, 2923, 1718, 1643, 1422, 1261, 1213, 1167, 1083, 891, 809 cm−1. HRMS: m/z = 531.2736 (M + Na)+. Calculated for C31H40O6Na: 531.2723.
LA17
Cross metathesis between LA14 (2 mg, 4 μmol, 1 equiv) and 2-vinyl-1,3-dioxolane (8 μL, 80 μmol, 20 equiv) using catalyst A (1 mg, 1.2 μmol, 0.3 equiv) provided analogue LA17 (0.8 mg, 41%) as a colorless oil. Rf: 0.10 (EtOAc:pentane 1:1). [α]D: −102.0° (c = 0.2, CDCl3). 1H NMR: (600 MHz, CDCl3) δ 6.48−6.42 (m, 1H), 5.94 (dd, J = 15, 5.4 Hz, 1H), 5.92−5.88 (m, 1H), 5.86−5.79 (m, 2H), 5.72−5.68 (m, 1H), 5.29 (d, J = 5.7 Hz, 1H), 5.20−5.16 (m, 1H), 4.86 (s, 1H), 4.85 (s, 1H), 4.33−4.28 (m, 1H), 4.28−4.25 (m, 1H), 4.09−4.05 (m, 1H), 4.01−3.94 (m, 2H), 3.93−3.86 (m, 2H), 3.78−3.67 (m, 2H), 3.08−3.04 (m, 1H), 2.90−2.88 (m, 1H), 2.39−2.33 (m, 2H), 2.26−2.20 (m, 1H), 2.14−2.09 (m, 1H), 2.06−1.98 (m, 2H), 1.97 (d, J = 6.6 Hz, 1H), 1.95−1.92 (m, 1H), 1.91−1.89 (m, 1H), 1.81−1.75 (m, 1H), 1.74−1.67 (m, 1H), 1.54−1.48 (m, 1H), 1.48−1.42 (m, 1H), 1.35−1.30 (m, 1H), 0.83 (d, J = 6.6 Hz, 2H) ppm. 13C NMR: (125 MHz, CDCl3) δ 165.9, 150.6, 144.8, 133.5, 129.5, 128.5, 125.2, 120.3, 112.6, 102.7, 73.1, 72.8, 72.0, 67.9, 66.5, 65.0, 64.9, 60.6, 52.0, 45.5, 43.4, 37.1, 33.8, 33.2, 31.6, 29.5, 20.7 ppm. IR (thin film) ν = 3435, 2918, 1719, 1642, 1420, 1263, 1213, 1165, 1060, 972, 892, 811, 774, 705 cm−1. HRMS: m/z = 513.2462 (M + Na)+. Calculated for C27H38O8Na: 513.2464.
LA18
Cross metathesis between LA14 (2 mg, 4 μmol, 1 equiv) and 4-vinyl-cyclohexene (racemic, 26 μL, 200 μmol, 50 equiv) using catalyst A (1 mg, 1.2 μmol, 0.3 equiv) provided analogue LA17 (1.1 mg, 55%, 1:1 mixture of diastereomers) as a colorless oil. Rf: 0.58 (EtOAc:pentane 1:1). [α]D: −53.7° (c = 0.15, CDCl3). 1H NMR: (600 MHz, CDCl3) δ 6.47−6.41 (m, 1H), 5.93−5.89 (m, 1H), 5.86−5.82 (m, 1H), 5.77 (dd, J = 15.6, 6.6 Hz, 1H), 5.72−5.68 (m, 1H), 5.68−5.63 (m, 2H), 5.45 (dd, J = 15.6, 6.6 Hz, 1H), 5.13 (ddd, J = 11.4, 5.4, 1.8 Hz, 1H), 4.86 (s, 1H), 4.85 (s, 1H), 4.33−4.28 (m, 1H), 4.15−4.11 (m, 1H), 4.09−4.04 (m, 1H), 3.79−3.68 (m, 2H), 3.09−3.05 (m, 1H), 2.91−2.89 (m, 1H), 2.40−2.32 (m, 2H), 2.32−2.25 (m, 1H), 2.25−2.19 (m, 1H), 2.15−1.97 (m, 6H), 1.95−1.87 (m, 2H), 1.86−1.67 (m, 5H), 1.52−1.42 (m, 2H), 1.42−1.30 (m, 2H), 0.83 (d, J = 6.6 Hz, 3H) ppm. 13C NMR: (125 MHz, CDCl3) δ 166.1, 150.3, 144.9, 128.5, 126.9, 126.2, 125.7, 125.2, 120.5, 116.6, 112.6, 76.7, 74.4, 72.5, 67.9, 66.6, 60.7, 52.1, 45.5, 43.3, 37.0, 36.0, 33.7, 33.6, 31.6, 29.5, 28.2, 24.6, 20.7 ppm. IR (thin film) ν = 3422, 3024, 2917, 1718, 1438, 1262, 1213, 1166, 1062, 874, 810 cm−1. HRMS: m/z = 521.2881 (M + Na)+. Calculated for C30H42O6Na: 521.2879.
LA13
Rf: 0.23 (EtOAc:pentane 2:1). [α]D: −41.7° (c = 0.1, CDCl3). 1H NMR: (600 MHz, CDCl3) δ 6.48−6.42 (m, 1H), 5.93−5.81 (m, 3H), 5.75−5.68 (m, 2H), 5.15 (ddd, J = 10.8, 4.8, 1.2 Hz, 1H), 4.86 (s, 1H), 4.85 (s, 1H), 4.33−4.28)m, 1H), 4.23−4.19 (m, 1H), 4.09−4.04 (m, 1H), 3.93 (ap. d., J = 6 Hz, 2H), 3.78−3.67 (m, 2H), 3.33 (s, 3H), 3.08−3.04 (m, 1H), 2.89 (t, J = 3 Hz, 1H), 2.40−2.34 (m, 2H), 2.26−2.20 (m, 1H), 2.15−2.10 (m, 1H), 2.06−7.97 (m, 2H), 1.96−1.88 (m, 2H), 1.81−1.75 (m, 1H), 1.74−1.68 (m, 1H), 1.54−1.47 (m, 1H), 1.45 (dd, J = 14.4, 7.2 Hz, 1H), 1.36−1.27 (m, 2H), 0.83 (d, J = 6.6 Hz, 3H) ppm. 13C NMR: (150 MHz, CDCl3) δ 166.0, 150.2, 144.5, 130.2, 130.1, 128.3, 125.0, 120.2, 112.4, 73.2, 72.9, 72.1, 71.8, 67.6, 66.4, 60.4, 57.9, 52.0, 45.3, 43.2, 36.9, 33.5, 33.2, 31.4, 29.5, 20.6 ppm. IR (thin film) ν = 3368, 2918, 2849, 1719, 1643, 1421, 1260, 1166, 1083, 890, 806 cm−1. HRMS: m/z = 485.2508 (M + Na)+. Calculated for C26H38O7Na: 485.2515.
LA15
Rf: 0.30 (EtOAc:pentane 1:1). [α]D: −106..9° (c = 0.25, CDCl3). 1H NMR: (600 MHz, CDCl3, major diastereomer) δ 6.50−6.44 (m, 1H), 5.97−5.90 (m, 1H), 5.87−5.82 (m, 1H), 5.73−5.67 (m, 1H), 5.36−5.32 (m, 1H), 4.87 (s, 1H), 4.85 (s, 1H), 4.34−4.28 (m, 1H), 4.11−4.05 (m, 1H), 3.92−3.88 (m, 1H), 3.49−3.70 (m, 2H), 3.10−3.06 (m, 1H), 2.93−2.87 (m, 2H), 2.85−2.82 (m, 1H), 2.07−1.96 (m, 3H), 1.96−1.87 (m, 2H), 1.85−1.58 (m, 8H), 1.49−1.42 (m, 1H), 1.37−1.30 (m, 1H), 1.25−1.13 (m, 3H), 1.11−1.00 (m, 2H), 0.95−0.86 (m, 1H), 0.83 ppm (d, J = 6.6 Hz, 3H). 1H NMR: (600 MHz, CDCl3, minor diastereomer, characteristic signals) δ 5.32−5.28 (m, 1H), 3.68−3.63 (m, 1H), 2.71−2.69 (m, 1H) ppm.13C NMR: (125 MHz, CDCl3, major diastereomer) δ 165.8, 150.8, 144.8, 128.4, 125.2, 120.4, 112.7, 72.9, 71.2, 70.6, 69.8, 67.9, 66.5, 60.6, 59.3, 56.5, 52.0, 45.5, 43.4, 39.2, 37.0, 33.7, 33.3, 31.6, 29.6, 28.8, 26.0, 25.7, 25.5, 20.8 ppm. 13C NMR: (125 MHz, CDCl3, minor diastereomer, characteristic signals) δ 150.7, 120.2, 112.5, 66.4, 60..6, 59.2, 56.8, 51.9, 39.3, 33.8, 29.7, 29.2, 26.1, 25.6 ppm. IR (thin film) ν = 3436, 2924, 2853, 1717, 1643, 1450, 1421, 1213, 1167, 1083, 892, 811, 732 cm−1. HRMS: m/z = 539.2993 (M + Na)+. Calculated for C30H44O6Na: 539.2985.
Conversion of LA13 to LA14
A solution of methoxy side chain analogue LA13 (1.3 mg, 2.8 μmol, 1 equiv) in dichloromethane (0.8 mL) was degassed using the freeze–pump–thaw method. To this solution was added vinylcy-clohexane (19 μL, 0.14 mmol, 50 equiv) followed by Grubbs's second generation metathesis catalyst A (0.2 mg, 0.28 μmol, 0.1 equiv). The resulting solution was stirred at room temperature for 20 h. The solvent was evaporated and the residue was purified by flash chromatography (silica gel, EtOAc:pentane 1:1) to provide side chain analogue LA14 (0.7 mg, 50%) as a clear oil. Physical and spectral data of the product were identical to known values.16
Cell Culture
A-10 smooth muscle cells and the cancer cell lines MDA-MB-435, NCI/ADR, 1A9 and PTX10 were obtained and cultured as previously reported.18
Inhibition of Proliferation
The sulforhodamine B (SRB) assay was used to measure the antiproliferative effects of the laulimalide analogues.19 Cells were plated into 96-well plates at predetermined densities, and 24 h later the test compounds were added in triplicate wells. Cells were treated for a total of 48 h, fixed, and stained with SRB, and the absorbance was read at 560 nM. IC50 values for inhibition of proliferation were calculated for each of 3 independent experiments (which each utilized triplicate data points), as previously described.18 The data presented represent the means of these 3 independent experiments.
Indirect Immunofluorescence
Cellular microtubule structures were visualized in A-10 cells using indirect immunofluorescence techniques with a β-tubulin antibody as previously described.2,18 Cells were treated for 24 h, and then they were fixed with cold methanol and incubated with appropriate antibodies.
Flow Cytometry
MDA-MB-435 cells were treated with a range of concentrations of the each of the laulimalides or vehicle (EtOH) for 24 h, and then the cells were stained with Krishan's Reagent and analyzed using Becton Dickinson FACScan flow cytometer as previously described. The propidium iodide intensity was plotted versus the number of events.18
Immunoblotting
MDA-MB-435 cells were treated for 24 h with the approximate IC85 concentration of each laulimalide. The cells were harvested in cell lysis buffer (Biosource Camarillo, CA) containing a cocktail of protease inhibitors. The protein concentrations of the lysates were determined, lysates containing equal amounts of protein were separated by PAGE and transferred to Immobilon P membrane, and the expression levels of cleaved p85 PARP and Bcl-2 were evaluated by Western blotting techniques.2,18 The p85 cleaved PARP antibody was purchased from Promega (Madison, WI), and the Bcl-2 antibody was obtained from BD Biosciences (San Diego, CA).
Results
Last Step Access to New Side Chain Analogues Using Cross Metathesis
Our previous work showed that side chain modified laulimalide analogues such as LA14 (Figure 1) could be synthesized using the cross metathesis reaction to install the new side chain fragment.16 However, the process, beginning from a macrolactone intermediate bearing a C20 vinyl group, required four new synthetic steps to produce each new analogue. This served the anticipated need at its inception, but with the realization that the side chain has an unusual effect on activity, a more step economical route was sought. Ideally, the point of diversification in accessing new analogues would be the final step of the synthesis. In order for this new strategy to be successful, the cross metathesis reaction would therefore need to occur without disruption of functionality previously determined to be labile, namely the C16–C17 epoxide and the C2–C3 Z-enoate. Further complicating matters, in the course of our previous work initial investigations determined that the C20 terminal vinyl group did not survive to the end of the synthesis. In fact, this monosubstituted alkene was competitively hydrogenated under the Lindlar conditions required to install the C2–C3 olefin, thus eliminating its intended role as diversification handle. However, in the synthesis of C22-cyclohexane analogue LA14, the more sterically encumbered 1,2-disubstituted C21–C22 olefin was found to be less prone to hydrogenation. Thus, as a solution to this problem, it was envisioned that the C22 cyclohexane could serve as a “protecting group” for the C21–C22 olefin during the required Lindlar hydrogenation of the C2–C3 alkyne and then be pressed into service in a last step metathesis process, provided that it did not “protect” the C21–C22 olefin against the desired cross metathesis reaction.
Figure 1.

Structure of laulimalide, isolaulimalide and side chain modified laulimalide analogues.
In an effort to test the above plan to establish a single step diversification route, the cross metathesis reaction was attempted directly on C22-cyclohexane analogue LA14 (Scheme 1) using the Grubbs second-generation ruthenium alkylidene (A) as the catalyst (0.2 or 0.3 equiv) and a variety of commercially available alkenes as the metathesis partner (20 or 50 equiv). Remarkably, despite the structural complexity of LA14, particularly the acid-sensitive C16–C17 epoxide and the labile Z-C2–C3 bond, and the proximity of the latter to the reaction site, selective cross metathesis reactions were observed with a variety of alkene substrates, leading to the formation of new analogues in only one synthetic step from our diversification node. The generality of this reaction and the structural assignment of the new analogues are supported by the conversion of C23-methoxy analogue LA13 (synthesized via our first-generation strategy) in one step to the parent C22 cyclohexane analogue LA14 (Scheme 1).
Scheme 1.

One Step Diversification of Laulimalide Side Chain Analoguesa
Antiproliferative Effects of the Side Chain Modified Laulimalides
The antiproliferative activities of the laulimalide side chain analogues (Figure 1) were evaluated in the MDA-MB-435 cell line and the dose response curves and IC50 values are shown in Figure 2 and Table 1, respectively. A wide range of potencies were observed among the six analogues tested. Significantly, all the new compounds, with the exception of the dioxolane analogue LA17, retained the ability to inhibit cancer cell proliferation at concentrations between 233 nM and 7.9 μM. Consistent with earlier observations and recent studies on new natural laulimalides, the potency of an individual analogue is highly dependent on the structure of its side chain.16,17,20 For example, the seemingly modest change of the pyran in laulimalide to a methyl ether in analogue LA13 results in a surprisingly significant drop in potency of over 3 orders of magnitude, indicating the importance of the carbon content of the side chain and a role for the alkene pi-system in activity. Aliphatic or aromatic side chains such as the aryl ring in LA16 or the cyclohexane in LA14 provide analogues with micromolar potency, suggesting that a six-membered ring alone or a non-alkene pi-system is not a suitable replacement for the side chain. LA15, isolated as a minor side product from the final step in the synthesis of LA14 (as a 1.5:1 mixture of epoxide diastereomers), demonstrates that the C21–C22 olefin can be epoxidized with little to no effect on potency. Most significantly, the greatly improved potency of the C23-cyclohexene analogue LA18 (233 nM for a 1:1 mixture of diastereomers) when compared to the C23-cyclohexane analogue LA14 indicates a critical contribution of the olefin of the laulimalide side chain to the natural product's exceptional potency. This is an important new finding.
Figure 2.
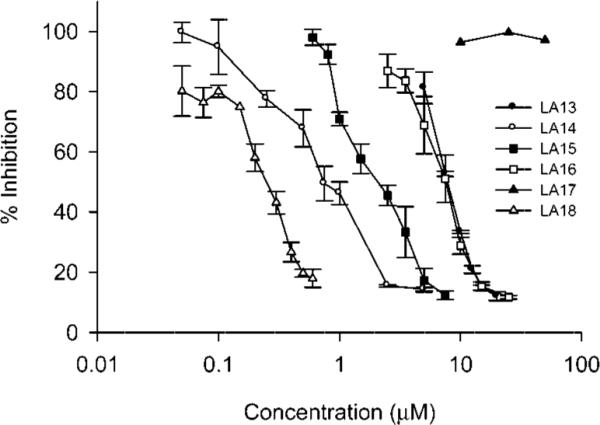
Effects of the laulimalide analogues on drug sensitive and multidrug resistant cancer cells. The antiproliferative effects of the laulimalide analogues were measured in MDA-MB-435 cells using the SRB assay in 3 independent experiments that each utilized triplicate data points (n = 3, ± SE).
Table 1.
Antiproliferative Effects of Laulimalide Analogues (n = 3)
| IC50 ± SD (μM) |
||||
|---|---|---|---|---|
| laulimalide analogue | MDA-MB-435 | NCI/ADRa | 1A9 | PTX10b |
| LA13 | 7.9 ± 0.320 | 22.9 ± 0.090(2.9) | NDc | ND |
| LA14 | 1.2 ± 0.091 | 1.2 ± 0.099(1.0) | 1.0 ± 0.021 | 1.8 ± 0.027(1.8) |
| LA15 | 1.35 ± 0.05 | 2.38 ± 0.09 (1.8) | 1.42 ± 0.32 | 3.49 ± 0.16 (2.5) |
| LA16 | 7.30 ± 1.27 | 12.4 ± 1.4 (1.7) | 7.91 ± 0.64 | 16.4 ± 4.4 (2.1) |
| LA17 | >50 | ND | ND | ND |
| LA18 | 0.233 ± 0.02 | 0.433 ± 0.04 (1.9) | 0.424 ± 0.02 | 1.11 ± 0.10 (2.6) |
The relative resistance values shown in parentheses were calculated by dividing the IC50 values of the resistant line NCI/ADR by the sensitive cell line MDA-MB-435.
The relative resistance values shown in parentheses were calculated by dividing the IC50 values of resistant cell line PTX10 by the IC50 obtained in the parental 1A9.
Not determined.
Significantly, all new analogues that are effective against the drug-sensitive MDA-MB-435 cell line also retain the ability to circumvent Pgp-mediated multidrug resistance exhibited by NCI/ADR cells (Table 1). While the potency of paclitaxel drops by more than 3 orders of magnitude in the resistant cells, this cell line demonstrates very little resistance to the laulimalide side chain analogues and, in the case of LA14, no resistance at all, suggesting that modifying the side chain region does not alter the ability of the laulimalides to overcome Pgp-mediated drug efflux. This is of paramount importance in selecting clinical lead compounds because the expression of Pgp is linked with clinical resistance to the taxanes and is a cause for the failure of many drugs and for the onset of cross-resistance to other drugs.21–23 Point mutations in β-tubulin within the paclitaxel binding site provide another mechanism of taxane resistance in cancer cell lines. The PTX10 cell line was generated by serial exposure of the parental 1A9 cell line to paclitaxel in the presence of verapamil.24 This cell line is 24- to 30-fold more resistant to paclitaxel because of a mutation in the paclitaxel binding region of β1 tubulin.24 The sensitivity of the taxane-resistant cell line PTX10 to the laulimalide analogues was evaluated. The data (Table 1) show that the PTX10 cell line was quite sensitive to the effects of the laulimalide analogues with calculated relative resistance values of 1.8 to 2.6. These results are consistent with relative resistance values that were obtained with other laulimalide analogues and for other drugs including 2-methoxyestradiol, which has a relative resistance of 2.5. These low relative resistance values were not unexpected because laulimalide binds to tubulin at a site that does not overlap the taxane binding site.
Effects of the Laulimalide Analogues on Interphase Microtubule, Mitotic Spindles and Cell Cycle Distribution
Microtubule stabilizers cause a striking increase in the density of cellular microtubules, and these effects are readily observed in the A-10 cell line because smooth muscle cells arrest in interphase in response to microtubule-disrupting agents.25 A normal array of interphase microtubules radiating from the microtubule organizing center was observed in vehicle-treated A-10 cells (Figure 3A). All of the side chain modified analogues with antiproliferative actions caused a concentration-dependent increase in the density of interphase microtubules. Interphase microtubules filled the cytoplasm in analogue-treated cells (Figure 3B–F), and the microtubules appear to radiate normally from the microtubule organizing center. Laulimalide caused the formation of similar interphase microtubule arrays at low concentrations (2 μM), and with very high concentrations (20 μM) it caused overt microtubule bundling and the formation of short thick microtubule tufts that nucleated independent of the microtubule organizing center.2 While these tufted microtubule structures were not observed with the concentrations of side chain modified analogues tested, the effects observed are consistent with potency differences between the analogues and laulimalide. The breakdown of nuclei into micronuclei was seen in A-10 cells treated with any of the antiproliferative analogues, and this effect is reminiscent of the effects of the natural product on interphase nuclei (data not shown).
Figure 3.

Effects of the laulimalide analogues on interphase microtubules. A-10 cells were treated for 24 h with vehicle (A), or a range of concentrations of the laulimalide analogues including 20 μM LA13 (B), 10 μM LA14 (C), 15 μM LA15 (D), 20 μM LA16 (E) or 1 μM LA18 (F). The cells were fixed and microtubules and DNA were visualized using indirect immunofluorescence techniques.
The effects of the laulimalide analogues on mitotic spindles were evaluated. A large body of evidence shows that it is the ability of microtubule stabilizers to disrupt mitosis that leads to antiproliferative and cytotoxic effects.26 Normal mitotic spindles are bipolar and appear to radiate from the centrosomes although some spindles nucleate from the kinetochores. A normal bipolar mitotic spindle functions to align the chromosomes in the metaphase plate to ensure even distribution of chromosomes into daughter cells. A normal bipolar mitotic spindle is shown in Figure 4A. One hallmark of the laulimalides is their propensity to cause circular mitotic spindle structures in multiple cell types.2,13 These circular spindles contain a ring of microtubules that radiate to the cell periphery where they appear to orient the DNA in a circular pattern. The orientation of the DNA in the cell periphery suggests that the circular mitotic spindles are functional, albeit in a highly abnormal manner. Laulimalide-induced circular mitotic spindles differ from monopolar spindles that are sometimes seen in cells treated with other microtubule disrupting agents, including paclitaxel, and mitotic kinesin inhibitors such as monastral. Laulimalide-induced circular spindles have a center devoid of spindles, and they contain two or more centrosomes within the central hub of spindles. The antiproliferative laulimalide analogues all caused the formation of circular mitotic spindles (Figure 4B–F). No differences were noted among these analogues in their ability to cause these highly aberrant mitotic spindles, and circular spindles were observed, suggesting that the analogues and laulimalide share the specific signaling effects that lead to circular mitotic spindles.
Figure 4.

Effects of the laulimalide analogues on mitotic spindles. A-10 cells were treated for 24 h with the laulimalide analogues. Representative pictures of controls (A), 20 μM LA13 (B), 10 μM LA14 (C), 10 μM LA15 (D), 20 μM LA16 (E) and 1 μM LA18 (F) are shown.
Microtubule stabilizers cause the formation of nonfunctional mitotic spindles, interrupting normal mitotic progression. The effects of the side chain modified analogues on cell cycle distribution were investigated to determine whether aberrant mitotic spindles would prevent cell cycle progression. A normal cell cycle distribution was seen in vehicle-treated cells in which the majority of cells were in the G1 phase of the cell cycle and some cells were in S and G2/M phases (Figure 5). Treatment of the MDA-MB-435 cells with the approximate IC85 concentration of the laulimalide analogues caused an accumulation of cells in G2/M within 24 h. LA13 caused significant mitotic accumulation, but a small population of cells remaining in G1 was observed with a 15 μM concentration. A 5 μM concentration of LA14 caused total G2/M accumulation with no evidence of any cells in G1. Complete G2/M accumulation was also noted in MDA-MB-435 cells treated with 10 μM LA15, 20 μM LA16 or 750 nM LA18. In each of the laulimalide-analogue-treated cell populations a pre-G1 peak, indicative of apoptosis, was present after a 24 h exposure. A small population of cells with tetraploid DNA content was also detected in cells treated with the laulimalide analogues. These data, together with the abnormal mitotic spindles formed in the presence of the side chain modified laulimalide analogues, suggest that the new laulimalide analogues interrupt normal mitotic spindle formation leading to G2/M accumulation, which results ultimately in the initiation of apoptosis. Further cellular consequences of the laulimalide-treated cells were investigated.
Figure 5.

The laulimalide analogues cause mitotic accumulation. MDA-MB-435 cells were incubated with vehicle (A), 15 μM LA13 (B), 5 μM LA14 (C), 10 μM LA15 (D), 20 μM LA16 (E), or 0.75 μM LA18 (F) for 24 h and then stained with Krishan's reagent and cell cycle distribution determined by flow cytometry.
Initiation of Apoptosis and Bcl-2 Phosphorylation
The poly(ADP-ribose) polymerase PARP is specifically cleaved between Asp214 and Gly215 during the process of apoptosis by activated caspases 3 and 7. To measure apoptosis, we investigated the ability of the laulimalide analogues to initiate caspase activation. Whole cell lysates of MDA-MB-435 cells treated with the approximate IC85 concentration for 24 h were evaluated for the expression of caspase-cleaved PARP. The results (Figure 6) show caspase-dependent PARP cleavage in cells treated with the laulimalide analogues. These data, together with the flow cytometry data showing a sub-G1 population of cells, show that the C20 side chain modified laulimalide analogues initiate apoptosis, consistent with the results obtained with laulimalide and other laulimalide analogues.
Figure 6.

The laulimalide analogues initiate apoptosis and Bcl-2 phosphorylation. MDA-MB-435 cells were treated for 24 h with LA13 (15 μM), LA14 (5 μM), LA15 (10 μM), LA16 (20 μM), or LA18 (0.75 μM) and whole cell lysates made. Aliquots containing equal amounts of protein were separated by PAGE and transferred and probed with antibodies for cleaved PARP and Bcl-2. The controls corresponded to lysates from vehicle-treated cells that were treated and harvested at the same time as each laulimalide analogue designated.
Bcl-2 is an antiapoptotic protein that is phosphorylated in cells treated with either microtubule depolymerizers or diverse microtubule stabilizers. The phosphorylation of Bcl-2 is thought to inhibit its antiapoptotic actions. The ability of a microtubule disruptor to initiate Bcl-2 phosphorylation confirms common antimitotic signaling events. Unlike some cell lines, MDA-MB-435 cells do not exhibit Bcl-2 phosphorylation in a cell-cycle-dependent manner and throughout normal mitosis no Bcl-2 phosphorylation is detected.27 The ability of the laulimalide analogues to initiate Bcl-2 phosphorylation was evaluated using Western blotting techniques. Bcl-2 phosphorylation is detected by the appearance of slower migrating Bcl-2 bands. Treatment of cells with the IC85 concentration of the laulimalide analogues caused robust Bcl-2 phosphorylation within 24 h (Figure 6). Several bands were observed, consistent with the ability of the laulimalide analogues to induce Bcl-2 phosphorylation at several sites. These effects are consistent with the effects of laulimalide and other laulimalide analogues, suggesting that modification of the C20 side chain does not alter the antimitotic and proapoptotic signaling events initiated by the laulimalides.
Discussion
The taxanes are extraordinarily useful anticancer drugs, but the development of multidrug resistance remains a major problem in the clinic. New classes of microtubule stabilizers with properties superior to the taxanes might have a significant clinical impact in the treatment of tumors with intrinsic and acquired multidrug resistance. The epothilone ixabepilone (Ixempra) was the first nontaxane microtubule stabilizer approved by the FDA for cancer therapy. It was approved in October 2007 for the treatment of multidrug resistant metastatic or locally advanced breast cancer alone or in combination with capcitabine.28 Ixabepilone is a semisynthetic analogue of epothilone B which is produced by the myxobacterium Sorangium cellulosum. The laulimalides also have excellent potential for treating taxane resistant tumors, and they have advantages over the epothilones because the laulimalides bind to a different tubulin/microtubule binding site. It is interesting to note that the relative resistance values of the laulimalide analogues in the PTX10/1A9 cell lines (1.8 to 2.6) are the same range as that observed for peloruside A in these same cell lines (3.3).7 Some level of resistance is probably due to the deregulation of survivin in the PTX10 cell line.29 The ability of the laulimalides and peloruside A to bind to another tubulin/microtubule binding site provides new opportunities for effective synergistic drug combinations. The use of a laulimalide together with paclitaxel caused synergistic antiproliferative and antimitotic actions, and two laulimalide analogues were each more synergistic than laulimalide itself.9 Peloruside A in combination with taxane site binding drugs caused synergistic effects on tubulin polymerization,6 inhibition of cell proliferation and G2/M blockade;30 however, peloruside A and laulimalide were not synergistic when combined, which is expected from a combination of drugs binding to the same binding site.6 A laulimalide in combination with paclitaxel, docetaxel or ixabepilone in early cancer treatment could prevent the generation of multidrug resistance and provide for cures or longer disease free survival.
Structural complexity is a hallmark of microtubule stabilizers, and the supply of laulimalide for continued preclinical research and development presents challenges because the natural product is rare and semisynthetic production options are not on the horizon. Function-oriented synthesis is a key to addressing this problem because, once the functionality required for biological activity is established, it would allow for the design and synthesis of simplified agents that could be made in a practical fashion.14 This approach has proven to be exceptionally effective in studies on bryostatin and has led to simplified but more active analogues that can be prepared in a practical fashion (40 steps fewer than the natural lead).14 The laulimalide analogues described here were synthesized using commercially available olefins in the cross metathesis reaction. This represents an improvement in step economy relative to laulimalide itself, in which the chiral dihydropyran side chain fragment must be synthesized in no fewer than five steps.15 Our in-depth evaluation of the functional consequences of the C23–C27 dihydropyran side chain, together with the work of others in this area, shows that the side chain plays a central and surprising role in determining biologic potency and that this feature or its close structural mimic, such as the cyclohexene in LA18, would need to be retained in designed analogues. However (and significantly), for the first time a specific simplification of the side chain that retains nanomolar potency has been achieved. Our new olefin-containing side chain analogue (LA18) is on par with the leading structurally simplified and more stable analogues, LA1 and LA2,13 in terms of potency, suggesting that exploration of this area of the molecule might yet yield analogues superior to the natural product, in contrast to previous reports. Studies to determine the reason for the greatly improved potency of this analogue, and most importantly whether the olefin contributes to enhanced binding of the analogue through a specific interaction such as a CH-π bond interaction,31 are in progress. It is clearly a region of fundamental and significant importance. Overall, the loss of potency observed with the C23–C27 dihydropyran side chain modified analogues provides a strong indication that this side chain is very important for potency and binding to the as-yet uncharacterized laulimalide binding site on tubulin/microtubules. It is also possible that the effects observed could be attributable to the existence of multiple orientations in a single binding site or of multiple binding sites that exhibit different degrees of occupancy.
Our last step metathesis diversification strategy for the synthesis of laulimalide side chain analogues has identified the central and unanticipated critical role of the C23–C27 dihydropyran side chain in the biological potency and mechanism of action of this class of microtubule stabilizers. The constituents of the side chain are critical for potency, but interestingly and mechanistically, modifications in the side chain are tolerated. One side chain modified analogue, the most potent analogue of the series, by nearly an order of magnitude, contains the C25–C26 olefin found in the natural product. This structurally simplified laulimalide, which can be obtained in a one-step late stage diversification strategy, provides a new avenue for the design and synthesis of structurally simplified laulimalide analogues with nanomolar potency that retain the promising biological activities of the natural product. The identification of the favorable potency of this side chain simplified laulimalide analogue and its ability to retain the promising actions of the natural product provide new opportunities for structural simplification. The new synthetic approach and identification of a mechanism to simplify the side chain offer a new capacity to tune the molecule to create simplified analogues to achieve potent microtubule stabilizing activities together with optimal toxicity profiles. Given the promising clinical performance of microtubule disruptors that offer advantages relative to paclitaxel, the relatively underexplored laulimalide class holds much clinical potential. Integrated synthetic and biological efforts to identify simpler agents with comparable or superior biological activity are key to advancement toward clinical studies.
Acknowledgment
This work was supported by the National Institutes of Health, CA31841 (P.A.W.) and the William Randolph Hearst Foundation (S.L.M.).
References
- 1.Rowinsky EK, Tolcher AW. Antimicrotubule Agents. In: DeVita VTJ, Hellman S, Rosenberg SA, editors. Cancer Principles and Practice of Oncology. Lippincott, Williams and Wilkins; Philadelphia, PA: 2001. pp. 431–447. [Google Scholar]
- 2.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Laulimalide and Isolaulimalide, New Paclitaxel-like Microtubule-stabilizing Agents. Cancer Res. 1999;59:653–660. [PubMed] [Google Scholar]
- 3.Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. Laulimalides: New Potent Cytotoxic Macrolides from a Marine Sponge and a Nudibranch Predator. J. Org. Chem. 1988;53:3644–3646. [Google Scholar]
- 4.Quinoa E, Kakou Y, Crews P. Fijianolides, Polyketide Structures from a Marine Sponge. J. Org. Chem. 1988;53:3642–3644. [Google Scholar]
- 5.Pryor DE, O'Brate A, Bilcer G, Díaz JF, Wang Y, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakon P, Hamel F. The Microtubule Stabilizing Agent Laulimalide Does Not Bind in the Taxoid Site, Kills Cells Resistant to Paclitaxel and Epothilones, and May Not Require Its Epoxide Moiety for Activity. Biochemistry. 2002;41:9109–9115. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 6.Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, Ghosh AK, Curran DP, Cushman M, Nicolaou KC, Paterson I, Sorensen EJ. Synergistic Effects of Peloruside A and Laulimalide with Taxoid Site Drugs, But Not With Each Other, on Tubulin Assembly. Mol. Pharmacol. 2006;70:1555–1564. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- 7.Gaitanos TN, Buey RM, Diaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH. Peloruside A Does Not Bind to the Taxoid Site on Beta-tubulin and Retains Its Activity in Multidrug-resistant Cell Lines. Cancer Res. 2004;64:5063–5067. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- 8.Gapud EJ, Bai R, Ghosh AK, Hamel E. Laulimalide and Paclitaxel: A Comparison of Their Effects on Tubulin Assembly and Their Synergistic Action When Present Simultaneously. Mol. Pharmacol. 2004;66:113–121. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 9.Clark EA, Hills PM, Davidson BS, Wender PA, Mooberry SL. Laulimalide and Synthetic Laulimalide Analogues Are Synergistic with Paclitaxel and 2-Methoxyestradiol. Mol. Pharmaceutics. 2006;3:457–467. doi: 10.1021/mp060016h. [DOI] [PubMed] [Google Scholar]
- 10.Lu H, Murtagh J, Schwartz EL. The Microtubule Binding Drug Laulimalide Inhibits Vascular Endothelial Growth Factor-induced Human Endothelial Cell Migration and Is Synergistic When Combined with Docetaxel (Taxotere). Mol. Pharmacol. 2006;69:1207–1215. doi: 10.1124/mol.105.019075. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, Murphy EA, Kuznetsov G, Johannes CW, Tremblay MR, Zhao H, Pesant M, Fang FG, Vermeulen MW, Gallagher BM, Jr., Littlefield BA. In Vitro and In Vivo Anticancer Activities of Synthetic (–)-Laulimalide, a Marine Natural Product Microtubule Stabilizing Agent. Anticancer Res. 2007;27:1509–1518. [PubMed] [Google Scholar]
- 12.Wender PA, Hegde SG, Hubbard RD, Zhang L, Mooberry SL. Synthesis and Biological Evaluation of (–)-Laulimalide Analogues. Org. Lett. 2003;5:3507–3509. doi: 10.1021/ol035339f. [DOI] [PubMed] [Google Scholar]
- 13.Mooberry SL, Randall-Hlubek DA, Leal RM, Hegde SG, Hubbard RD, Zhang L, Wender PA. Microtubule-stabilizing Agents Based on Designed Laulimalide Analogues. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8803–8808. doi: 10.1073/pnas.0402759101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wender PA, Verma VA, Paxton TJ, Pillow TH. Function-oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 15.Wender PA, Hilinski MK, Soldermann N, Mooberry SL. Total Synthesis and Biological Evaluation of 11-Desmethyllaulimalide, a Highly Potent Simplified Laulimalide Analogue. Org. Lett. 2006;8:1507–1510. doi: 10.1021/ol060233g. [DOI] [PubMed] [Google Scholar]
- 16.Wender PA, Hilinski MK, Skaanderup PR, Soldermann NG, Mooberry SL. Pharmacophore Mapping in the Laulimalide Series: Total Synthesis of a Vinylogue for a Late-Stage Metathesis Diversification Strategy. Org. Lett. 2006;8:4105–4108. doi: 10.1021/ol061619u. [DOI] [PubMed] [Google Scholar]
- 17.Johnson TA, Tenney K, Cichewicz RH, Morinaka BI, White KN, Amagata T, Subramanian B, Media J, Mooberry SL, Valeriote FA, Crews P. Sponge-derived Fijianolide Polyketide Class: Further Evaluation of Their Structural and Cytotoxicity Properties. J. Med. Chem. 2007;50:3795–3803. doi: 10.1021/jm070410z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr., Hemscheidt TK, Mooberry SL. Taccalonolides E and A: Plant-derived Steroids with Microtubule-stabilizing Activity. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
- 19.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-drug Screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 20.Paterson I, Menche D, Håkansson AE, Longstaff A, Wong D, Barasoain I, Buey RM, Díaz JF. Design, Synthesis and Biological Evaluation of Novel, Simplified Analogues of Laulimalide: Modification of the Side Chain. Bioorg. Med. Chem. Lett. 2005;15:2243–2247. doi: 10.1016/j.bmcl.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 21.Gottesman MM, Fojo T, Bates SE. Multidrug Resistance in Cancer: Role of ATP-dependent Transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 22.Yeh JJ, Hsu WH, Wang JJ, Ho ST, Kao A. Predicting Chemotherapy Response to Paclitaxel-based Therapy in Advanced Non-small-cell Lung Cancer With P-glycoprotein Expression. Respiration. 2003;70:32–35. doi: 10.1159/000068411. [DOI] [PubMed] [Google Scholar]
- 23.Chiou JF, Liang JA, Hsu WH, Wang JJ, Ho ST, Kao A. Comparing the Relationship of Taxol-based Chemotherapy Response with P-glycoprotein and Lung Resistance-related Protein Expression in Non-small Cell Lung Cancer. Lung. 2003;181:267–273. doi: 10.1007/s00408-003-1029-7. [DOI] [PubMed] [Google Scholar]
- 24.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS. Paclitaxel-resistant Human Ovarian Cancer Cells Have Mutant Beta-tubulins That Exhibit Impaired Paclitaxel-driven Polymerization. J. Biol. Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 25.Blagosklonny MV, Darzynkiewicz Z, Halicka HD, Pozarowski P, Demidenko ZN, Barry JJ, Kamath KR, Herrmann RA. Paclitaxel Induces Primary and Postmitotic G1 Arrest in Human Arterial Smooth Muscle Cells. Cell Cycle. 2004;3:1050–1056. [PubMed] [Google Scholar]
- 26.Jordan MA, Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 27.Weiderhold KN, Randall-Hlubek DA, Polin LA, Hamel E, Mooberry SL. CB694, a Novel Antimitotic With Antitumor Activities. Int. J. Cancer. 2006;118:1032–1040. doi: 10.1002/ijc.21424. [DOI] [PubMed] [Google Scholar]
- 28.Conlin A, Fornier M, Hudis C, Santwana K, Kirkpatrick P. Ixabepilone. Nat. Rev. Drug Discovery. 2007;6:953–954. [Google Scholar]
- 29.Zhou J, O'Brate A, Zelnak A, Giannakakou P. Survivin Deregulation in Beta-tubulin Mutant Ovarian Cancer Cells Underlies Their Compromised Mitotic Response to Taxol. Cancer Res. 2004;64:8708–8714. doi: 10.1158/0008-5472.CAN-04-2538. [DOI] [PubMed] [Google Scholar]
- 30.Wilmes A, Bargh K, Kelly C, Northcote PT, Miller JH. Peloruside A Synergizes with Other Microtubule Stabilizing Agents in Cultured Cancer Cell Lines. Mol. Pharmaceutics. 2007;4:269–280. doi: 10.1021/mp060101p. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa Y, Irie K, Yanagita RC, Ohigashi H, Tsuda K. Indolactam-V Is Involved in the CH/π Interaction with Pro-11 of the PKCδ C1B Domain: Application for the Structural Optimization of the PKCδ Ligand. J. Am. Chem. Soc. 2005;127:5746–5747. doi: 10.1021/ja050447d. [DOI] [PubMed] [Google Scholar]