

Abstract
Development of effective neuroprotective drugs for Alzheimer's disease (AD) is a formidable challenge because this disease is multifactorial and heterogeneous. Although AD is characterized histopathologically by the presence of numerous amyloid-β plaques and neurofibrillary degeneration of abnormally hyperphosphorylated tau in the brain, these two hallmark lesions do not exist in any fixed proportion in this disease. Furthermore, in the brains of some normal aged individuals, there are as many amyloid-β plaques seen as in typical cases of AD. On the other hand, extensive neurofibrillary degeneration of abnormally hyperphosphorylated tau and dementia but in the absence of amyloid-β plaques occur in several related neurodegenerative disorders called tauopathies. More than one molecular mechanism has been described for the development of amyloid-β as well as neurofibrillary degeneration of abnormally hyperphosphorylated tau. Thus, AD apparently results from several different etiopathogenic mechanisms and offers numerous rational therapeutic targets. We have discovered that there are at least five different subgroups of AD, and future studies are likely to identify additional subgroups. The employment of these subgroups of AD in clinical trials can markedly increase the success in developing specific and potent therapeutic drugs.
Keywords: Alzheimer's disease, Alzheimer's disease subgroups, amyloid-β, Lewy bodies, neurofibrillary pathology, tau, tauopathies, ubiquitin
INTRODUCTION
Alzheimer's disease (AD) was described approximately one hundred years ago, yet, to date, there is no effective, long-term treatment available for this major cause of dementia of the middle to old age individuals. Discoveries of amyloid-β (Aβ) as the major component of plaque and cerebrovascular amyloids in 1984 [10,11] and abnormally hyperphosphorylated tau as the major protein subunit of paired helical filaments (PHF)/neurofibrillary tangles in 1986 [13,14] generated enormous excitement in the field and laid down the foundations for the studies on mechanisms of AD and related disorders. During the last ∼25 years, an impressive amount of progress has been made in understanding the mechanisms of β-amyloidosis and neurofibrillary degeneration of abnormally hyperphosphorylated tau, the two hallmark neurobiological abnormalities of AD. Appropriately, for the last several years a large number of labs, both in the pharmaceutical industry and in academia, have been working hard to develop the first generation of rational neuroprotective drugs for AD. However, given the multifactorial nature and comorbidity, AD presents some formidable challenges when it comes to development of rational therapeutic treatments of this disease.
In this article, we discuss the complexities involved in the development of neuroprotective/disease modifying drugs and the need for stratification of AD patients into identifiable subgroups for clinical trials.
POLYETIOLOGY AND HETEROGENEITY OF AD
AD is characterized by relatively slow chronic but progressive neurodegeneration and loss of cognition. The disease is both multifactorial and heterogeneous and, thus, offers a large number of rational therapeutic targets.
In less than 1% of the cases, AD cosegregates with certain mutations in amyloid-β protein precursor (AβPP), presenilin-1, and presenilin-2 [4]. Over 99% of AD cases are not associated with any known mutations and the nature of the etiological agents is not yet understood, but might involve metabolic and signal transduction abnormalities [19]. These different etiological factors apparently lead to some common pathogenic events and vicious cycles that ultimately produce cerebral neurodegeneration associated with plaques and tangles and the dementia syndrome.
Age is the single largest risk factor for AD. The second known major risk factor for late onset AD is the presence of APOE4 allele; the presence of one and two copies of APOE4 increases the risk for AD by ∼3.5-fold and ∼10-fold, respectively [27].
Neither of the two histopathological brain lesions of AD, Aβ plaques and neurofibrillary tangles of abnormally hyperphosphorylated tau, are unique to AD. Some normal aged individuals have as many Aβ plaques as in a typical case of AD. In addition to AD, the neurofibrillary changes of abnormally hyperphosphorylated tau, but without Aβ plaques, are seen in several related tauopathies which include frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), corticobasal degeneration, Pick disease, supranuclear palsy, dementia pugilistica, and Guam Parkinsonism dementia complex, among several other neurodegenerative disorders. While AD is characterized by the presence of both Aβ plaques and neurofibrillary tangles of abnormally hyperphosphorylated tau, there is no fixed proportion in the number of these two lesions from AD case to case. AD can be neocortical type that occurs relatively early, i.e., at around age 60, or limbic type which peaks at around age 80−90, or plaque predominant type occurring predominantly in the tenth decade of life [25,26]. This heterogeneity in the proportion of these two hallmark lesions in AD brain might be due to the involvement of different upstream etiopathogenic mechanisms.
Plaque amyloid in AD brain contains a heterogeneous mixture of various fragments of Aβ, i.e., 1−40, 1−42, 1−39, 1−43, x-40, x-42, etc. [24]. Increase in Aβ levels and consequently Aβ load can result from both higher amyloidogenic processing of AβPP as well as decrease in turnover of Aβ, for instance, by decrease in insulin degrading enzyme and or neprolysin activity [21,28].
The abnormally hyperphosphorylated tau which makes PHF/neurofibrillary tangles in AD brain is phosphorylated at over 30 sites [16]. Both proline directed and non-proline directed sites are involved in this abnormal hyperphosphorylation of tau [17]. The AD type abnormal hyperphosphorylation of tau, which results in its self-assembly into bundles of PHF, can be achieved with different combinations of tau protein kinases, suggesting that more than one different etiopathogenic mechanism could be involved in leading to neurofibrillary degeneration [30]. Thus, the multifactorial nature of AD, while offering a large number of rational therapeutic targets, also suggests that any specific therapeutic drug might benefit only a subgroup of patients. Identification of various subgroups of AD can substantially facilitate the development of therapeutic drugs.
Subgroups of AD
We postulate: 1) that different signal transduction and metabolic factors through different disease mechanisms apparently lead to the same two common disease characteristic lesions, neurofibrillary degeneration of abnormally hyperphosphorylated tau and β-amyloidosis (Fig. 1); and 2) that identification of different AD subgroups which probably represent different etiopathogenic mechanisms will improve the accuracy of the diagnosis as well as serve as a useful outcome measure for the development of effective neuroprotective drugs.
Fig. 1.
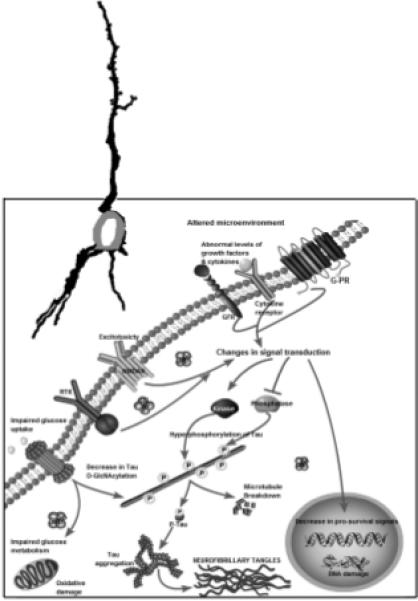
A schematic showing different major steps of the “Metabolic/Signal Transduction Hypothesis”. AD and other tauopathies require a genetic predisposition and are triggered by a variety of environmental factors, affecting one or more specific signal transduction pathways which result in a protein phosphorylation/dephosphorylation imbalance and the abnormal hyperphosphorylation of tau that leads to neurofibrillary degeneration and dementia. In AD, the protein phosphorylation/dephosphorylation imbalance in the affected neurons is generated at least in part by a decrease in the activities of tau phosphatases, i.e., PP-2A and PP-1; the activities of tau kinases such as cdk5, GSK-3, CaM kinase II and PKA might also be increased in the affected neurons. This protein phosphorylation/dephosphorylation imbalance probably involves an alteration of a specific signal transduction pathway(s) produced by an increase in the levels of an extracellular signal, e.g., FGF2 or an alteration in the molecular topology of the neuronal cell membrane or both. With age, the molecular topology of the cell membranes is altered due to a decrease in membrane fluidity. The mutations in transmembrane proteins, such as AβPP, PS1 and PS2, increase the vulnerability of the cell membrane to alteration in pathological signal transduction. The increased risk for AD in the carriers of APOE4 allele as opposed to APOE2 or APOE3 alleles might also involve alteration of signal transduction through the interaction of APOE4 with the neuronal cell membrane. Any mutation or posttranslational modification of tau that will make it a better substrate for abnormal hyperphosphorylation will also increase the risk for the disease. High cholesterol might be involved in decreasing membrane fluidity. Decreased glucose metabolism/uptake might lead to the abnormal hyperphosphorylation of tau through a decrease in its O-GlcNAcylation. RTK, receptor tyrosine kinase; NMDAR, NMDA receptor; GFR, growth factor receptor; G-PR, G-protein receptor; P-Tau, phosphorylated Tau.
Because of clinical heterogeneity, the diagnosis of AD remains probable until postmortem histopathological examination and is made primarily by exclusion of other causes of dementia [23]. AD histopathology shows considerable qualitative and quantitative heterogeneity. AD can be neocortical type, limbic type and plaque-dominant type, and it may present with numerous neurofibrillary tangles exclusively confined to the hippocampus and entorhinal cortex [26]. The two most common confounding diagnoses are cerebral vascular disease (multi-infarct dementia) and dementia with Lewy bodies. Increased rates of ventricular volume and whole brain atrophy have been demonstrated in AD [29]. The whole brain atrophy in AD brain results in a loss of brain mass of as much as ∼2−3% per year compared with ∼0.4−0.5% in age-matched control subjects [20].
There is growing evidence that cerebrospinal fluid (CSF) reflects the state of protein metabolism in the brain. A number of animal and human studies have suggested that Aβ1−42 levels in CSF reflect the Aβ pathology in the brain. Reduction of Aβ1−40 and Aβ1−42 in the brain of adult rats treated orally with γ-secretase inhibitors have been found to result in decreased levels of Aβ in both brain and CSF [2,8]. An inverse relation between in vivo amyloid load and CSF levels of Aβ1−42 has been found in humans [9]. Antemortem CSF levels of Aβ1−42, total tau and phosphotau-Thr231 have been reported to reflect the histopathological changes observed postmortem in the brains of AD cases [3,7]. The CSF levels of tau have been shown to be markedly increased in patients with diffuse axonal injury in head trauma which revert on clinical improvement [31].
Development of therapeutic drugs requires our ability to accurately diagnose the disease and its specific subtypes, as well as the availability of specific outcome measures. We postulate that more than one disease mechanism and signaling pathway are involved in producing the AD pathology, especially the neurofibrillary degeneration of abnormally hyperphosphorylated tau, and that various subgroups of AD can be identified based on the CSF levels of proteins associated with senile (neuritic) plaques and neurofibrillary tangles. Towards testing this hypothesis, we immunoassayed the levels of tau, ubiquitin and Aβ1−42 in retrospectively collected lumbar CSFs of 468 patients clinically diagnosed as AD (353 CSFs) and as non-AD neurological and non-neurological cases (115 CSFs) and, based on the level of these molecular markers, all subjects were subjected to the latent profile analysis to determine the assignment of each subject to a particular cluster [18]. We found that AD subdivides into at least five subgroups based on the CSF levels of Aβ1−42, tau and ubiquitin, and that each subgroup presented a different clinical profile (Fig. 2). Furthermore, we found strong associations between these patterns of biomarkers and other observed characteristics related to AD and its symptomatic manifestations. Subgroups AELO, ATEO, HARO and ATURO accounted for approximately 50%, 22%, 5% and 1%, respectively, of the AD cases studied. Subgroup LEBALO, which contained a majority of AD cases with Lewy bodies, accounted for ∼19% of the AD cases. These findings suggested the use of CSF biomarkers to discriminate AD with Lewy bodies from AD. The accuracy of the clinical diagnosis of AD mixed with vascular dementia is relatively low. Thus, identification of this subgroup of AD based on CSF levels of Aβ1−42, tau, and ubiquitin will require studies where postmortem histopathological diagnosis confirmation is available.
Fig. 2.
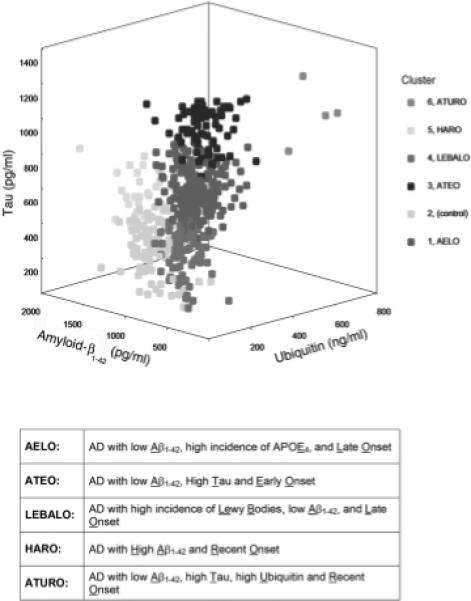
Three-dimensional representation of the five AD subgroups (Clusters 1, 3, 4, 5 and 6) and the controls (Cluster 2). (Reproduced with permission from Iqbal et al., 2005, Annals of Neurology, 58, 748−757.)
To classify diagnosed AD cases into the proposed subgroups, we sought a simple set of rules using the level of only one indicator protein at any stage in the classification process. Ideally, it would classify cases with a sensitivity and a specificity of no less than 90% of each category and a comparable overall level of correct classification. The algorithm must unambiguously categorize all cases. A decision tree based on the algorithm was derived based on examination of cluster characteristics and experimental runs that came closest to fulfilling those criteria (see Fig. 3). The respective sensitivities (ability to detect a true positive case) and specificities (ability to detect a true negative case) were: AELO – 90%, 92%; ATEO – 90%, 95%; LEBALO – 88%, 99%; HARO – 100% 99%; and ATURO – 100%, 100%. This study demonstrated that CSF levels of Aβ1−42, tau and ubiquitin could diagnose AD in five different subgroups at sensitivities and specificities of greater than 88% and, overall, 86% of cases were classified correctly. This rate of diagnostic accuracy not only is superior to using one of these markers individually or in combination of twos, but also exceeds the biomarker criteria of the Consensus Report [1]. Furthermore, assay of phospholipase-2 activity, a well known inflammatory key enzyme, in 141 of the above AD and 34 control CSFs showed no differences between the five subgroups, which validates the specificity of the identified subgroups [5].
Fig. 3.
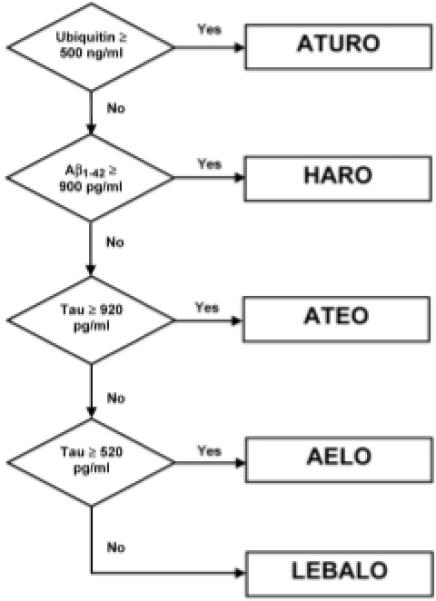
Decision tree for identifying various subgroups of Alzheimer's disease based on CSF levels of ubiquitin, Aβ1−42 and tau. (Reproduced with permission from Iqbal et al., 2005, Annals of Neurology, 58, 748−757.)
Our recent studies have revealed that more than one signaling pathway is involved in neurofibrillary degeneration. We have found that tau can be abnormally hyperphosphorylated to self-assemble into bundles of paired helical filaments with more than one combination of protein kinases and that this phosphorylation of tau can be regulated by protein phosphatases (PP), especially PP-2A [30]. Thus, it is likely that, in future, additional subgroups of AD may be identified from phosphorylation patterns of CSF tau of AD patients.
The CSF analysis not only helps identify a specific AD subgroup of a patient but also can serve as the outcome measure of a drug treatment. We discovered that memantine inhibited abnormal hyperphosphorylation of tau in rat hippocampal slices in culture [22], and that this effect of the drug was through disinhibition of PP-2A activity [6] which we previously showed to be down regulated in AD brain [12]. Based on our finding on the restoration of the PP-2A activity by memantine, Gunnarrsson et al. [15] investigated and found a significant decrease in phosphotau level in the CSF of patients one year after treatment with memantine.
In conclusion, because of the involvement of different etiopathogenic mechanisms in AD, the identification of different subgroups of this single major cause of age-associated dementia is critical for development of potent and specific drugs that can prevent and cure this disease. Currently, several hundred drugs for AD are under development by the pharmaceutical industry. Stratification of the test subjects in clinical trials by disease subgroups may increase the chance of success up to several fold. For instance, in a 24-week randomized clinical trial of rosiglitazone maleate which increases brain glucose, APOE4 non-carriers but not APOE4 carriers were found to show clinical improvement. The future of therapeutic drugs for AD may depend on recognition of different subgroups of the disease.
ACKNOWLEDGMENTS
We are grateful to Janet Murphy for secretarial assistance. Studies in our laboratories were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities and NIH grants AG019158 and AG028538, and Alzheimer's Association (Chicago, IL) grant IIRG-06-25836.
References
- 1.Anonymous Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer's Disease”. The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Neurobiol Aging. 1998;19:109–116. [PubMed] [Google Scholar]
- 2.Best JD, Jay MT, Otu F, Churcher I, Reilly M, Morentin-Gutierrez P, Pattison C, Harrison T, Shearman MS, Atack JR. In vivo characterization of Abeta(40) changes in brain and cerebrospinal fluid using the novel gamma-secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1, 1-trifluoromethanesulfonamide (MRK-560) in the rat. J Pharmacol Exp Ther. 2006;317:786–790. doi: 10.1124/jpet.105.100271. [DOI] [PubMed] [Google Scholar]
- 3.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2006;129:3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 4.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalbot S, Blennow K, Grundke-Iqbal I, Iqbal K. CSF phospholipase A2 activity is a biomarker of blood brain barrier permeability but not Alzheimer's Disease. 11th International Conference on Alzheimer's Disease & Related Disorders; 2008; in press. [Google Scholar]
- 6.Chohan MO, Khatoon S, Grundke-Iqbal I, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006;580:3973–3979. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 7.Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, Morris JC, McKeel DW, Jr., Farlow M, Weitlauf SL, Quinn J, Kaye J, Knopman D, Arai H, Doody RS, DeCarli C, Leight S, Lee VM, Trojanowski JQ. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60:1696–1702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 8.El Mouedden M, Vandermeeren M, Meert T, Mercken M. Reduction of Abeta levels in the Sprague Dawley rat after oral administration of the functional gamma-secretase inhibitor, DAPT: a novel non-transgenic model for Abeta production inhibitors. Curr Pharm Des. 2006;12:671–676. doi: 10.2174/138161206775474233. [DOI] [PubMed] [Google Scholar]
- 9.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 10.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 11.Glenner GG, Wong CW, Quaranta V, Eanes ED. The amyloid deposits in Alzheimer's disease: their nature and pathogenesis. Appl Pathol. 1984;2:357–369. [PubMed] [Google Scholar]
- 12.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 13.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 14.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunnarsson M, Kilander L, Sudelof J, Basun H, Lannfelt L. Reduction of hyperphosphorylated tau during memantine treatment of Alzheimer's disease. Alzheimers Dementia. 2006;2:S63–S64. [Google Scholar]
- 16.Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, Reynolds CH, Ward MA, Anderton BH. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282:23645–23654. doi: 10.1074/jbc.M703269200. [DOI] [PubMed] [Google Scholar]
- 17.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Iqbal K, Flory M, Khatoon S, Soininen H, Pirttila T, Lehtovirta M, Alafuzoff I, Blennow K, Andreasen N, Vanmechelen E, Grundke-Iqbal I. Subgroups of Alzheimer's disease based on cerebrospinal fluid molecular markers. Ann Neurol. 2005;58:748–757. doi: 10.1002/ana.20639. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal K, Grundke-Iqbal I. Metabolic/signal transduction hypothesis of Alzheimer's disease and other tauopathies. Acta Neuropathol (Berl) 2005;109:25–31. doi: 10.1007/s00401-004-0951-y. [DOI] [PubMed] [Google Scholar]
- 20.Karas GB, Scheltens P, Rombouts SA, Visser PJ, van Schijndel RA, Fox NC, Barkhof F. Global and local gray matter loss in mild cognitive impairment and Alzheimer's disease. Neuroimage. 2004;23:708–716. doi: 10.1016/j.neuroimage.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Kim M, Hersh LB, Leissring MA, Ingelsson M, Matsui T, Farris W, Lu A, Hyman BT, Selkoe DJ, Bertram L, Tanzi RE. Decreased catalytic activity of the insulin-degrading enzyme in chromosome 10-linked Alzheimer disease families. J Biol Chem. 2007;282:7825–7832. doi: 10.1074/jbc.M609168200. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–269. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 23.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 24.Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 25.Mizutani T, Kasahara M. Hippocampal atrophy secondary to entorhinal cortical degeneration in Alzheimer-type dementia. Neurosci Lett. 1997;222:119–122. doi: 10.1016/s0304-3940(97)13365-1. [DOI] [PubMed] [Google Scholar]
- 26.Mizutani T, Sakata M, Enemoto M, Yamada S. Pathological heterogeneity of Alzheimer type dementia. In: Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, editors. Alzheimer Disease: Biology, Diagnosis and Therapeutics. Vol. 33. John Wiley & Sons Ltd.; Chichester, UK: 1997. pp. 247–255. [Google Scholar]
- 27.Roses AD. On the discovery of the genetic association of Apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–366. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- 28.Saido TC, Iwata N. Metabolism of amyloid beta peptide and pathogenesis of Alzheimer's disease. Towards presymptomatic diagnosis, prevention and therapy. Neurosci Res. 2006;54:235–253. doi: 10.1016/j.neures.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Schott JM, Price SL, Frost C, Whitwell JL, Rossor MN, Fox NC. Measuring atrophy in Alzheimer diseaseL a serial MRI study over 6 and 12 months. Neurology. 2005;65:119–124. doi: 10.1212/01.wnl.0000167542.89697.0f. [DOI] [PubMed] [Google Scholar]
- 30.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, Cohen JA, Rudick RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–750. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]