

Abstract
Animals with a neonatal ventral hippocampal lesion (NVHL) develop abnormal behaviors during or after adolescence, suggesting that early insults can have delayed consequences. Many of these behaviors depend on the prefrontal cortex (PFC), and we have reported that PFC pyramidal neurons of adult rats with an NVHL respond to stimulation of the ventral tegmental area with an increase in firing instead of the characteristic decrease. As the dopamine modulation of cortical interneurons matures during adolescence, these findings raise the possibility that maturation of local inhibitory circuits within the PFC may have been altered in NVHL rats. Here, we assessed the state of PFC interneurons in NVHL rats with in situ hybridization measures of the mRNAs for the calcium binding protein parvalbumin (PV) and the GABA synthesizing enzyme GAD67, as well as with electrophysiological measures of interneuron function. Although no differences were observed with PV or GAD67, whole-cell recordings in slices revealed abnormal responses to the D2 agonist quinpirole in interneurons from NVHL rats. The loss of D2 modulation of local inhibition in slices from NVHL rats was also evident in the absence of a lasting component in the D2 attenuation of excitatory synaptic responses in pyramidal neurons, which in sham treated rats was picrotoxin sensitive. The results suggest that the neonatal lesion causes improper maturation, but not loss, of PFC interneurons during adolescence, a finding consistent with current interpretations of imaging data in schizophrenia that suggest a hyperactive, “noisy” cortex underlying dysfunction in the PFC and other cortical areas.
Keywords: prefrontal cortex, dopamine, interneurons, GABA, neonatal ventral hippocampal lesion, schizophrenia
Introduction
Anomalies in cortical interneurons are consistently found in schizophrenia. Postmortem studies have shown reduced markers of interneuron function in the prefrontal cortex (PFC) (Akbarian et al., 1995; Lewis, 1995; Beasley and Reynolds, 1997; Beasley et al., 2002), cingulate cortex (Benes et al., 1991), temporal lobe (Chance et al., 2005), hippocampus (Benes, 1999), amygdala (Benes and Berretta, 2001), and even the motor and visual cortices (Hashimoto et al., 2008a) in brains from schizophrenia patients. The high reproducibility of these findings supports the possibility that interneuron deficits are a central feature in schizophrenia pathophysiology (Benes and Berretta, 2001; Hashimoto et al., 2008b; Lewis and González-Burgos, 2008; O'Donnell, 2008). Many functional deficits are interpreted as arising from interneuron alterations. For example, reduced gamma (i.e., 30–50 Hz) activation in the EEG of schizophrenia patients (Spencer et al., 2003; Cho et al., 2006) is thought to be an expression of altered inhibition in cortical areas because interneurons are critical for this type of activity (Szabadics et al., 2001; Traub et al., 2001; Frankle et al., 2008). The reduced capacity in working memory functions and the inefficient PFC activity suggested by functional imaging studies (Callicott et al., 2000; Manoach, 2003) could also be the consequence of impaired inhibitory circuits. Furthermore, noncompeting NMDA antagonists, a well established pharmacological model of schizophrenia (Javitt and Zukin, 1991), increased glutamate levels (Moghaddam et al., 1997) and pyramidal cell firing (Jackson et al., 2004) in the PFC, likely attributable to blockade of NMDA receptors in interneurons (Homayoun and Moghaddam, 2007). A recent clinical trial with a metabotropic glutamate receptor subunit 2/3 agonist, which would reduce glutamate release, proved almost as effective as olanzapine in treating schizophrenia symptoms (Patil et al., 2007). Thus, preclinical and clinical studies converge remarkably in highlighting interneuron deficits in schizophrenia (Lewis and Moghaddam, 2006).
Because it has become widely accepted that schizophrenia has a strong developmental component (Weinberger, 1987; Harrison and Weinberger, 2005), the need for developmental animal models has become stronger. Many such models have been studied, including the neonatal ventral hippocampal lesion (NVHL) (Lipska et al., 1993; Brake et al., 1999; O'Donnell et al., 2002), prenatal exposure to an antimitotic (Flagstad et al., 2004; Gourevitch et al., 2004; Moore et al., 2006), prenatal stress (Koenig et al., 2005), and many genetic models (Miyakawa et al., 2003; Pillai-Nair et al., 2005; Kellendonk et al., 2006; Hikida et al., 2007). The behavioral phenotype in all these models is quite similar: hyperlocomotion, enhanced sensitivity to stress and stimulants, prepulse inhibition deficits, reduced social interactions, and, in some cases, reversal with antipsychotic drugs. Most of these anomalies emerge during adolescence, therefore including a time course similar to the human disease, and in many cases there is also evidence of reduced GABAergic markers (Lipska et al., 2003; Pillai-Nair et al., 2005; Penschuck et al., 2006; Endo et al., 2007). Because we have shown recently that the dopamine (DA) modulation of interneurons matures during adolescence (Tseng and O'Donnell, 2007a,b), it is possible that this maturation is altered in rodent developmental models of schizophrenia. In vivo intracellular recordings from adult rats with an NVHL revealed that ventral tegmental area stimulation causes abnormal firing of PFC pyramidal neurons in this model (O'Donnell et al., 2002) instead of the decrease typically observed in naive or sham rats (Lewis and O'Donnell, 2000; O'Donnell et al., 2002). This suggests that inhibitory mechanisms in the PFC may have been altered by the neonatal lesion and the alteration became apparent with the transition to a young adult age. Here, we directly tested that possibility with molecular measures of interneuron-related markers and whole-cell recordings from PFC pyramidal neurons and interneurons in brain slices from NVHL and sham rats.
Materials and Methods
All experimental procedures were approved by the Albany Medical College and the University of Maryland School of Medicine Institutional Animal Care and Use Committees and were conducted according to the United States Public Health Service Guide for Care and Use of Laboratory Animals.
Neonatal ventral hippocampal lesion.
Pregnant Sprague Dawley rats were obtained at 18 d of gestation from Taconic Farms. At postnatal day 6 (P6), male pups (15–19 g) were randomly separated into two groups to receive vehicle injection (sham) or ibotenic acid injection (lesion). Pups (P6–P7) were anesthetized with hypothermia for 10–15 min and secured on a Styrofoam platform mounted on a stereotaxic frame (David Kopf Instruments). A cannula was lowered into the ventral hippocampus (anteroposterior, −3.0 mm; lateral, +3.5 mm; 5.0 mm below cortical surface) and 0.3 μl of ibotenic acid [10 μg/μl (in mm): 148 NaCl, 3 KCl, 0.2 NaH2PO4, 1.5 Na2HPO4, 1.4 CaCl2, and 0.8 MgCl2, pH 7.4] was delivered using a minipump at a rate of 0.15 μl/min. The cannula was left in place for 3 additional minutes before being removed. This procedure was repeated in the contralateral hemisphere. Sham animals received the same volume of vehicle on each side. After surgery, pups were warmed up and returned to their cages, where they remained undisturbed until weaning except for husbandry. The extent of damage induced by ibotenic acid (i.e., areas with cell loss and cell disorganization) was estimated in all animals by Nissl staining. All rats were maintained on a 12 h light/dark cycle with food and tap water available ad libitum until the time of the experiment.
In situ hybridization for GAD67 and parvalbumin.
Rats were anesthetized with chloral hydrate (400 mg/kg, i.p.) 15 min before being decapitated. Brains were immediately frozen after removal from the skull and stored at −80°C. Serial coronal sections (12 μm) were cut from +2.7 to +2.2 bregma (Paxinos and Watson, 1998), and three sections evenly spaced at ∼160 μm intervals were selected from each animal and used for in situ hybridization studies for each mRNA of interest.
For synthesis of riboprobes, we used a 311 bp fragment for GAD67 mRNA and a 339 bp fragment for parvalbumin (PV) mRNA, corresponding to bases 151–461 of the mouse GAD67 mRNA (Y12257) and bases 256–594 of the mouse PV mRNA (X59382). Amplified fragments were subcloned into the plasmid pSTBlue-1 (Novagen), and antisense and sense probes were transcribed in vitro in the presence of [35S]CTP (GE Healthcare), using T7 or SP6 RNA polymerase.
Hybridization was performed as described previously (Hashimoto et al., 2005). After fixation with 4% paraformaldehyde in PBS, the sections were acetylated, dehydrated through a graded ethanol series, and defatted in chloroform for 10 min. The sections were then hybridized with 35S-labeled riboprobes in hybridization buffer at 56°C for 16 h. The sections were washed in solution containing 0.3 m NaCl, 20 mm Tris-HCl, pH 8.0, 1 mm EDTA, pH 8.0, and 50% formamide at 63°C, treated with RNase A (20 μg/ml) at 37°C, and washed in 0.1× SSC (150 mm NaCl and 15 mm sodium citrate) at 67°C. Sections were then dehydrated through a graded ethanol series, air dried, and exposed to BioMax MR film (Eastman Kodak).
Quantification was performed without knowledge of subject condition by random coding of the sections. Trans-illuminated autoradiographic film images were captured by a video camera under precisely controlled conditions, digitized, and analyzed using a microcomputer imaging device MCID system (Imaging Research). Images of the sections were also captured and superimposed onto the autoradiographic images to draw contours of the full thickness of the prefrontal cortex, including the cingulate, prelimbic, and infralimbic cortices. Expression levels of each mRNA were determined as optical densities within the contours and expressed as nanocuries per gram of tissue by reference to radioactive standards (Carbon-14 standards; ARC Inc.) exposed on the same film.
Because bilateral lesions were made and the mRNA levels were quantified in the PFC of each hemisphere independently, we used a two-factor design ANOVA with manipulation (lesion vs sham) as a between-subject factor and laterality (left vs right) as repeated measures for each mRNA.
Brain slice preparation.
Rats were anesthetized with chloral hydrate (400 mg/kg, i.p.) 15 min before being decapitated. Brains were quickly removed from the skull into ice-cold artificial CSF (aCSF) oxygenated with 95% O2–5% CO2 and containing the following (in mm): 125 NaCl, 25 NaHCO3, 10 glucose, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 3 MgCl2, pH 7.45 (295–300 mOsm). Coronal slices (350 μm thick) containing prelimbic and infralimbic regions of the medial PFC were obtained with a Vibratome in ice-cold aCSF and incubated in warm (∼35°C) aCSF solution constantly oxygenated with 95% O2–5% CO2 for at least 60 min before recording. The recording aCSF solution was delivered to the recording chamber with a minipump at the rate of 2 ml/min, and CaCl2 and MgCl2 were adjusted to 2 and 1 mm, respectively. Patch electrodes (6–9 MΩ) were obtained from 1.5 mm borosilicate glass capillaries (World Precision Instruments) with a Flaming-Brown horizontal puller (P97; Sutter Instruments) and filled with a solution containing 0.125% Neurobiotin and the following (in mm): 115 K-gluconate, 10 HEPES, 2 MgCl2, 20 KCl, 2 MgATP, 2 Na2-ATP, and 0.3 GTP, pH 7.25–730 (280–285 mOsm). All chemicals and drugs (quinpirole, eticlopride, CNQX, and picrotoxin) were purchased from Sigma, and they were mixed into oxygenated recording aCSF solution in known concentrations.
Whole-cell patch-clamp recordings.
All experiments were conducted at 33–35°C. Medial PFC pyramidal cells and interneurons from layers V–VI were identified under visual guidance using infrared (IR) differential interference contrast video microscopy with a 40× water-immersion objective (Olympus BX51-WI). The image was detected with an IR-sensitive CCD camera (Dage-MTI) and displayed on a monitor. Whole-cell current-clamp recordings were performed with a computer-controlled amplifier (MultiClamp 700A; Molecular Devices), digitized (Digidata 1322; Molecular Devices), and acquired with Axoscope 8.1 (Molecular Devices) at a sampling rate of 10 kHz. The liquid junction potential was not corrected, and electrode potentials were adjusted to zero before obtaining the whole-cell configuration.
Interneuron excitability was assessed by counting the number of action potentials evoked by a 500 ms duration constant-amplitude depolarizing current pulse before and after drug treatment. Typically, current intensity was adjusted to elicit between 5 and 15 action potentials during baseline. In each neuron, input resistance and membrane potential were also monitored throughout the entire recording session.
Synaptic responses were tested in pyramidal neurons with electrical stimulation of layers I–II with a bipolar electrode made from a pair of twisted Teflon-coated nichrome wires (tips separated by ∼200 μm) and placed ∼1 mm lateral to the vertical axis of the recorded neuron. Stimulation pulses (0.4–0.8 mA; 0.3 ms) were delivered every 20 s, and the intensity was adjusted to half the current required to evoke an action potential. If synaptic responses exhibited >15% variation in amplitude during the initial 10 min of recording or the current intensity required was larger than 0.8 mA, the neuron was discarded. Input resistance (measured with hyperpolarizing square pulses), membrane potential, and evoked synaptic responses were analyzed before, during, and after drug application.
Histology.
Standard histochemical techniques were used to verify morphology and location of the neurons recorded with Neurobiotin. After completion of the recording session, the entire slices were fixed in 4% paraformaldehyde for 2 h. After a series of rinses in 0.1 m PBS, slices were incubated in 2% Triton X-100 in PBS for 2 h to enhance penetration, followed by 10–12 h in Vectastain Elite ABC reagent (Vector Laboratories) at 4°C. After another series of rinses, slices were reacted with 3,3′ diaminobenzidine and urea-hydrogen peroxide (FAST DAB set; Sigma). Slices were then rinsed, mounted on gelatin-coated slides, air dried for 24 h, cleared in xylene, coverslipped in Permount, and examined on an Olympus CH30 microscope.
Statistics.
All measures are expressed as mean ± SD. Drug effects were compared using Student's t test or repeated-measures ANOVA, and the differences between experimental conditions were considered statistically significant when p < 0.05. In some cases, a two-way ANOVA was used to compare the interactions between experimental groups and the time course of synaptic changes obtained throughout the recording.
Results
Bilateral infusion of ibotenic acid into the ventral hippocampus of neonate rats caused local neuronal loss and cellular disarray that persisted in adulthood. Only rats that showed bilateral anatomical disruption restricted to the ventral hippocampus (Fig. 1), which comprised ∼85% of the lesioned animals, were included in the study.
Figure 1.

Neonatal ventral hippocampal lesion. A, Drawings showing the extension of the ventral hippocampal damage induced by neonatal ibotenic acid observed in adulthood. Gray and dark areas indicate maximal and minimal extents of damage, respectively. B, Coronal Nissl-stained sections showing the ventral hippocampus of a sham rat (top, Sham) and a typical neonatal ventral hippocampal lesion (bottom, NVHL), characterized by cell loss (thick arrows) and enlarged ventricles (asterisks).
GAD67/PV mRNA expression in the prefrontal cortex of NVHL rats
Ten sham and 10 NVHL rats were used for in situ hybridization analyses of GAD67 and PV mRNA expression in the PFC. In control animals, the cortical expression patterns of GAD67 and PV mRNAs were consistent with previous observations in rodents (de Lecea et al., 1995; Cochran et al., 2003; Lipska et al., 2003; Hashimoto et al., 2005) (Fig. 2A,B). In all animals, the expression patterns of both mRNAs in the PFC appeared symmetrical between hemispheres. Neither manipulation nor laterality had a significant effect on the expression levels of GAD67 (F(1,20) = 1.649, p = 0.214 for manipulation; F(1,20) = 0.206, p = 0.655 for laterality) or PV (F(1,20) = 1.686, p = 0.209 for manipulation; F(1,20) = 0.005, p = 0.942 for laterality) mRNAs.
Figure 2.
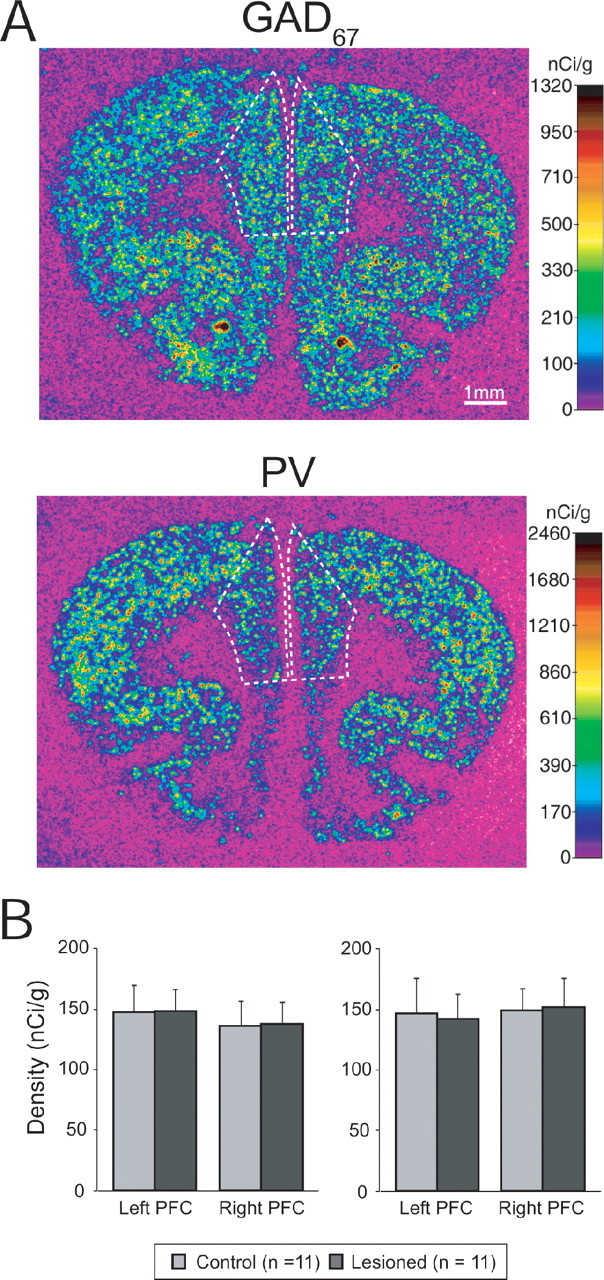
Interneuron markers in the PFC of NVHL rats. A, Representative autoradiograms illustrating the expression of GAD67 (top) and PV (bottom) mRNAs in a control rat. The densities of hybridization signals are presented in a pseudocolor manner according to the calibration scales. The densities were quantified in the area indicated by the broken lines. B, Graphs summarizing the mean ± SD for GAD67 (left) and PV (right) mRNA levels in right and left PFC of control (gray bars) and NVHL (black bars) rats.
Electrophysiological properties of PFC interneurons in NVHL rats
In another set of sham (n = 22) and NVHL (n = 16) rats, brain slices were prepared for electrophysiological recordings from interneurons and pyramidal neurons. All nonpyramidal neurons (n = 62) included in the study were from layers V–VI of the medial PFC (prelimbic and infralimbic regions). Based on their firing pattern, afterhyperpolarization (AHP), and spike-frequency adaptation characteristics (Cauli et al., 1997; Gibson et al., 1999), 27 cells were classified as fast-spiking interneurons (FSIs); those that did not (n = 35) were labeled non-fast spiking (NFS) (Fig. 3). FSIs typically responded with constant firing throughout the current pulse (Fig. 3A,B) and exhibited a larger AHP (19.2 ± 2.9 mV) and faster spike kinetics (0.58 ± 0.08 ms half-width duration) than NFS interneurons (9.4 ± 2.6 mV AHP and 0.9 ± 0.15 ms spike half-width; p < 0.00001, unpaired t test). FSIs recorded from sham (n = 14) and NVHL (n = 13) rats exhibited similar resting membrane potential (−62.0 ± 2.0 vs −62.2 ± 1.8 mV, sham vs NVHL) and input resistance (230.5 ± 61.8 vs 231.5 ± 65.5 MΩ, sham vs NVHL). No differences in FSI AHP amplitude (19.2 ± 3.0 vs 19.2 ± 2.9 mV, sham vs NVHL) or half-width duration (5.4 ± 1.7 vs 4.4 ± 1.5 ms, sham vs NVHL) or in action potential kinetics (half-width, 0.58 ± 0.08 vs 0.65 ± 0.09 ms; amplitude, 69.9 ± 5.8 vs 71.3 ± 8.7 mV; spike threshold, −45.5 ± 1.8 vs −47.0 ± 1.4 mV, sham vs NVHL) were observed between sham and NVHL rats (Fig. 3C). Similarly, NFS interneurons recorded from sham (n = 19) and NVHL (n = 16) rats exhibited similar resting membrane potential (-61.6 ± 4.1 vs. −61.9 ± 3.5 mV, Sham vs. NVHL), input resistance (259.7 ± 55.9 vs 249.7 ± 81.0 MΩ, sham vs NVHL), AHP (half-width duration, 3.2 ± 0.9 vs 3.1 ± 1.0 ms; amplitude, 9.4 ± 2.6 vs 9.9 ± 2.4 mV, sham vs NVHL) and spike kinetics (half-width, 0.91 ± 0.15 vs 0.90 ± 0.17 ms; amplitude, 74.5 ± 6.7 vs 73.4 ± 9.3 mV; spike threshold, −46.1 ± 2.9 vs −46.6 ± 2.6 mV, sham vs NVHL). These results indicate that membrane properties and action potential firing in late adolescent PFC interneurons were not affected by the NVHL.
Figure 3.

Basic membrane properties of PFC interneurons in NVHL and sham rats. A, Traces illustrating the firing pattern of FSIs in PFC slices from developmentally mature (older than P60) sham and NVHL rats. FSIs exhibited a pronounced fast AHP (middle panel inset) and responded with a constant instantaneous firing rate throughout the current pulse (middle panel: no spike-frequency adaptation). Right, Neurobiotin staining of the FSIs from which the traces were obtained. B, Traces from an NFS interneuron, which instead show increasingly longer interspike intervals when activated with current injection (left and middle panels). Right, Neurobiotin staining of the interneuron recorded. C, Plots of AHP amplitude, half-width, and action potential amplitudes and duration for both cell types.
Postpubertal disruption of PFC interneurons response to quinpirole in NVHL rats
We have shown recently that the D2 agonist quinpirole enhances FSI excitability in PFC slices from late adolescent and adult (older than P55) but not pre-pubertal (younger than P35) rats (Tseng and O'Donnell, 2007b). In slices from adult rats with a neonatal sham surgery, bath application of quinpirole enhanced interneuron excitability by near 40% (n = 11; six FSI and five NFS) (Fig. 4A), as observed in naive postpubertal rats (Tseng and O'Donnell, 2007b). The number of spikes evoked by somatic current injection increased from 9.3 ± 2.2 (baseline) to 12.7 ± 2.8 after 10 min of quinpirole incubation (p < 0.0001, paired t test) (Fig. 4A). This effect was completely blocked by the D2 antagonist eticlopride (20 μm; n = 5), confirming that quinpirole-induced excitability increase was D2 dependent. Interneuron excitability remained elevated after quinpirole was removed; typically, a washout period of 15–25 min was required to recover to baseline activity. In slices from adult rats with an NVHL, quinpirole failed to increase PFC interneuron excitability (n = 13; six FSI and seven NFS). The number of evoked spikes was 10.8 ± 3.1 during baseline and 10.3 ± 3.7 after 10 min of drug application (NS, paired t test) (Fig. 4B). Although the majority of interneurons (70%) recorded from NVHL rats did not change excitability after quinpirole, 4 of 13 showed a decrease (Fig. 4B). No effects of the D2 agonist were observed in slices from preadolescent rats with an NVHL or sham operation (Fig. 4D,E), indicating that the abnormal responses emerge during adolescence (Fig. 4F).
Figure 4.

Abnormal maturation of PFC interneurons in NVHL rats. A, B, Plot showing the effect of quinpirole (1 μm) on PFC interneuron excitability in slices from developmentally mature [older than P60 (PD>60)] sham (A) and NVHL (B) rats. The excitatory action of quinpirole was observed only in the PFC of adult sham rats. In the lesioned animals, quinpirole failed to increase PFC interneuron excitability. Instead, some neurons were inhibited with quinpirole. C, Representative traces illustrating the increase in excitability in a neuron from a sham rat (top) and a neuron from an NVHL rat (bottom). D, E, Plot showing the lack of effect of quinpirole (1 μm) in prepubertal (younger than P35) sham (D) and NVHL (E) rats. F, Plot of normalized responses in cell excitability changes by quinpirole in all groups. An excitatory action of quinpirole was observed only in the PFC of adult sham rats. In contrast, the majority of adult NVHL interneurons (70%) recorded remained unchanged after quinpirole, resembling the response observed in the PFC of prepubertal rats, whereas others showed a decrease in excitability.
NVHL causes the loss of a GABA component contributing to the D2-mediated inhibition of PFC pyramidal neuron excitatory synaptic response
The attenuation of local excitatory inputs to PFC pyramidal neurons by quinpirole also acquires a mature profile during adolescence attributable to the emergence of a D2-dependent enhancement of interneuron excitability (Tseng and O'Donnell, 2007a). If the D2 modulation of PFC interneurons is compromised in NVHL animals, such late component would not be present in late adolescent or adult PFC slices. We examined the effect of quinpirole on synaptic responses evoked in pyramidal neurons by stimulation of layers I–II in slices from sham and NVHL rats. All pyramidal neurons included here were labeled with Neurobiotin to confirm their location and morphology. Stimulation of superficial layers with an electrode placed ∼1 mm lateral to the axis of the apical dendrite of the recorded neuron evoked a fast depolarizing AMPA-mediated EPSP. The monosynaptic nature of the evoked response was identified by the short and constant latency (∼5 ms) to postsynaptic potential onset. The AMPA antagonist CNQX eliminated the evoked response (data not shown). In similar experiments performed in slices from naive rats, the evoked response remained unchanged after perfusing the slice with the GABAA antagonists picrotoxin or bicuculline, indicating absence of a detectable fast GABAergic component in this response (Tseng and O'Donnell, 2007a). Bath application of quinpirole (2 μm) reduced EPSP amplitude by ∼20%, in both postpubertal sham (n = 6) and NVHL (n = 7) rats (Fig. 5B,C). As reported previously (Tseng and O'Donnell, 2007a), this effect was completely blocked with the D2 receptor antagonist eticlopride (n = 7), confirming that quinpirole-induced synaptic attenuation is mediated by D2 receptors (data not shown). However, the duration of this inhibition was significantly longer in sham rats compared with the lesioned group (Fig. 5D). Pyramidal neurons recorded from NVHL rats recovered to baseline amplitude by ∼10 min after quinpirole was removed, whereas a washout period of at least 25 min was required in the sham group. This persistent post-quinpirole effect in sham rats could be reduced to ∼10 min with the GABAA antagonist picrotoxin (10 μm; n = 6). The data suggest that part of the inhibitory action of D2 receptors on pyramidal neuron excitatory transmission is also compromised in the PFC of late adolescent and adult NVHL rats, particularly the activation of local GABAergic interneurons.
Figure 5.
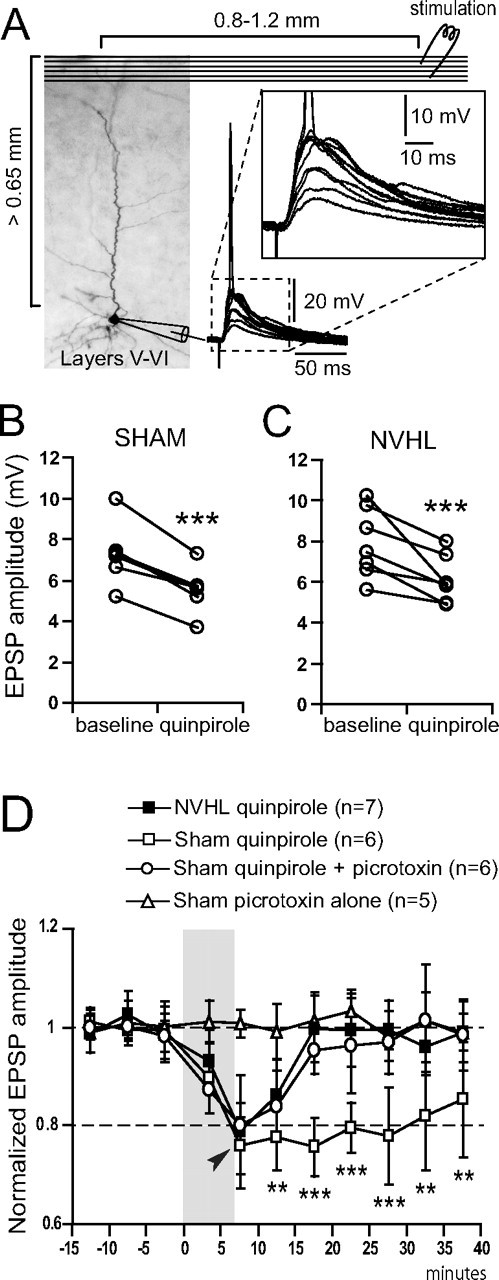
Loss of a D2-dependent GABA modulation of synaptic responses of pyramidal neurons in NVHL rats. A, Electrical stimulation of layers I–II in the medial PFC of NVHL elicited a primarily AMPA-dependent EPSP in deep layer pyramidal neurons. Diagram illustrating the arrangement of stimulating electrode (top right; layers I–II) and recording sites (bottom left; layers V–VI), along with an overlay of EPSPs recorded in the pyramidal neuron. Inset shows EPSPs at higher magnification. B, Diagram illustrating the decrease in EPSP amplitude by quinpirole (2 μm) in slices from sham rats. C, Plot illustrating the effects of quinpirole in slices from NVHL rats. D, Plot summarizing the time course of these effects. The D2-dependent synaptic attenuation in pyramidal neurons was persistent in the PFC of sham rats (open squares) but not in NVHL rats (filled squares). The GABAA antagonist picrotoxin blocked the persistent component of this D2 effect in slices from sham rats (open circles) but did not affect the response when given alone (open triangles). Thus, the D2 modulation of corticocortical synaptic responses in adult NVHL rats lacks the recruitment of interneurons. *p < 0.05, **p < 0.005, ***p < 0.0005; quinpirole versus quinpirole plus picrotoxin.
Discussion
Although adolescent and adult rats with an NVHL did not show reduced levels of GAD67 or PV, their PFC interneurons had abnormal dopaminergic modulation. The D2 agonist quinpirole failed to increase cell excitability in FSIs and NFS interneurons from adult rats with an NVHL, and, in many cases, the neurons showed an abnormal decrease in excitability. The lack of activation of GABA interneurons by quinpirole was also reflected in the absence of a late, GABAA-dependent component in the D2 modulation of synaptic responses within the PFC in slices from NVHL rats. The data suggest that the periadolescent maturation of interneuron function is altered in NVHL rats, even if there is no actual loss of this cell population.
The use of a lesion as a schizophrenia model does require some clarification. If the model has any claim to construct validity, it cannot reside in the lesion as schizophrenia patients to not present a lesion. Because many studies suggest an abnormal PFC in NVHL rats and inactivating the medial PFC reverses behavioral and electrophysiological deficits (Lipska et al., 1998; Goto and O'Donnell, 2004), it is possible that the critical aspect of the NVHL model is an altered PFC function. Our data indicate that the periadolescent maturation of PFC interneurons becomes abnormal as a consequence of the lesion; therefore, we propose that the construct validity in this model arises from the cortical hyperactivity it causes via impaired inhibitory mechanisms. These changes could alter the spatial selectivity of PFC neuronal response to excitatory inputs by setting inappropriate coordination between pyramidal neurons and GABAergic interneurons, which in turn may be associated with the abnormal behavioral performance in NVHL animals.
The deficits in PFC interneurons we observed in slices from NVHL rats are functional. There were no differences in PV and GAD67 mRNA expression between lesion and sham rats, suggesting that the full complement of GABA neurons, and of PV-containing FSIs in particular, are present in the lesion animals. This finding contrasts with a previous report of reduced GAD67 in the PFC of NVHL rats (Lipska et al., 2003). It is possible that the lesion affects PFC interneurons, rendering them into a vulnerable state. Because the expression of both GAD67 and PV is activity dependent, it could change with certain conditions (e.g., housing status of the animal and other factors). Many developmental models have reported reductions in PV and/or GAD67 without actual interneuron loss (Pillai-Nair et al., 2005; Cabungcal et al., 2006). Although neither the total number of PFC neurons (Thune et al., 2001) nor the densities of PV-immunoreactive (Woo et al., 1997) and PV mRNA-containing (Hashimoto et al., 2003) neurons are reduced in the PFC in schizophrenia, only 55% of PV mRNA-positive neurons expressed detectable levels of GAD67 mRNA (Hashimoto et al., 2003). Together, these findings suggest that the interneurons may be present but do not express the normal profile of GABA-related markers. Thus, because these markers are activity dependent, they may be reduced to different extents depending on the condition of the system. The altered modulation by the D2 agonist without detectable changes in GAD67 or PV mRNA expression reported here makes a strong case for abnormal functionality in PFC circuits in this developmental model.
Why does an early hippocampal alteration affect postnatal maturation of PFC interneurons? Although mechanisms can only be speculated on at this time, evidence suggests that altered prefrontal cortical maturation could be attributable to an early downregulation of neurotrophic factors that may depend on hippocampal inputs. First, unpublished observations from our laboratory using DiI injections into the early postnatal ventral hippocampus indicate that the time at which the lesions were done here is when hippocampal fibers are arriving into the PFC, suggesting that both formation of new synapses and the impact of trophic factors released by these fibers may be altered in NVHL. Indeed, the NVHL reduces cortical levels of BDNF (Lipska et al., 2001) and nurr77 (Bhardwaj et al., 2003). Any detrimental effect the impairment of hippocampal inputs may have on PFC circuitry may not be evident until interneurons become fully functional. Our recent work revealed that responses of PFC interneurons to dopamine do not mature until late adolescence (Tseng and O'Donnell, 2007b), which may explain the delayed onset of prefrontal deficits in NVHL rats.
The cellular mechanisms responsible for the abnormal D2 response in NVHL interneurons are not clear. Although speculative, the abnormal D2 response could be attributable to altered expression of D2-like (i.e., D4) receptors, to recruitment of a different type of G-protein (Gq vs Gi), or to changes in the heterodimerization of receptors in these neurons. DA (in particular D1 and D4) receptor expression changes postnatally in rodents (Tarazi and Baldessarini, 2000) and humans (Weickert et al., 2007). Furthermore, the activity of catechol-O-methyl transferase, the enzyme responsible for inactivating DA in the PFC, also increased postnatally into the young adult ages (Tunbridge et al., 2007). More research is needed to unveil the mechanisms that may cause abnormal maturation of interneurons in NVHL animals.
The loss of functionality in cortical interneurons seems to be a common finding in schizophrenia research. Individuals with schizophrenia do present reduced GABA-related transcripts (Hashimoto et al., 2008b), and clinical neurophysiology studies reveal abnormal patterns associated with poor interneuron performance, such as a loss or reduction in evoked or induced gamma oscillations (Spencer et al., 2003, 2008; Hong et al., 2004; Cho et al., 2006; Ferrarelli et al., 2008). Similar loss of beta/gamma frequency bands is observed with noncompeting NMDA antagonists (Roopun et al., 2008), a model known to target primarily interneuron function (Homayoun and Moghaddam, 2007). Many of those findings revealed absence of high-frequency oscillations that were task dependent. Our finding that abnormal interneuron function is evident when DA agonists are applied implies that, in NVHL rats, when DA cells fire in response to unexpected reward or reward-predicting stimuli (Schultz et al., 1993), the balance between excitation and inhibition in PFC circuits will be altered. Thus, the lack of activation of interneurons during brief high-DA epochs (typically related to the presence of salient stimuli) would contribute to noisy information processing in the PFC and poor assignment of saliency to sensory information associated with the surge in DA as well as impaired working memory functions. Because the symptoms of schizophrenia are broad, it is conceivable that whether cognitive deficits or positive or negative symptoms dominate depends on the cortical region primarily affected by the developmental alteration of interneurons. The cortical functional deficits in NVHL rats do bear some resemblance to current views on schizophrenia pathophysiology, and what makes this model appealing is the emergence of these deficits during adolescence. In summary, the altered periadolescent maturation of PFC interneurons in NVHL rats may reproduce the impact of deleterious combination of predisposing genes and environmental factors that may be present in schizophrenia.
Footnotes
This work was supported by National Institutes of Health Grants MH57683 (P.O.) and MH51234 (D.A.L.). We thank Katrina Emmerich and Holly Bazmi for excellent technical assistance.
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res. 1997;24:349–355. doi: 10.1016/s0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Benes FM. Evidence for altered trisynaptic circuitry in schizophrenic hippocampus. Biol Psychiatry. 1999;46:589–599. doi: 10.1016/s0006-3223(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- Bhardwaj SK, Beaudry G, Quirion R, Levesque D, Srivastava LK. Neonatal ventral hippocampus lesion leads to reductions in nerve growth factor inducible-B mRNA in the prefrontal cortex and increased amphetamine response in the nucleus accumbens and dorsal striatum. Neuroscience. 2003;122:669–676. doi: 10.1016/j.neuroscience.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Brake WG, Sullivan RM, Flores G, Srivastava LK, Gratton A. Neonatal ventral hippocampal lesions attenuate the nucleus accumbens dopamine response to stress: an electrochemical study in the adult rat. Brain Res. 1999;831:25–32. doi: 10.1016/s0006-8993(99)01477-8. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Nicolas D, Kraftsik R, Cuénod M, Do KQ, Hornung JP. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: relevance to schizophrenia. Neurobiol Dis. 2006;22:624–637. doi: 10.1016/j.nbd.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, Goldberg TE, Weinberger DR. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex. 2000;10:1078–1092. doi: 10.1093/cercor/10.11.1078. [DOI] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17:3894–3906. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance SA, Walker M, Crow TJ. Reduced density of calbindin-immunoreactive interneurons in the planum temporale in schizophrenia. Brain Res. 2005;1046:32–37. doi: 10.1016/j.brainres.2005.03.045. [DOI] [PubMed] [Google Scholar]
- Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci U S A. 2006;103:19878–19883. doi: 10.1073/pnas.0609440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- de Lecea L, del Río JA, Soriano E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Mol Brain Res. 1995;32:1–13. doi: 10.1016/0169-328x(95)00056-x. [DOI] [PubMed] [Google Scholar]
- Endo K, Hori T, Abe S, Asada T. Alterations in GABA(A) receptor expression in neonatal ventral hippocampal lesioned rats: comparison of prepubertal and postpubertal periods. Synapse. 2007;61:357–366. doi: 10.1002/syn.20393. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Massimini M, Peterson MJ, Riedner BA, Lazar M, Murphy MJ, Huber R, Rosanova M, Alexander AL, Kalin N, Tononi G. Reduced evoked gamma oscillations in the frontal cortex in schizophrenia patients: a TMS/EEG study. Am J Psychiatry. 2008;165:996–1005. doi: 10.1176/appi.ajp.2008.07111733. [DOI] [PubMed] [Google Scholar]
- Flagstad P, Mørk A, Glenthøj BY, van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- Frankle WG, Cho RY, Narendran R, Mason NS, Vora S, Litschge M, Price JC, Lewis DA, Mathis CA. Tiagabine increases [(11)C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology. 2008 doi: 10.1038/npp.2008.104. Advance online publication. Retrieved July 9, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Goto Y, O'Donnell P. Prefrontal lesion reverses abnormal mesoaccumbens response in an animal model of schizophrenia. Biol Psychiatry. 2004;55:172–176. doi: 10.1016/s0006-3223(03)00783-2. [DOI] [PubMed] [Google Scholar]
- Gourevitch R, Rocher C, Le Pen G, Krebs MO, Jay TM. Working memory deficits in adult rats after prenatal disruption of neurogenesis. Behav Pharmacol. 2004;15:287–292. doi: 10.1097/01.fbp.0000135703.48799.71. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 5. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, Sun Z, Sampson AR, Lewis DA. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 2005;25:372–383. doi: 10.1523/JNEUROSCI.4035-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry. 2008a;165:479–489. doi: 10.1176/appi.ajp.2007.07081223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, Unger T, Maldonado-Avilés JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008b;13:147–161. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andradé M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong LE, Summerfelt A, McMahon RP, Thaker GK, Buchanan RW. Gamma/beta oscillation and sensory gating deficit in schizophrenia. Neuroreport. 2004;15:155–159. doi: 10.1097/00001756-200401190-00030. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phenciclidine model of schizophrenia. Am J Psychiat. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49:603–615. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Koenig JI, Elmer GI, Shepard PD, Lee PR, Mayo C, Joy B, Hercher E, Brady DL. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res. 2005;156:251–261. doi: 10.1016/j.bbr.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O'Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D1 dopamine receptors. Cereb Cortex. 2000;10:1168–1175. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- Lewis DA. Neural circuitry of the prefrontal cortex in schizophrenia. Arch Gen Psychiat. 1995;52:269–273. doi: 10.1001/archpsyc.1995.03950160019004. discussion 277–278. [DOI] [PubMed] [Google Scholar]
- Lewis DA, González-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpuberal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, al-Amin HA, Weinberger DR. Excitotoxic lesions of the rat medial prefrontal cortex. Effects on abnormal behaviors associated with neonatal hippocampal damage. Neuropsychopharmacology. 1998;19:451–464. doi: 10.1016/S0893-133X(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Khaing ZZ, Weickert CS, Weinberger DR. BDNF mRNA expression in rat hippocampus and prefrontal cortex: effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur J Neurosci. 2001;14:135–144. doi: 10.1046/j.1460-9568.2001.01633.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia: effects of antipsychotic drugs. Eur J Neurosci. 2003;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Manoach DS. Prefrontal cortex dysfunction during working memory performance in schizophrenia: reconciling discrepant findings. Schizophr Res. 2003;60:285–298. doi: 10.1016/s0920-9964(02)00294-3. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci U S A. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P. Increased cortical excitability as a critical element in schizophrenia pathophysiology. In: O'Donnell P, editor. Cortical deficits in schizophrenia: from genes to function. New York: Springer; 2008. pp. 219–236. [Google Scholar]
- O'Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. San Diego: Academic; 1998. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci. 2006;23:279–284. doi: 10.1111/j.1460-9568.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- Pillai-Nair N, Panicker AK, Rodriguiz RM, Gilmore KL, Demyanenko GP, Huang JZ, Wetsel WC, Maness PF. Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J Neurosci. 2005;25:4659–4671. doi: 10.1523/JNEUROSCI.0565-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopun AK, Cunningham MO, Racca C, Alter K, Traub RD, Whittington MA. Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophr Bull. 2008;34:962–973. doi: 10.1093/schbul/sbn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Apicella P, Ljungberg T. Responses of monkey dopamine neurons to reward and conditioned stimuli during successive steps of learning a delayed response task. J Neurosci. 1993;13:900–913. doi: 10.1523/JNEUROSCI.13-03-00900.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME, McCarley RW. Abnormal neural synchrony in schizophrenia. J Neurosci. 2003;23:7407–7411. doi: 10.1523/JNEUROSCI.23-19-07407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Niznikiewicz MA, Shenton ME, McCarley RW. Sensory-evoked gamma oscillations in chronic schizophrenia. Biol Psychiatry. 2008;63:744–747. doi: 10.1016/j.biopsych.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Lorincz A, Tamás G. Beta and gamma frequency synchronization by dendritic gabaergic synapses and gap junctions in a network of cortical interneurons. J Neurosci. 2001;21:5824–5831. doi: 10.1523/JNEUROSCI.21-15-05824.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazi FI, Baldessarini RJ. Comparative postnatal development of dopamine D(1), D(2) and D(4) receptors in rat forebrain. Int J Dev Neurosci. 2000;18:29–37. doi: 10.1016/s0736-5748(99)00108-2. [DOI] [PubMed] [Google Scholar]
- Thune JJ, Uylings HB, Pakkenberg B. No deficit in total number of neurons in the prefrontal cortex in schizophrenics. J Psychiatr Res. 2001;35:15–21. doi: 10.1016/s0022-3956(00)00043-1. [DOI] [PubMed] [Google Scholar]
- Traub RD, Kopell N, Bibbig A, Buhl EH, LeBeau FE, Whittington MA. Gap junctions between interneuron dendrites can enhance synchrony of gamma oscillations in distributed networks. J Neurosci. 2001;21:9478–9486. doi: 10.1523/JNEUROSCI.21-23-09478.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. D2 dopamine receptors recruit a GABA component for their attenuation of excitatory synaptic transmission in the adult rat prefrontal cortex. Synapse. 2007a;61:843–850. doi: 10.1002/syn.20432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007b;17:1235–1240. doi: 10.1093/cercor/bhl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunbridge EM, Weickert CS, Kleinman JE, Herman MM, Chen J, Kolachana BS, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase enzyme activity and protein expression in human prefrontal cortex across the postnatal lifespan. Cereb Cortex. 2007;17:1206–1212. doi: 10.1093/cercor/bhl032. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Webster MJ, Gondipalli P, Rothmond D, Fatula RJ, Herman MM, Kleinman JE, Akil M. Postnatal alterations in dopaminergic markers in the human prefrontal cortex. Neuroscience. 2007;144:1109–1119. doi: 10.1016/j.neuroscience.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Woo TU, Miller JL, Lewis DA. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry. 1997;154:1013–1015. doi: 10.1176/ajp.154.7.1013. [DOI] [PubMed] [Google Scholar]