

Abstract
Background:
PINK1 loss-of-function causes recessive, early-onset parkinsonism. In Tunisia there is a high rate of consanguineous marriage but PINK1 carrier frequency and disease prevalence have yet to be assessed.
Objectives:
The frequency of PINK1 mutations in familial parkinsonism, community-based patients with idiopathic Parkinson disease (PD) (non-familial PD), and control subjects was determined. Demographic and clinical characteristics of individuals with PINK1 homozygous or heterozygous variants, or without PINK1 mutations, were compared.
Methods:
A total of 92 kindreds (with 208 affected and 340 unaffected subjects), 240 nonfamilial PD, and 368 control participants were recruited from the Institut National de Neurologie, Tunis. Clinical examinations included Hoehn &Yahr, UPDRS, and Epworth scales. PINK1 sequencing and dosage analysis was performed in familial index patients, the variants identified screened in all subjects. Parkin and LRRK2 genes were also examined.
Results:
Four PINK1 homozygous mutations, three novel (Q129X, Q129fsX157, G440E, and one previously reported; Q456X), segregate with parkinsonism in 46 individuals in 14 of 92 families (15%). Six of 240 patients with nonfamilial PD were found with either homozygous Q456X or Q129X (2.5%) substitutions. In patients with familial disease, PINK1 homozygotes were younger at disease onset (36 ± 12 years) than noncarriers (57 ± 15 years) and more often had an akinetic-rigid presentation at examination and slow progression.
Conclusions:
Segregation of PINK1 mutations with parkinsonism within families, and frequency estimates within population controls, suggested only four PINK1 mutations were pathogenic. Several PINK1 sequence variants are potentially benign and there was no evidence that PINK1 heterozygosity increases susceptibility to idiopathic Parkinson disease.
GLOSSARY
- AAO
= age at onset;
- CRF
= case report forms;
- ET
= essential tremor;
- PD
= Parkinson disease;
- UPDRS
= Unified Parkinson's Disease Rating Scale.
Parkinson disease (PD) is a neurodegenerative syndrome with a complex, multifactorial etiology, although causal mutations in several genes have been implicated in families with a Mendelian pattern of disease inheritance.1 The PARK6 locus was linked to chromosome 1p35-36 in a Sicilian kindred2 and mutations in the PTEN induced putative kinase 1 (PINK1) were subsequently identified.3 PINK1 encodes a mitochondrial serine-threonine protein kinase4 and is one of three genes including parkin (PRKN) and DJ-1 implicated in autosomal recessive, early-onset forms of parkinsonism.
The frequency of neurodegenerative disorders including idiopathic PD is higher in Tunisia than in most other countries.5 In this region unique geographic and sociocultural factors, including large pedigrees, low rates of migration, and high rates of consanguinity, facilitate genotype-phenotype correlations in genetic disease.
Current findings originated as a genome-wide linkage study of familial parkinsonism in Tunisia that identified a significant linkage peak on chromosome 1p35-36. The PINK1 gene was subsequently sequenced and the segregation and frequency of any variants identified were examined within families, nonfamilial PD, and control subjects. The clinical characteristics of individuals with and without PINK1 mutations are described.
METHODS
Study population.
The Institut National de Neurologie, Tunis, provides a specialized neurologic service to the entire country of Tunisia.6 A total of 92 families were recruited, comprised of 76 multiplex kindreds which include clinical data and DNA samples from 208 patients with parkinsonism, 340 unaffected subjects, and 27 with an uncertain diagnosis. The remaining 16 familial index cases (singleton families) were derived from pedigrees consisting of affected siblings, parent-offspring pairs, avuncles, first cousins, or half-first cousins once removed. In these 16 kindreds DNA was only available from one affected subject and unaffected family members. Subjects in the study also included 240 nonfamilial patients with PD and 368 control participants. The site obtained local ethics committee approval before beginning subject recruitment. Subjects were informed of all aspects pertaining to their participation in the study, and gave either written or proxy consent, prior to their inclusion.
Physical examinations were performed by neurologists specialized in movement disorders. Patients with PD and control subjects without a family history of parkinsonism were collected from all regions of Tunisia, through the Institut National de Neurologie, Tunis. For our family-based study the proband was examined at the study site, and additional family members were recruited via the proband. Inclusion criteria were age at assessment older than 18 years, with at least one other affected first- to third-degree blood relative (excluding a monozygotic twin).
Individuals were diagnosed as “affected” if they satisfied the United Kingdom Parkinson's Disease Society Brain Bank criteria,7 “unaffected” if all signs of parkinsonism were absent, “controls” if all signs of parkinsonism were absent and there was no family history of parkinsonism, and “uncertain” if only one parkinsonian sign or abnormal feature was present. Most of the latter subjects were diagnosed with essential tremor (ET). Standardized case report forms (CRF) were used for clinical and demographic data collection and included data on Hoehn and Yahr staging,8 the Unified Parkinson's Disease Rating Scale (UPDRS)9, and the Epworth scale.10 Approximately half of the patients (105/208) completed a simplified CRF including section III of the UPDRS but without Hoehn and Yahr staging. Duration of disease was calculated by subtracting the age at onset (AAO) in medical records from the age at examination. For a minority of patients AAO was not documented (n = 42/208, primarily as past medical records were not available for 33/208 [16%] of affected family members), and was estimated either from their clinical history or by self-report. Confirmatory diagnoses of 35 affected members from 18 pedigrees with familial parkinsonism and 6 patients with nonfamilial PD were performed by independent movement disorders specialists and were completely concordant.
Mutation detection.
DNA was extracted by standard procedures from a peripheral venous blood sample.11 The original genome scan was carried out using 1116 microsatellite markers spaced an average of 4 centiMorgans across the genome.12 MERLIN was used for nonparametric multipoint linkage analysis.13
All eight PINK1 exons were subsequently sequenced in one familial index case from each of the families recruited. Quantitative analysis of PINK1 exon dosage was also performed by real-time PCR, controlled using a DNA sample hemizygous for chromosome 1p35-36,14 a gift from Dr. Enza-Maria Valente (CSS-Mendel Institute, Rome, Italy). ABI probes were designed for the 14 nonsynonymous coding changes that were found in <10% of sequenced individuals. These probes were then run through all remaining family members, 240 nonfamilial patients and 372 control subjects. Parkin (PRKN) sequencing and dosage analysis was also performed for all exons for any families with PINK1 homozygous mutations using published methods.15,16 In addition, LRRK2 point mutations including 6055G>A (G2019S) were assayed as previously described.17
RESULTS
The linkage analysis included 69 multiplex families with 174 affected individuals genotyped at <5 cM resolution. The maximum lod score on chromosome 1 was 5.7 from multipoint nonparametric linkage analysis. The lod-1 interval spanned 7 MB from D1S199 to D1S2749 which corresponds to the physical position of the PINK1 locus. To determine if the linkage signal was due to mutations within PINK1, the gene was successfully sequenced in 89 familial index cases. Fourteen nonsynonymous coding changes were identified and assessed within other family members from 92 families (76 multiplex families and an additional 16 singleton families), 240 nonfamilial patients with PD and 368 control subjects (table 1). Eight substitutions have been identified previously, including Q115L, P196L, A340T, A383T, G411S, E476K, Q456X, and N521T. Novel variants identified in this study included Q129X, Q129fsX157, T145M, R152W, G227R, and G440E. Of these changes, Q129X, Q129fsX157, G440E, and Q456X appear to be pathogenic as they were found in a homozygous state in 46 out of 50 (92%) of the affected individuals within 14 families (figure). Excluding these kindreds from linkage analysis abolished the chromosome 1p35-36 signal.
Table 1 Frequency of PINK1 mutations in Parkinson disease family probands, nonfamilial patients, and control subjects
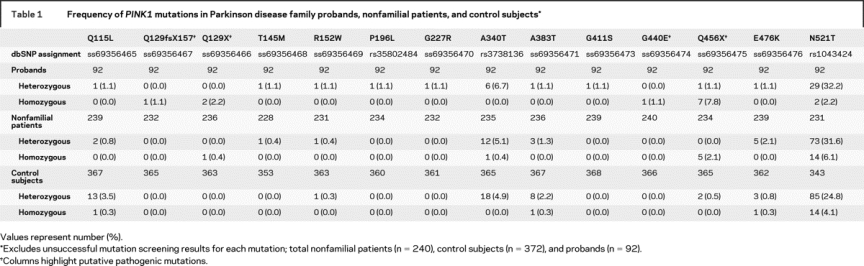
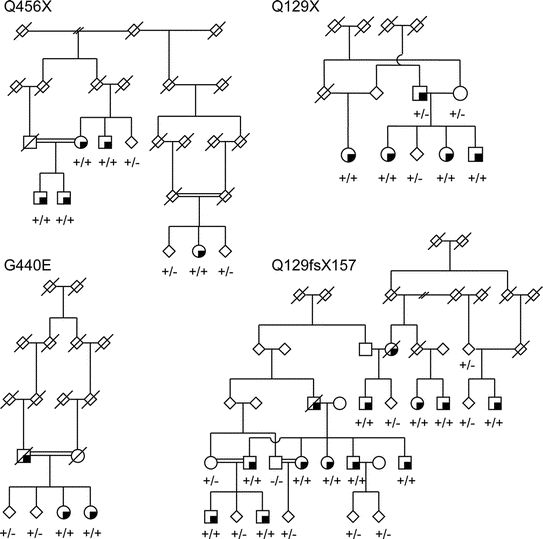
Figure Four pedigrees are shown representative of families with homozygous PINK1 Q129X, Q129fsX157, G440E, or Q456X mutations
Squares represent males, circles are females, diamonds are where gender has been disguised. Deceased individuals are shown with a diagonal line, affected subjects have a quarter-filled quadrant, and consanguineous marriages have a double horizontal line between parents. +/+ and +/− represent homozygous and heterozygous PINK1 mutation carriers.
Evidence for pathogenicity of the other 10 PINK1 mutations was equivocal. One coding substitution N521T was common (heterozygous frequency of 24.8%, homozygous frequency 4.1% in controls) and did not segregate with parkinsonism in the families. This was excluded from further analyses of the demographic and clinical characteristics of PINK1 carriers. Other substitutions were uncommon (<5% frequency) in controls and generally found in a heterozygous state with the exception of homozygous inheritance of Q115L, A383T, and E476K in three control subjects aged 64, 49, and 49 years at examination. Nonfamilial patients were also identified with homozygous substitutions Q129X (n = 1, AAO 38 years) and Q456X (n = 5, average AAO 38 ± 9) which are putatively pathogenic.
Demographic characteristics are reported for the affected and unaffected members of families with parkinsonism, nonfamilial patients with PD, and control subjects, stratified by PINK1 status (table 2). All participants were of Arab-Berber ethnicity with the exception of one family which originated from Turkey and two from Southern Europe. All PINK1 mutations were in individuals of Arab-Berber descent. Within families, unaffected individuals with homozygous PINK1 mutations were marginally younger at examination (34 ± 5 years) than the onset age of affected homozygous PINK1 individuals (36 ± 12 years); therefore some may yet develop parkinsonism.
Table 2 Demographic characteristics of subjects focused on four putatively pathogenic PINK1 mutations
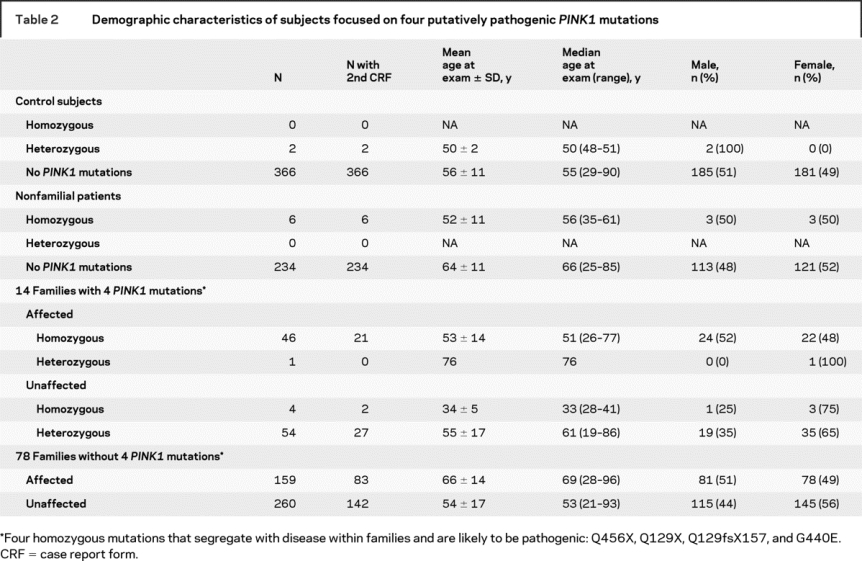
Subsequent analyses focus on subjects with pathogenic mutations, Q129X, Q129fsX157, G440E, and Q456X. Clinical characteristics of patients with parkinsonism with PINK1 homozygous and heterozygous mutations were compared to those without PINK1 variants (table 3). The mean age at onset of parkinsonism in familial PINK1 homozygous patients (36 ± 12) was younger than in familial PD with heterozygous PINK1 mutations (69 ± 8 years, Wilcoxon rank sum test p < 0.005) or affected family members without any of the four potentially pathogenic PINK1 mutations (57 ± 15 years, Wilcoxon rank sum test p < 0.0001). The mean age at onset of PD in six individuals with nonfamilial PD and PINK1 homozygous mutations (38 ± 9 years) was younger than in nonfamilial PD without any of the four mutations (58 ± 12 years, Wilcoxon rank sum test p < 0.001).
Table 3 Characteristics of patients with Parkinson disease (PD) with PINK1 homozygous and heterozygous mutations compared to those without PINK1 mutations
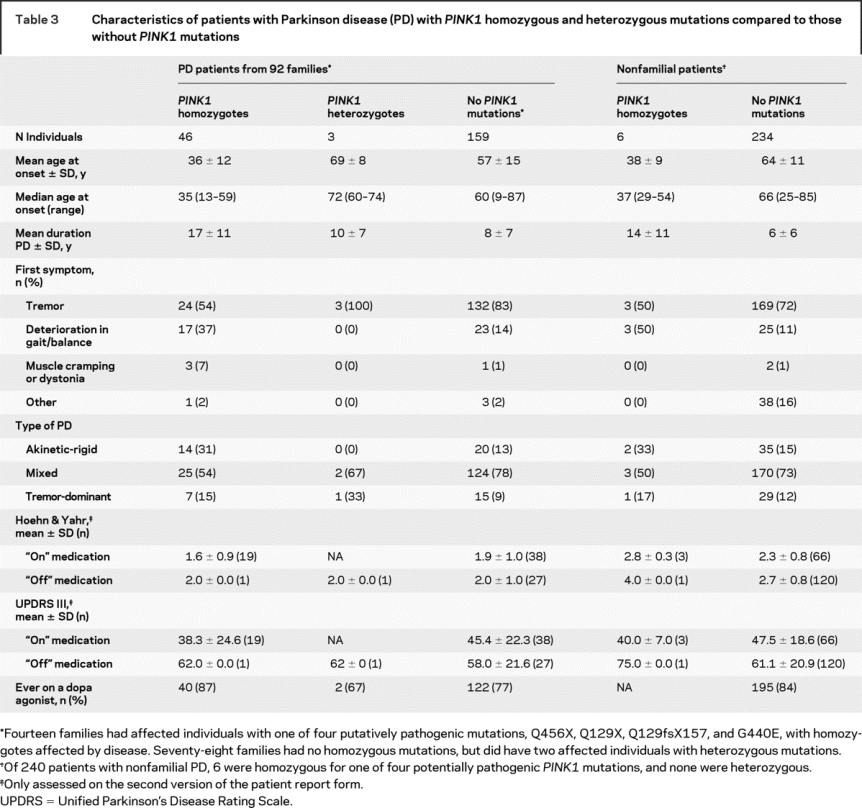
In familial patients with homozygous PINK1 mutations, in contrast to patients without any PINK1 mutation, the duration of parkinsonism was longer (Wilcoxon rank sum test p < 0.0001) and Hoehn and Yahr scores were not significantly different (table 3). The notable exception were higher mean Hoehn & Yahr scores in nonfamilial patients with homozygous PINK1 mutations and with a very long duration of disease, although the sample size was too small for significance (n = 6, p = 0.1). The majority of familial patients without PINK1 mutations recalled tremor as a first feature (83%) whereas gait or balance problems were infrequent (14%). In contrast, familial PINK1 homozygotes noted their first symptom as either tremor (54%) or deterioration in gait or balance (37%). At examination, the type of parkinsonism in most affected individuals were classified as mixed, with both rigidity and tremor present, but PINK1 homozygotes more often had akinetic-rigid parkinsonism (31%), compared to heterozygotes (0%) or individuals without PINK1 mutations (13%). Clinical differences on first presentation and subtype at examination between PINK1 homozygotes and PINK1 heterozygote or patients without PINK1 mutations were statistically significant (p < 0.0002). Data on cognition and dementia are limited to neurologist observations as more than 60% of the study population was unable to read and therefore their MMSE scores were not valid.18
Some families carried more than one PINK1 sequence variation. One familial affected individual (onset age 74) was a compound heterozygote for both Q129X and A340T substitutions; another familial patient (onset age 60) was a compound heterozygote for Q456X and A383T, whereas two unaffected individuals (ages 44 and 69) from two families were compound heterozygous for both Q456X and A340T. In two of the eight families with the Q456X mutation, the proband did not screen positive for the mutation but it was identified in other family members.
Within the 14 families with PINK1 mutations, PRKN sequencing and dosage analysis was performed and LRRK2 point mutations including 6055G>A (G2019S) were assayed. There are seven heterozygous PINK1 carriers who also inherited either M192L (n = 2), P153R (n = 4), or D394N (n = 1) heterozygous PRKN substitutions. However, all were unaffected with a mean age at examination of 54 ± 19, range 26–73 years. Of three homozygous PINK1 carriers with heterozygous P153R PRKN substitutions, two were affected at 30 and 36 years while the other remains unaffected at examination at 35 years. In one family, one affected (age 56 years at onset) and one unaffected individual (age 62 years at examination) also carried a heterozygous Lrrk2 G2019S substitution.
DISCUSSION
This report describes the largest group of PINK1 carriers identified to date and all are from a single population background. A total of 14 PINK1 variants were found in Tunisian patients with familial parkinsonism. Of these four PINK1 mutations, Q129X, Q129fsX157, G440E, and Q456X, were found to segregate with disease and were homozygous in affected individuals within 14 families. Three of these are novel substitutions. All four substitutions were rarely found in 368 control subjects and never in a homozygous state. Segregation of parkinsonism within these 14 families explains the linkage signal on chromosome 1p35-36 and provides genetic evidence that Q129X, Q129fsX157, G440E, or Q456X inherited in a recessive homozygous fashion are pathogenic. Two of these four mutations, Q129X and Q456X, were also present in patients with seemingly nonfamilial PD (Q129X 0.4%, Q456X 2.1%). Of the other 10 variants N521T is a common polymorphism but the rest are uncommon; within nonfamilial patients with PD only A340T was found in a homozygous state, and homozygous Q115l, A383T, and E476K substitutions were identified in control subjects.
Our subsequent analysis focused on the four pathogenic homozygous PINK1 substitutions identified in 14 families, as other mutations are rare, do not segregate with disease within pedigrees, and may be benign. Overall, PINK1 homozygotes had a younger mean onset age than heterozygotes and those without any of the four potentially pathogenic PINK1 mutations. However, despite longer disease duration, familial PINK1 homozygotes did not show greater symptom severity based on Hoehn & Yahr or UPDRS III scales. PINK1 homozygotes were less likely to have tremor as a first symptom or at examination than other patients with PD, and deterioration in gait and balance were noted more frequently; PINK1 homozygous patients were also more likely to develop akinetic-rigid parkinsonism, compared with other patients with PD. In idiopathic PD, deterioration in gait and balance are often associated with a faster rate of cognitive decline and may be considered a risk factor for incident dementia.19 Cognitive decline and dementia were difficult to gauge but were not pronounced within the 14 families with PINK1 substitutions. PINK1 heterozygous mutations may be associated with the presence of psychosis20; however, while psychiatric variables were assessed through a self-report of medical history, there were a large number of missing responses. Further study of cognitive and psychiatric aspects of PINK1 parkinsonism may be worthwhile.
In the literature, it remains controversial whether PINK1 heterozygous mutations are a risk factor for idiopathic PD.21,22 Cross-sectional imaging studies provide support that PINK1 haploinsufficiency may contribute to nigrostriatal dysfunction but it is not clear whether the impairment is developmental or the consequence of a slowly progressive neurodegenerative process.23 Similarly, many studies have suggested PRKN heterozygous mutations are a risk factor for idiopathic PD, but community-based studies remain equivocal.16,24 In our study, of the many heterozygous PINK1 carriers with one of four pathogenic mutations (including parents of homozygous affected individuals who must be at least obligate heterozygotes) few had parkinsonism (table 2). This suggests that heterozygote mutations may not be a risk factor for PD, at least within the age range that this study was able to sample.
In good agreement with our observations, functional data from RNAi knockdown of PINK1 expression in mice to less than 10% of the endogenous gene and protein does not lead to dopaminergic neuronal death or dysfunction, whereas targeted, complete germ-line deletion of PINK1 can lead to impaired dopamine release and reduced synaptic plasticity in the striatum.25,26 In Drosophila removal of the PINK1 homologue results in a more dramatic albeit nondopaminergic phenotype, including male sterility, apoptotic muscle degeneration, and mitochondrial fragmentation that may be rescued by parkin overexpression suggesting PINK1 and parkin function, at least in part, in the same pathway.27 In our study there was no evidence for a joint effect of PINK1 and PRKN on disease susceptibility or age at onset although the number of subjects with mutations in both genes was limited. Nevertheless, it will be important to elucidate the potential interactions between these proteins and the convergent pathways in which they function.
The frequency of PINK1 mutations described may be atypical and limited to the Arab-Berber sample. However, as clinical studies of the PRKN gene illustrate, genetic mutations generally have the same effects in different ethnicities.28 Testing for PINK1 mutations in patients in Tunisia may have practical utility in diagnosis and patient care. Within affected kindreds, PINK1 carrier status for pathogenic mutations might contribute to genetic counseling. However, it is important to note that not all PINK1 coding variants are pathogenic, only when inherited in a homozygous state, and that the majority of mutations identified appear benign. Similarly, within families or within the general community, there was no evidence that PINK1 heterozygous carriers are at increased risk of idiopathic PD.
Affected and symptomatic individuals within the PINK1 pedigrees described may now enable biomarker identification and validation. Assuming findings are generalizable such biomarkers would facilitate the design of future neuroprotection (delayed disability) trials in idiopathic PD, and the development of novel therapeutic strategies aimed at more than symptomatic relief.
ACKNOWLEDGMENT
The authors thank the patients and their families, GSK PD Programme Team, GSK Bioinformatics; Siwan Oldham for managing the study site; Mark Hall, Donna Backshall, Rodney Winkler, Santhi Subramanian, and Link McGaughey for software and database support; Tina Stapleton and Carole Stapleton for sample management; Allen Roses for project guidance; and David Burn, Jina Swartz, and Ray Watts for their neurologic expertise.
Address correspondence and reprint requests to Dr. Matthew J. Farrer, Department of Neuroscience, Division of Neurogenetics, Mayo Clinic Florida, Birdsall Building, 4500 San Pablo Road, Jacksonville, Florida 32224 farrer.matthew@mayo.edu
e-Pub ahead of print on August 6, 2008, at www.neurology.org.
GlaxoSmithKline financially supported the patient recruitment and initial genetic analysis. Molecular genetic analysis was supported by the Neurogenetic Core of a Morris K. Udall Center (National Institute of Neurological Disorders and Stroke P50 NS40256).
Disclosure: The authors report no disclosures.
Received September 18, 2007. Accepted in final form May 20, 2008.
REFERENCES
- 1.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006;7:306–318. [DOI] [PubMed] [Google Scholar]
- 2.Valente EM, Bentivoglio AR, Dixon PH, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 2001;68:895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160. [DOI] [PubMed] [Google Scholar]
- 4.Silvestri L, Caputo V, Bellacchio E, et al. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 2005;14:3477–3492. [DOI] [PubMed] [Google Scholar]
- 5.Gouider-Khouja N, Belal S, Hamida MB, Hentati F. Clinical and genetic study of familial Parkinson's disease in Tunisia. Neurology 2000;54:1603–1609. [DOI] [PubMed] [Google Scholar]
- 6.Ishihara L, Gibson RA, Warren L, et al. Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson's disease families. Mov Disord 2007;22:55–61. [DOI] [PubMed] [Google Scholar]
- 7.Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson's disease: a clinicopathologic study. Neurology 1992;42:1142–1146. [DOI] [PubMed] [Google Scholar]
- 8.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 9.Fahn S, Elton RL. Unified Parkinson's Disease Rating Scale. Florham Park, NJ: Macmillan Healthcare Information, 1987. [Google Scholar]
- 10.Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 1991;14:540–545. [DOI] [PubMed] [Google Scholar]
- 11.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakonarson H, Gulcher JR, Stefansson K. deCODE genetics, Inc. Pharmacogenomics 2003;4:209–215. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin– rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 14.Marongiu R, Brancati F, Antonini A, et al. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat 2007;28:98. [DOI] [PubMed] [Google Scholar]
- 15.Gouider-Khouja N, Larnaout A, Amouri R, et al. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family: clinical, genetic and pathological study. Parkinsonism Relat Disord 2003;9:247–251. [DOI] [PubMed] [Google Scholar]
- 16.Lincoln SJ, Maraganore DM, Lesnick TG, et al. Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord 2003;18:1306–1311. [DOI] [PubMed] [Google Scholar]
- 17.Mata IF, Kachergus JM, Taylor JP, et al. Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics 2005;6:171–177. [DOI] [PubMed] [Google Scholar]
- 18.Sarangmath N, Rattihalli R, Ragothaman M, et al. Validity of a modified Parkinson's disease screening questionnaire in India: effects of literacy of participants and medical training of screeners and implications for screening efforts in developing countries. Mov Disord 2005;20:1550–1556. [DOI] [PubMed] [Google Scholar]
- 19.Burn DJ, Rowan EN, Allan LM, Molloy S, O'Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson's disease, Parkinson's disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2006;77:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ephraty L, Porat O, Israeli D, et al. Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Mov Disord 2007;22:566–569. [DOI] [PubMed] [Google Scholar]
- 21.Abou-Sleiman PM, Muqit MM, McDonald NQ, et al. A heterozygous effect for PINK1 mutations in Parkinson's disease? Ann Neurol 2006;60:414–419. [DOI] [PubMed] [Google Scholar]
- 22.Djarmati A, Hedrich K, Svetel M, et al. Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord 2006;21:1526–1530. [DOI] [PubMed] [Google Scholar]
- 23.Khan NL, Valente EM, Bentivoglio AR, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol 2002;52:849–853. [DOI] [PubMed] [Google Scholar]
- 24.Kay DM, Moran D, Moses L, et al. Heterozygous parkin point mutations are as common in control subjects as in Parkinson's patients. Ann Neurol 2007;61:47–54. [DOI] [PubMed] [Google Scholar]
- 25.Kitada T, Pisani A, Porter DR, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA 2007;104:11441–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci 2007;3:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- 28.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]