

Abstract
Objective:
To examine the association between treatment for diabetes and Alzheimer disease (AD) neuropathology.
Methods:
This postmortem study matched 124 subjects with diabetes to 124 without diabetes from the Mount Sinai School of Medicine Brain Bank, on age (mean = 81.2 + 9.3), sex (57.3% F), and severity of dementia (Clinical Dementia Rating [CDR] 2.4 + 1.7). Densities of neuritic plaques (NPs) and of neurofibrillary tangles (NFTs) were assessed in several neocortical regions and in the hippocampus, entorhinal cortex, and amygdala. Diabetic subjects were classified according to their recorded lifetime antidiabetic medications: none (n = 29), insulin only (n = 49), diabetes medications other than insulin only (n = 28), or concomitant use of both insulin and any oral antidiabetic medications (n = 18). For each dependent variable, analysis of covariance controlling for age at death, sex, and CDR distinguished among the nondiabetic patients and four diabetic subgroups.
Results:
There were differences among the five groups for NP ratings in the entorhinal cortex (p = 0.003), amygdala (p = 0.009), and overall NP (p = 0.014) as well as counts of NPs in all regions examined (p values ranging from 0.009 to 0.04). NP ratings in the hippocampus (p = 0.057) and the combined neocortical measure (p = 0.052) approached significance. In each analysis, the concomitant medication group had significantly fewer NPs (∼20%) than any of the other groups, which were relatively similar. No significant NFT differences were found.
Conclusion:
The results of this study suggest that the combination of insulin with other diabetes medication is associated with substantially lower neuritic plaque density consistent with the effects of both on the neurobiology of insulin.
GLOSSARY
- AD
= Alzheimer disease;
- ANCOVA
= analysis of covariance;
- CDR
= Clinical Dementia Rating;
- CERAD
= Consortium to Establish a Registry for Alzheimer’s Disease;
- IR
= insulin receptors;
- JHH
= Jewish Home and Hospital;
- NFT
= neurofibrillary tangle;
- NP
= neuritic plaque.
Type 2 diabetes has been shown to increase risk for both vascular dementia and Alzheimer disease (AD), in most,1–4 but not all,5,6 epidemiologic studies. It has been estimated that type 2 diabetes or abnormal fasting blood glucose might be present in up to 80% of patients with a diagnosis of AD.3 Diabetes has also been associated with mild cognitive impairment.7 A systematic review of effects of diabetes on dementia and cognitive decline led some investigators to conclude that the latter conditions should be considered among the consequences and disabling manifestations of the former.8 Recently, an association was also shown between prediabetes and dementia and AD.9 Some investigations have proposed that AD constitutes a “type 3 diabetes.”10
The evidence for an association between diabetes and the neuropathology of AD is less clear. We reported that subjects with diabetes had less AD neuropathology.11 Brains of subjects with diabetes had significantly fewer neuritic plaques (NPs) in the hippocampus and cerebral cortex, and significantly fewer neurofibrillary tangles (NFTs) in the cerebral cortex than nondiabetic subjects. Some investigations have observed a trend to less AD neuropathology in diabetic subjects12 consistent with our report while others failed to detect any association at all.4,13 To complicate matters further, there are conflicting findings regarding the effects of antidiabetic medication on the pathogenesis of clinical AD. Diabetic patients, especially those taking insulin, and to a lesser extent those taking oral antidiabetic medications, had the highest risk for developing clinical signs of AD and all dementias compared to nondiabetic patients in the Rotterdam Study.14 In contrast, diabetic patients on antidiabetic monotherapy (insulin or oral), and more so on any combination therapy, had less cognitive decline, especially among those with a longer duration of the disease in the SALSA study.15
Studying the interaction between medications for diabetes and AD neuropathology directly may clarify the relationships among diabetes, diabetes medications, and AD. This might imply therapeutic avenues and suggest neurobiologic processes involved in dementia and AD. A large autopsy sample from the Mount Sinai Brain Bank provided the opportunity to investigate the relationship between diabetes medication and the severity of AD neuropathology. This study was designed to test the hypothesis that treatment of diabetes is associated with lower extents of the cardinal neuropathologic lesions of AD. We compared the extent of AD neuropathology (NPs and NFTs) in the hippocampus, entorhinal cortex, amygdala, and cerebral cortex of five groups of elderly subjects: diabetic patients who were not taking antidiabetic medication; diabetic patients receiving insulin, oral antidiabetic medications, or both; and nondiabetic subjects.
METHODS
Subjects.
Postmortem brains, donated by the next of kin of deceased residents of the Jewish Home and Hospital (JHH) in Manhattan, NY, and Bronx, NY, participating in studies of aging and early dementia, were received over a period of 20 years by the Mount Sinai School of Medicine Department of Psychiatry Brain Bank. Analyses were based on the brains of 124 diabetic patients and 124 matched nondiabetic patients (described in Statistical Analysis), selected from 519 nondiabetic patients. Sixty subjects had a diagnosis of normal brain according to Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuropathologic criteria,16 74 had definite AD, 11 probable AD, 20 possible AD, 41 cerebrovascular disease, and 42 had other neuropathologies. Subjects with severe psychiatric disorders (e.g., schizophrenia) were excluded. All assessments were approved by the institutional review boards of both the JHH and the Mount Sinai School of Medicine.
The Clinical Dementia Rating (CDR) scale assesses cognitive and functional impairments associated with dementia and provides specific severity criteria for classifying subjects as without dementia (CDR = 0), with questionable dementia (CDR = 0.5), or increasing levels of severity of dementia from CDR = 1 to CDR = 5.17,18 A previously described19,20 postmortem assignment of CDR scores was based on medical, neurologic, psychiatric, cognitive, and functional status during the final 6 months of life. Research staff, blind to the hypotheses being tested and to the neuropathology findings, reviewed detailed medical records, and whenever possible conducted in-depth interviews with staff and family caregivers.
Assessment of diabetes.
The JHH maintains extensive medical records on all residents,20,21 with a complete medical history at admission and a complete medical examination monthly and a fasting blood glucose level at least yearly. Patients were diagnosed with diabetes by a geriatrician or an internist (e.g., G.L.) based on the American Diabetes Association criteria (symptoms of diabetes plus casual plasma glucose concentration >200 mg/dL; fasting plasma glucose >126 mg/dL; 2 hours plasma glucose >200 mg/dL during OGTT. Impaired fasting glucose was defined as blood level of 110–125 mg/dL (6.1–7.0 mmol/L). Diagnoses of diabetes were ascertained from detailed review of all medical records and medical history. The analysis included only subjects with type 2 diabetes or no record of diabetes (absence of reported history and failure to meet blood chemistry-based criteria). Measures of duration of diabetes prior to JHH admission were not generally available.
Diabetic subjects were further classified according to their recorded lifetime medications: none (n = 29), only insulin (n = 49), only diabetes medication other than insulin (n = 28), or concomitant use of both insulin and some oral antidiabetic medication (n = 18). Too few subjects had specific oral antidiabetic medications, preponderantly sulfonylureas, for further subdivision.
Neuropathologic assessment.
The neuropathologic assessment procedures employed have been described extensively.19,20 Standardized representative blocks from superior and midfrontal gyrus, orbital cortex, basal ganglia with basal forebrain, amygdala, hippocampus (rostral and caudal levels with adjacent parahippocampal and inferior temporal cortex), superior temporal gyrus, parietal cortex (angular gyrus), calcarine cortex, hypothalamus with mammillary bodies, thalamus, midbrain, pons, medulla, cerebellar vermis, and lateral cerebellar hemisphere were examined using hematoxylin and eosin, modified Bielschowsky, and modified thioflavin S. Any case showing evidence of Lewy body formation in the substantia nigra or locus ceruleus underwent anti-ubiquitin staining of representative cerebral cortical sections. Neuropathologists were blinded to all dementia-associated clinical and psychometric data.
The extent of neuropathologic lesions was assessed using the CERAD neuropathologic battery.16 Sections from each of the tissue blocks described above were rated for the extent of NPs and NFTs using the CERAD four-point scale of 0 = none, 1 = sparse, 3 = moderate, or 5 = severe, as described previously.20 Additionally, quantitative data regarding the density of NPs were collected in five cortical regions using previously published methods20: the midfrontal gyrus, orbital frontal cortex, superior temporal gyrus, inferior parietal cortex, and calcarine cortex. Five representative high power fields (0.5 mm2) were examined in each cortical region and a mean density score per mm2 was calculated. Ancillary exploratory analyses revealed that the inclusion of NP and NFT density estimates from the subcortical fields assessed as part of the CERAD neuropathology battery did not substantively contribute to the results described.19,20
NP and NFT ratings of the neocortical sections were summed into respective summary variables. The rating variables reported here were NPs and NFTs in the entorhinal cortex, hippocampus, amygdala, and neocortex, and also totals of ratings of NPs and NFTs in these four regions. The five neocortical quantitative measures of NPs and their mean were also reported.
Statistical analyses.
For each diabetic subject, a nondiabetic subject was matched for sex and for CDR, and for the closest age at death. The mean absolute difference in age at death was 0.9 years (SD = 1.4, maximum = 6). Analyses were limited to the 124 diabetic patients and their respective 124 matched nondiabetic subjects. The distributions of dependent variables were reviewed for non-normality. As expected for a neuropathology series, the cortical NP counts were skewed and kurtotic. Square root transformations normalized their distributions; none of the neuropathologic ratings had a substantially non-normal distribution. Due to the limit to four possible neuropathologic ratings, groups with small means had small SDs. Nonetheless, these variables were not transformed.
For each dependent variable, analysis of covariance (ANCOVA) controlling for age at death, sex, and CDR was performed to distinguish among the nondiabetic patients and four diabetic subgroups. In addition, four preplanned comparisons of groups were performed. 1) Since in our prior findings11 nondiabetic patients had more neuropathology than diabetic patients in general (medicated or unmedicated), we compared nondiabetic patients to unmedicated diabetic patients. 2) To examine whether antidiabetic medication had a protective effect within diabetic patients, we compared unmedicated diabetic patients to all other diabetic patients. 3) To examine the possible effect of combination therapy, we compared diabetic patients who had taken only insulin or only oral agents with those who had taken both concomitantly. 4) Finally, to examine the specific effects of diabetic monotherapies, we compared diabetic patients taking only insulin to those taking only oral antidiabetic medication.
Since we identified the four planned comparisons a priori, each test of significance was evaluated separately, p ≤ 0.05.
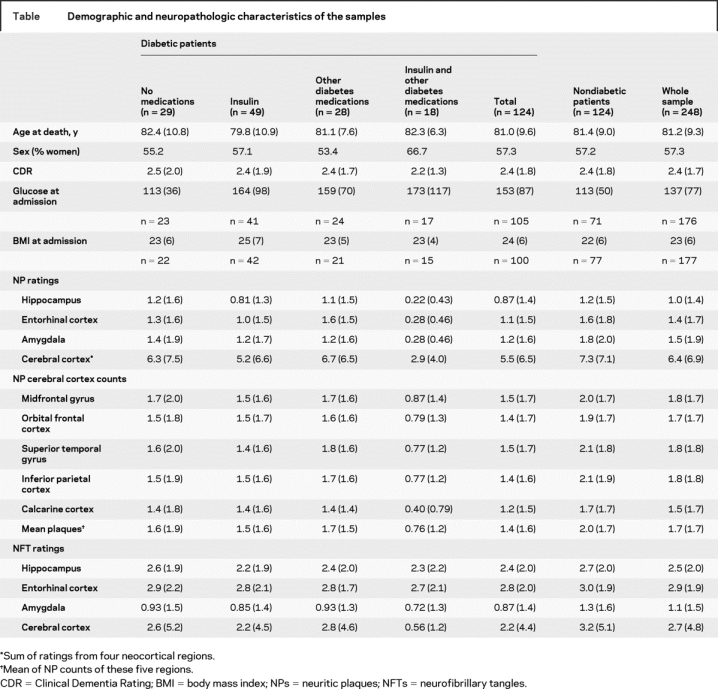
RESULTS
The table presents the demographic and neuropathologic characteristics of the sample. The sample averaged 81.2 (SD = 9.3) years of age at death, 57.3% were women, and the CDR average score was 2.4 (SD = 1.7), reflecting generally moderate to severe dementia. The insulin plus oral medication subgroup was a year older and had a higher proportion of women, but the five subgroups did not differ significantly in age at death, sex, or CDR score.
Table Demographic and neuropathologic characteristics of the samples
There were differences among the five groups for NP ratings in the entorhinal cortex [F(4,236) = 4.22, p = 0.003], amygdala [F(4,224) = 3.48, p = 0.009], and for the summed NP scores [F(4,218) = 3.20, p = 0.014]. The differences for NP ratings in the hippocampus [F(4,236) = 2.33, p = 0.057] and for the neocortex sum [F(4,232) = 2.38, p = 0.052] approached significance. In each region, the group taking both insulin and other antidiabetic medication had fewer NPs than the other groups, which were relatively similar. The planned comparison of diabetic patients on combination therapy to diabetic patients on monotherapy had p values ranging from 0.009 to 0.04 on the five regional assessments of NPs, but none of the other three planned comparisons showed significant differences. There were no differences for any of the NFT ANCOVAs (p values 0.12–0.89) or planned comparisons (p values 0.21–0.97). The figure depicts the means and standard errors of NPs and NFTs for the five groups and in the four regions studied.

Figure Means and standard deviations of CERAD neuropathologic ratings of NPs and NFTs in the five groups and the four regions of interest
(A) ANCOVA comparing the five groups on NPs in the hippocampus, controlling for age at death, severity of dementia, and sex, approached significance (p = 0.057). Rate of NPs for those on combination therapy was lower than for the two monotherapy groups (p = 0.04). NFTs in the hippocampus did not differ between the groups (p = 0.66). (B) ANCOVA comparing the five groups on NPs in the entorhinal cortex had p = 0.003. Rate of NPs for those on combination therapy was lower than the two monotherapy groups (p = 0.009). NFTs did not differ between the groups in the entorhinal cortex (p = 0.89). (C) ANCOVA comparing the five groups on NPs in the amygdala had p = 0.009. Rate of NPs for those on combination therapy was lower than for the two monotherapy groups (p = 0.02). NFTs did not differ between the groups in the amygdala (p = 0.25). (D) ANCOVA comparing the five groups on NP sums in the cerebral cortex approached significance (p = 0.052). Rate of NPs for those on combination therapy was lower than for the two monotherapy groups (p = 0.04). NFTs did not differ between the groups in the cerebral cortex (p = 0.12). ANCOVA = analysis of covariance; CERAD = Consortium to Establish a Registry for Alzheimer’s Disease; NFTs = neurofibrillary tangles; NPs = neuritic plaques.
There were differences among the five groups for NP counts in all five neocortical regions and their mean (p values 0.002–0.02). Again, the group with both insulin and other diabetes medications had fewer NPs than the other groups, and its planned comparisons had p values 0.003–0.04. None of the other planned comparisons were significant.
When the analyses were performed also controlling for fasting glucose levels, weight, or BMI, on subsamples for which these data were available, all results were essentially unchanged, with the only significant differences for the insulin plus oral medications group. This was also the case including only subjects with CERAD diagnoses of normal or AD, excluding subjects with other significant neuropathologies.
DISCUSSION
The present neuropathology-based study of AD demonstrated significantly fewer NPs in diabetic subjects who received both insulin and oral antidiabetic medication, as compared to diabetic subjects with other medication statuses (none, or only insulin or oral antidiabetic medication) or nondiabetic subjects. The group taking both insulin and other antidiabetic medication had fewer NPs specifically in the entorhinal cortex and in the amygdala, and tended to have less NP pathology in the hippocampus and cerebral cortex. The other groups had, in all regions, NP densities that were similar to one another. Results for comparisons among these diabetes groups using quantitative NP density estimates were significant with similar patterns of group differences in all five cerebral cortex regions. Exclusion of cases with other than AD neuropathology, and controlling for fasting blood glucose levels, weight, or BMI did not substantially change any of these results. The diabetes medication effects were specific to NPs, since the extent of NFT pathology was not associated with diabetes medications.
In normal physiology, insulin facilitates memory as demonstrated when administered at optimal doses and in the context of sufficient glucose availability.22 However, the salutary effects of insulin on brain function are reversed under conditions that impair its functioning, such as insulin resistance.23 Type 2 diabetic patients are insulin resistant and have chronic hyperinsulinemia to compensate. The peripheral utilization of insulin reduces insulin transport into the brain, ultimately producing brain insulin deficiency,24 therefore abrogating the many beneficial influences of insulin.22 These observations led to the investigation of effects of antidiabetic medications in AD, which suggested that either monotherapy with insulin25 or with hypoglycemic medication26 can improve memory performance and protect against AD progression. Whether the observed association between combination therapy for diabetes and NP is through their respective biologic pathways or through another pathway remains speculative and invites further examination.
Insulin receptors (IR) are abundant throughout the brain27 and are expressed specifically in regions that support cognitive function.28 At the top of the brain insulin signaling pathway, Aβ, the main component of NPs, decreases insulin affinity reducing the binding of insulin to its receptor29 and preventing rapid activation of specific kinases required for multiple cellular functions, including LTP.30 Soluble Aβ oligomers were recently shown to significantly lower IR responses to insulin and to cause rapid and substantial loss of neuronal surface IRs.31 At the bottom of the signaling pathway, IR desensitization hampers the release of Aβ from the intracellular to the extracellular compartment.32 Insulin concentration is decreased in AD brains,33 further diminishing the functional effectiveness of the insulin signaling pathway. Since desensitization in AD34 can affect the entire insulin signaling pathway, one may speculate that exogenous insulin and insulin sensitizers address insulin deficits singly or simultaneously and help lower NP density in persons with AD.
In an earlier study from our group, we reported that diabetic patients had less AD neuropathology than nondiabetic patients.11 Almost half of the subjects in the current study were not in our former study, but the overall result was similar. Our results suggest that diabetes medication status may be a crucial factor in explaining discrepancies in the associations between diabetes and AD neuropathology noted in other studies.4,12,13
The results of this study also implicate combination therapy of diabetes as a modulator of the density of NPs. The density of NPs was profoundly lower in subjects receiving combination therapy with insulin and oral antidiabetic medication relative to the other age and dementia severity matched subjects in this study, or subjects of comparable age in other studies.35 This suggests that pharmacologic agents that modulate insulin-associated neurobiology may combat the severity of AD neuropathology. It is also possible that in addition to the primary effects on insulin and glucose levels, combination therapy with oral antidiabetic agents might directly attenuate the density of NP lesions of AD. Animal model studies could shed light on this therapeutically relevant possibility.
A parsimonious alternative explanation for the observed association between combination of diabetes medications and NPs is a survival effect in subjects who were relatively old (82 years old at death in average). Their survival might reflect an absence of other medical conditions, including neuropathologic burden. Another survival explanation is that the biologic mechanisms that necessitated the use of combination therapy to control diabetes were also protective against the development of AD neuropathology. This is counterintuitive but not unprecedented; for example, consider the protective effects of sickle cell anemia to malaria.
The analyses in this study found group differences in NPs to be much the same after controlling for differences in dementia severity (CDR). In fact, there were negligible differences between the groups in average dementia severity. A potential reason for the lack of an apparent effect of diabetes medication on CDR is the possibility that differential NP densities above a threshold no longer affect a relatively gross measure of cognition. This possibility is supported by our earlier finding19,20 that densities of NPs too low to fulfill diagnostic criteria for AD are sufficient to affect cognition adversely. An alternative hypothesis suggested by the observed results is that when NP ratings are very low (such as in the subjects with combined antidiabetic medications), dementia severity might be explained by neurodegenerative processes other than NPs that are associated with diabetes, such as degenerative changes of neurons, dendrites, and synapses,36 or by other mechanisms activated by oxidative stress,37 microglial activation,38 or blood–brain barrier overpermeability.39 Additionally, the potential permanent neurologic effects of multiple hypoglycemic episodes should be considered.40 It should be noted, however, that when subjects with CERAD neuropathologic diagnoses other than AD were excluded from the analysis, the results remained essentially unchanged.
In our previous study examining the relationship of neuropathology with diabetes,11 NFT relationships with diabetes were similar to, although weaker than, those for NPs. Therefore we also hypothesized NFT differences among the five groups in the current study but none were significant. Since this study replicates the previous study in finding an overall NFT difference between subjects with and without diabetes, the discrepancy between the NP and NFT results suggests that antidiabetes medications do not have the same effects on NFTs as NPs.
Strengths of the study were a relatively large sample size, systematic independent rating of the extent of AD pathology and assessment of comorbid neuropathology, quantitative counts of NPs in the cortex in addition to the qualitative CERAD neuropathologic ratings, and diagnosis of diabetes based on comprehensive blood examinations. The main limitation of this study was the lack of a full history of diabetes (duration, glucose control, complications, and serum insulin) that might modulate the effect of diabetes on the neuropathology of AD. Such information would address the question of whether the diabetic groups differ in terms of disease severity, and clarify whether the observed results are attributable to disease severity rather than medication effects. The single observation of fasting glucose at admission to the JHH, which was used in a secondary analysis, provided an indication of glucose control, but cannot be interpreted as a reliable summary of glucose control over time. Additionally, if some subjects died before the availability of hypoglycemic medication and the observed differences were due only to medication assignment strategy rather than to medication effects, statistical power would have been reduced by assigning subjects who should have been in the combination group to either insulin or no medication.
The present study provides evidence of substantially lower NP density in diabetic subjects taking both insulin and hypoglycemic medication consistent with the effects of both on the neurobiology of insulin. These pathways should be considered as potentially mechanistically important in the etiology of Aβ associated neuropathology and deposition of NPs.
Address correspondence and reprint requests to Dr. Michal Schnaider Beeri, Mount Sinai School of Medicine, Department of Psychiatry, One Gustave Levy Place, Box 1230, New York, NY 10029 michal.beeri@mssm.edu
Supported by NIA grants K01 AG023515-01A2 (Dr. Beeri), AG02219 (Dr. Haroutunian), and AG05138 (Dr. Sano) and by the Dextra Baldwin McGonagle Foundation and the Joseph E. and Norma G. Saul Foundation.
Disclosure: The authors report no disclosures.
Received January 17, 2008. Accepted in final form May 30, 2008.
REFERENCES
- 1. Schnaider BM Goldbourt U Silverman JM et al. Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology. 2004;63:1902–1907. doi: 10.1212/01.wnl.0000144278.79488.dd. [DOI] [PubMed] [Google Scholar]
- 2. Whitmer RA Sidney S Selby J Johnston SC Yaffe K Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:277–281. doi: 10.1212/01.WNL.0000149519.47454.F2. [DOI] [PubMed] [Google Scholar]
- 3. Janson J Laedtke T Parisi JE O’Brien P Petersen RC Butler PC Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 4. Peila R Rodriguez BL Launer LJ Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 5. MacKnight C Rockwood K Awalt E McDowell I Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14:77–83. doi: 10.1159/000064928. [DOI] [PubMed] [Google Scholar]
- 6. Hassing LB Johansson B Nilsson SE et al. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: a population-based study of the oldest old. Int Psychogeriatr. 2002;14:239–248. doi: 10.1017/s104161020200844x. [DOI] [PubMed] [Google Scholar]
- 7. Luchsinger JA Reitz C Patel B Tang MX Manly JJ Mayeux R Relation of diabetes to mild cognitive impairment. Arch Neurol. 2007;64:570–575. doi: 10.1001/archneur.64.4.570. [DOI] [PubMed] [Google Scholar]
- 8. Cukierman T Gerstein HC Williamson JD Cognitive decline and dementia in diabetes–systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- 9.Xu W, Qiu C, Winblad B, Fratiglioni L. The effect of borderline diabetes on the risk of dementia and Alzheimer’s disease. Diabetes 2007 January;56:211–216. [DOI] [PubMed]
- 10. de la Monte SM Tong M Lester-Coll N Plater MJr Wands JR Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 11. Beeri MS Silverman JM Davis KL et al. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60:471–475. doi: 10.1093/gerona/60.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heitner J Dickson D Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects: a retrospective postmortem immunocytochemical and histofluorescent study. Neurology. 1997;49:1306–1311. doi: 10.1212/wnl.49.5.1306. [DOI] [PubMed] [Google Scholar]
- 13. Arvanitakis Z Schneider JA Wilson RS et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960–1965. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- 14. Ott A Stolk RP van HF Pols HA Hofman A Breteler MM Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 15. Wu JH Haan MN Liang J Ghosh D Gonzalez HM Herman WH Impact of antidiabetic medications on physical and cognitive functioning of older Mexican Americans with diabetes mellitus: a population-based cohort study. Ann Epidemiol. 2003;13:369–376. doi: 10.1016/s1047-2797(02)00464-7. [DOI] [PubMed] [Google Scholar]
- 16. Mirra SS Heyman A McKeel D et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 17. Fillenbaum GG Peterson B Morris JC Estimating the validity of the clinical Dementia Rating Scale: the CERAD experience. Consortium to Establish a Registry for Alzheimer’s Disease. Aging (Milan) 1996;8:379–385. doi: 10.1007/BF03339599. [DOI] [PubMed] [Google Scholar]
- 18. Morris JC Ernesto C Schafer K et al. Clinical dementia rating training and reliability in multicenter studies: the Alzheimer’s Disease Cooperative Study experience. Neurology. 1997;48:1508–1510. doi: 10.1212/wnl.48.6.1508. [DOI] [PubMed] [Google Scholar]
- 19. Haroutunian V Purohit DP Perl DP et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- 20. Haroutunian V Perl DP Purohit DP et al. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- 21. Lesser G Kandiah K Libow LS et al. Elevated serum total and LDL cholesterol in very old patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:138–145. doi: 10.1159/000051248. [DOI] [PubMed] [Google Scholar]
- 22. Craft S Asthana S Cook DG et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28:809–822. doi: 10.1016/s0306-4530(02)00087-2. [DOI] [PubMed] [Google Scholar]
- 23. Craft S Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord. 2006;20:298–301. doi: 10.1097/01.wad.0000213866.86934.7e. [DOI] [PubMed] [Google Scholar]
- 24. Zhao Z Xiang Z Haroutunian V Buxbaum JD Stetka B Pasinetti GM Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol Aging. 2007;28:824–830. doi: 10.1016/j.neurobiolaging.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 25. Reger MA Watson GS Frey WH et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27:451–458. doi: 10.1016/j.neurobiolaging.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 26. Risner ME Saunders AM Altman JF et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 27. Unger JW Livingston JN Moss AM Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol. 1991;36:343–362. doi: 10.1016/0301-0082(91)90015-s. [DOI] [PubMed] [Google Scholar]
- 28. Craft S Watson GS Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 29. Xie L Helmerhorst E Taddei K Plewright B Van BW Martins R Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:RC221. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Townsend M Mehta T Selkoe DJ Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- 31. Zhao WQ De Felice FG Fernandez S et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- 32. Solano DC Sironi M Bonfini C Solerte SB Govoni S Racchi M Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14:1015–1022. doi: 10.1096/fasebj.14.7.1015. [DOI] [PubMed] [Google Scholar]
- 33. Frolich L Blum-Degen D Bernstein HG et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 34. Hoyer S Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490:115–125. doi: 10.1016/j.ejphar.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 35. Li G Larson EB Sonnen JA et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007;69:878–885. doi: 10.1212/01.wnl.0000277657.95487.1c. [DOI] [PubMed] [Google Scholar]
- 36. Li ZG Zhang W Sima AA The role of impaired insulin/IGF action in primary diabetic encephalopathy. Brain Res. 2005;1037:12–24. doi: 10.1016/j.brainres.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 37. Munch G Schinzel R Loske C et al. Alzheimer’s disease–synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105:439–461. doi: 10.1007/s007020050069. [DOI] [PubMed] [Google Scholar]
- 38. Krady JK Basu A Allen CM et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54:1559–1565. doi: 10.2337/diabetes.54.5.1559. [DOI] [PubMed] [Google Scholar]
- 39. Simpson IA Appel NM Hokari M et al. Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem. 1999;72:238–247. doi: 10.1046/j.1471-4159.1999.0720238.x. [DOI] [PubMed] [Google Scholar]
- 40. Cryer PE Hypoglycemia, functional brain failure, and brain death. J Clin Invest. 2007;117:868–870. doi: 10.1172/JCI31669. [DOI] [PMC free article] [PubMed] [Google Scholar]