

Abstract
Background:
Multiple lines of research suggest that increased cystatin C activity in the brain protects against the development of Alzheimer disease (AD).
Methods:
Serum cystatin C levels were analyzed at two examinations of the Uppsala Longitudinal Study of Adult Men, a longitudinal, community-based study of elderly men (age 70 years, n = 1,153 and age 77 years, n = 761, a subset of the age 70 examination). Cox regressions were used to examine associations between serum cystatin C and incident AD. AD cases were identified by cognitive screening and comprehensive medical chart review in all subjects.
Results:
On follow-up (median 11.3 years), 82 subjects developed AD. At age 70 years, lower cystatin C was associated with higher risk of AD independently of age, APOE4 genotype, glomerular filtration rate, diabetes, hypertension, stroke, cholesterol, body mass index, smoking, education level, and plasma amyloid-β protein 40 and 42 levels (hazard ratio [HR] for lowest [<1.12 μmol/L] vs highest [>1.30 μmol/L] tertile = 2.67, 95% CI 1.22–5.83, p < 0.02). The results were similar at age 77 years (43 participants developed AD during follow-up). Furthermore, a 0.1-μmol/L decrease of cystatin C between ages 70 and 77 years was associated with a 29% higher risk of incident AD (HR 1.29, 95% CI 1.03–1.63, p < 0.03).
Conclusions:
Low levels of serum cystatin C precede clinically manifest Alzheimer disease (AD) in elderly men free of dementia at baseline and may be a marker of future risk of AD. These findings strengthen the evidence for a role for cystatin C in the development of clinical AD.
GLOSSARY
- Aβ40
= amyloid-β protein 40;
- Aβ42
= amyloid-β protein 42;
- AD
= Alzheimer disease;
- BMI
= body mass index;
- GFR
= glomerular filtration rate;
- HR
= hazard ratio;
- ICD
= International Classification of Diseases;
- ULSAM
= Uppsala Longitudinal Study of Adult Men.
Cystatin C is an endogenous cysteine inhibitor, produced by nearly all human cells and available in all body fluids.1 During the past decade, experimental,2–7 genetic,8–10 and clinical data6,11,12 have suggested that cystatin C activity in the brain may protect against the development of Alzheimer disease (AD) by inhibition of Aβ aggregation, one of the pathologic hallmarks of AD.13 Recently, it was shown that in a transgenic mouse model, cystatin C binds to soluble Aβ, preventing its deposition in the brain.9,10
Given prior studies supporting a neuroprotective role for cystatin C in the development of AD, we hypothesized that serum levels of cystatin C are related to the activity of cystatin C in the brain and consequently would be associated with the risk of incident AD. Accordingly, we investigated the association between serum cystatin C and risk of incident AD in a large population-based cohort of 70-year-old men. Furthermore, we investigated the association between serum cystatin C and incident AD in a subset of the cohort when the subjects were aged 77 years and whether a change in serum cystatin C levels between ages 70 and 77 years was associated with the future development of AD.
METHODS
Study population.
The Uppsala Longitudinal Study of Adult Men (ULSAM) was initiated in 1970. All 50-year-old men born in 1920–1924 and living in Uppsala, Sweden, were invited to a health survey, initially focusing on identifying risk factors for cardiovascular disease (described in detail at http://www.pubcare.uu.se/ULSAM). The present analyses are based on the third and fourth examination cycles of the ULSAM cohort, when subjects were aged approximately 70 years (1991–1995, n = 1,221) and 77 years (1997–2001, n = 839).
Serum cystatin C was available in 1,193 men at age 70 years (97.7%) and in 792 men at age 77 years (94.4%). Subjects diagnosed with any type of dementia at baseline (n = 3 at age 70 years and n = 11 at age 77 years) and those who did not agree to have their medical records reviewed (n = 37 at age 70 years and n = 20 at age 77 years) were excluded. Thus, the present study sample comprised 1,153 subjects at age 70 years and 761 subjects at age 77 years. Of these, two serial samples of cystatin C (at both age 70 and age 77 years) were available in 678 individuals. The study was approved by the ethics committee at Uppsala University. Informed consent was obtained from all subjects.
Baseline measurements and definitions.
Serum samples were collected and stored at −70°C. Cystatin C measurements were performed with a latex-enhanced reagent (N Latex Cystatin C, Dade Behring, Deerfield, IL) using a Behring BN ProSpec analyzer (Dade Behring). The interassay coefficients of variation were 4.8% at 0.56 mg/L and 3.7% at 2.85 mg/L. Assessment of APOE genotype, serum creatinine–based glomerular filtration rate (GFR),14 diabetes, hypertension, serum cholesterol, body mass index (BMI), smoking status, education level, and plasma amyloid-β protein 40 (Aβ40) and 42 (Aβ42) levels was performed as previously described.15–17
Dementia evaluation.
Cases of AD or any other type of dementia were identified by cognitive screening and medical chart review as previously described in detail.15 In short, subjects were invited to cognitive testing at age 72, 77, and 82 years. Subjects with low test performance (Mini-Mental State Examination [MMSE] score ≤26; or at age 82 years, MMSE score ≤26 and/or 7-minute screen = high risk),18,19 were referred to the Geriatric Memory Clinic at the Uppsala University Hospital for a thorough clinical assessment. Further, all available medical records from the Uppsala University Hospital, all general practitioners in Uppsala (private and/or public), and all community nursing homes and dementia group living settings were reviewed on all subjects (n = 1,153) from 1990 up to December 31, 2005, regardless of the results on the cognitive screening. Diagnoses were assigned and validated according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association and Diagnostic and Statistical Manual of Mental Disorders, 4th Edition.20,21 The frequencies of different types of dementia are presented in table e-1 on the Neurology® Web site at www.neurology.org. Subjects with renal failure and stroke were identified by the Swedish hospital discharge register by using the following International Classification of Diseases (ICD) codes: renal failure: 584-588 (ICD-9), N17-N19 (ICD-10); stroke: 430-438 (ICD-9), I60-I69, G45-G46 (ICD-10). The compulsory Swedish hospital discharge register is updated annually, and the diagnoses have been shown to be of high validity.22,23 Only 16 individuals were lost to follow-up.
Statistical analyses.
Logarithmic transformation was used to achieve normal distribution for skewed variables (Aβ40, Aβ42, and cystatin C). Cox proportional hazards models were used to estimate hazard ratios (HRs) with 95% CIs for the associations of serum levels of cystatin C at ages 70 and 77 years to AD incidence. Proportional hazard assumptions were confirmed by examining Nelson–Aalen curves with log-rank tests and Schoenfeld tests. The assumption of proportional hazards was not violated in any model. Serum cystatin C was evaluated both as a continuous variable and as a categorical variable divided into tertiles. For each subject, the follow-up was defined as the time from the baseline examination (at ages 70 and 77 years) to the date of diagnosis of AD, the date of diagnosis of any other cognitive impairment making the subsequent diagnosis of AD impossible, the date of death, the date of move from the county of Uppsala (n = 16), or the end of the follow-up period (December 31, 2005). Dates of deaths and of moves were based on data from the annually updated and mandatory Swedish National Population Register and the Swedish Death Cause Register. Subjects who developed stroke but did not develop cognitive impairment were considered to be still at risk of AD, whereas subjects with stroke and cognitive impairment were considered to be not at risk for AD from the date of the stroke diagnosis. Three sets of Cox regression models were performed in a hierarchical fashion:
Unadjusted analyses
Multivariable-adjusted analyses adjusting for established risk factors for AD, age, and APOE genotype
Covariates as in model B with the addition of other potential confounders based on previous association with AD or cystatin C in the literature; serum creatinine-based GFR (only in the age 70 cohort, continuous), diabetes (binary), hypertension (binary), stroke at baseline (binary), stroke during follow-up (binary, modeled as a time dependent covariate), serum-cholesterol (continuous), BMI (continuous), smoking status (binary), education level (ordinal), and plasma Aβ40 and Aβ42 levels (continuous)
To investigate the importance of longitudinal change in circulating cystatin C, we investigated the association between changes of serum cystatin C between the examination at ages 70 and 77 years and the incidence of AD from age 77 years and up. Effect modification was examined by testing the statistical significance of two-way interaction terms in the multivariable models between cystatin C and all covariates in model C. None of the interaction terms were significant (all p > 0.10). To minimize the risk of competing risk from other comorbidities as an explanation for our findings, analyses were performed in samples where subjects who developed non-AD dementia (n = 62), stroke (n = 215), or renal failure (n = 52) or died (n = 407) during follow-up were excluded. In addition, analyses were performed, hypothesizing that the incidence rate of AD would be similar in subjects who survived the follow-up as compared with subjects who died, or developed stroke or non-AD dementia during follow-up. In these analyses, 10% of the subjects (subjects in the highest decile of cystatin C) who died or developed stroke or were diagnosed with non-AD dementia during follow-up were reclassified as AD cases. To adjust for the potential inclusion of false negatives in the baseline sample, additional analyses were performed after exclusion of all subjects who developed AD or any type of dementia within 2 years from baseline. To minimize the potential “healthy cohort effect,” analyses were performed introducing dummy variables for being alive but not participating. Missing data were handled such that only subjects with a missing covariate needed for that particular model were excluded from the analyses, to maximize the statistical power. The statistical software package STATA 8.2 (Stata Corp., College Station, TX) was used for all analyses.
RESULTS
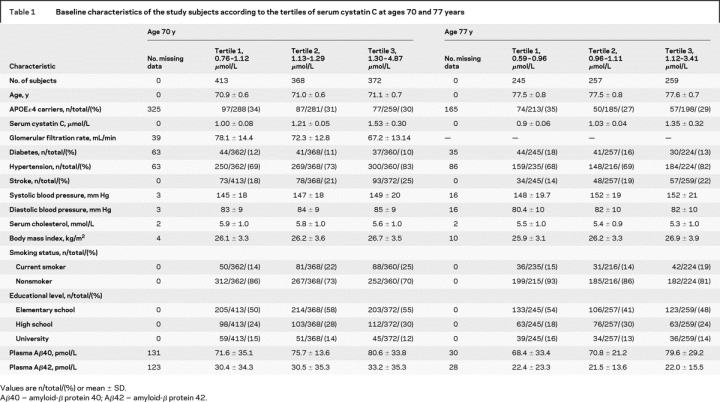
The clinical characteristics of the subjects by tertiles of cystatin C at baseline examinations at ages 70 and 77 years are shown in table 1.
Table 1 Baseline characteristics of the study subjects according to the tertiles of serum cystatin C at ages 70 and 77 years
Cystatin C and AD in subjects aged 70 years.
The median follow-up from the age 70 examination was 11.3 years (range 0.01–14.4 years), contributing to 11,505 person-years at risk. Eighty-two subjects were diagnosed with AD during follow-up (incidence rate 7.1/1,000 person-years at risk). Incidence rates of AD according to serum cystatin C levels are shown in figure 1.

Figure 1 Incidence rates (and 95% CIs) of Alzheimer disease according to the tertiles of serum cystatin C levels in the age 70 (A) and 77 (B) samples
A 1-SD decrease in serum cystatin C was significantly associated with a 28% to 38% higher risk of incident AD both in crude models (model A) and in multivariable models adjusting for established risk factors (model B) and additional potential confounders (model C) (table 2). Subjects in the lowest cystatin C tertile had a more than twofold higher risk of incident AD compared with the highest tertile in all models (models A–C). Nelson–Aalen curves demonstrating the cumulative incidence of AD by tertiles of cystatin C at age 70 years is shown in figure 2A.
Table 2 Relation of serum cystatin C level at ages 70 and 77 years to the incidence of Alzheimer disease
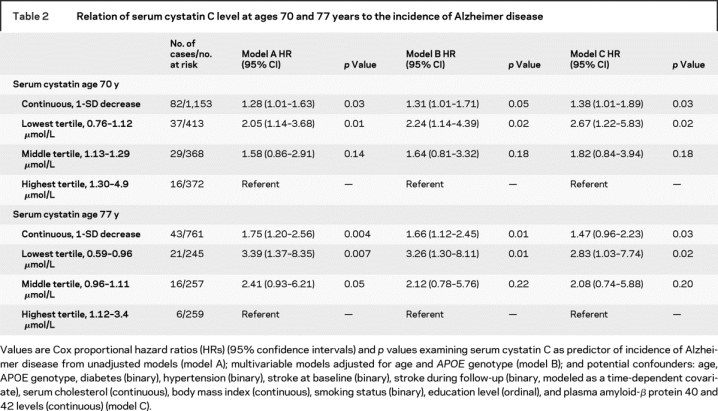

Figure 2 Nelson–Aalen curves on the cumulative incidence of Alzheimer disease by tertiles of serum cystatin C levels in the age 70 (A) and 77 (B) samples
Continuous line shows lowest tertile, dashed line shows middle tertile, and dotted line shows highest tertile.
Cystatin C and AD in subjects aged 77 years.
The median follow-up from the age 77 examination was 5.3 years (range 0.02–7.9 years), contributing to 3,516 person-years at risk. Forty-three subjects were diagnosed with AD during follow-up (incidence rate of 13.9/1,000 person-years at risk). Incidence rates of AD according to serum cystatin C levels are shown in figure 1.
A 1-SD decrease in serum cystatin C was significantly associated with a 47% to 75% higher risk of incident AD both in crude models (model A) and in multivariable models adjusting for established risk factors (model B) and additional potential confounders (model C) (table 2). Men in the lowest cystatin C tertile had a more than twofold higher risk of incident AD compared with the highest tertile models A through C (table 2). Nelson–Aalen curves demonstrating the cumulative incidence of AD by tertiles of cystatin C at age 77 years are shown in figure 2B.
Longitudinal change in serum cystatin C levels.
A 0.1-μmol/L decrease of serum cystatin C between ages 70 and 77 years was associated with a 29% higher risk of developing incident AD (HR 1.29, 95% CI 1.03–1.63, p < 0.03). This model was adjusted for the age of the subjects (at the examination at both age 70 and age 77 years), APOE genotype, and the baseline level of cystatin C at age 70 years (to limit the regression toward the mean).
Additional analyses.
The association between serum cystatin C and AD incidence remained robust in samples without subjects who developed non-AD dementia, stroke, or renal failure or died during follow-up (table e-2). Adjustments for being alive but not participating did not influence the results. Thus, a potential “healthy cohort effect” does not seem to influence the results. The results remained significant after exclusion of all subjects who developed any type of dementia within 2 years from baseline, which argues against inclusion of false negatives in the baseline sample as an explanation for our findings (data not shown). Moreover, the results remained robust when 10% of the subjects who died, developed stroke, or were diagnosed with non-AD dementia during follow-up were reclassified as AD cases (HR after reclassification of subjects who died = 1.41, 95% CI 1.03–1.94, p < 0.03; or developed stroke = 1.38, 95% CI 1.01–1.89, p < 0.04; or developed non-AD dementia = 2.0, 95% CI 0.98–4.12, p = 0.05). Furthermore, the association of serum cystatin C and AD incidence remained when AD cases with different levels of mixed disease (AD plus cerebral vascular disease and AD plus vascular dementia) were included in the AD endpoint (no. of AD cases at age 70 years = 97 and at age 77 years = 56, HR for lowest vs highest tertile of cystatin C = 3.12, 95% CI 1.25–7.80, p < 0.01 at age 70 years; HR 2.73, 95% CI 1.13–6.65, p < 0.04 at age 77 years, in fully adjusted models [model C]). Thus, the potential misclassification of the individuals with AD and mixed disease did not seem to substantially influence the results.
DISCUSSION
In this study, lower serum levels of cystatin C were associated with higher incidence of AD. Previous studies of the association between serum cystatin C and AD have reported conflicting results, one reporting no difference in cystatin C levels between AD cases and controls,24 one reporting high cystatin C levels in AD,25 and a third reporting low levels of cystatin C in AD.12 These studies may all be limited by a cross-sectional case–control design and limited adjustments for potential confounders. Also, the two first studies had smaller study samples (n = 41 and n = 66)24,25 compared with the third (n = 646).12
Multiple lines of research support a neuroprotective role for cystatin C in the development of AD. Immunohistochemical studies in AD brains have demonstrated that cystatin C colocalizes with Aβ6,7 and that staining with cystatin C is increased in AD brains, whereas it is absent or minimal in brains from individuals who remained cognitively intact during life.11 Furthermore, cystatin C has been reported to inhibit Aβ aggregation and deposition in a concentration-dependent manner by binding to the amyloid precursor protein molecule and the Aβ40 and Aβ42 peptides.4 Cystatin C is also believed to inhibit Aβ production and aggregation by the inhibition of cathepsins, a group of lysosomal proteases believed to promote Aβ-amyloidogenesis.26 Finally, there is genetic data that support a causal role for cystatin C in the development of AD,8-10,27 although there are conflicting findings.28 Interestingly, the Icelandic form of hereditary cerebral hemorrhage with amyloidosis is caused by a mutation in the cystatin C gene, and these patients are characterized by low levels of cystatin C in CSF as well as in the systemic circulation, increased deposition of Aβ in cerebral blood vessels, dementia and death before age 40 years.29 Recently, lower levels of cystatin C were reported in CSF in AD compared with controls.30 Our finding that low serum cystatin C levels predicted AD in subjects free of dementia at baseline suggests that low serum cystatin C levels precede clinical AD. Possibly, low serum cystatin C levels mirror a reduced ability to inhibit neuronal Aβ aggregation. However, it is not possible to establish causality between serum cystatin C levels and AD pathogenesis in this study.
Another possible interpretation of our finding is that cystatin C is not causally related to AD, i.e., that the present association merely is mediated by some other causal factors. There are several factors that potentially could confound the association of serum cystatin C and AD incidence, such as age,31 stroke,32 diabetes,33 hypertension,34 serum cholesterol,35 BMI,35 and smoking status.31 However, because the association between serum cystatin C and AD incidence remained robust after adjustment for both established risk factors (model B) and potential confounders (model C), it is not likely that confounding by these factors is the sole explanation for our findings.
A third possible explanation that needs to be considered is that the association between serum cystatin C and incident AD could be due to competing risk by comorbidities such as non-AD dementia, stroke, and renal failure or by cardiovascular mortality. Because individuals with higher serum cystatin C are at higher risk of dying prematurely of cardiovascular32,36 or renal disease,16,37 they are consequently likely to be at lower risk of developing AD. It may be difficult to fully exclude the influence of competing risks in this study. However, arguing against competing risk as an explanation for our findings is the fact that the association between serum cystatin C and AD incidence remained robust both after exclusion of subjects who developed these comorbidities or died during follow-up and in analyses where we assumed a similar incidence rate of AD in subjects who died or were diagnosed with non-AD dementia or stroke during follow-up as in subjects who survived the follow-up.
Currently, our ability to predict future AD is limited. Apart from age and APOE ɛ4 genotype, there are no widely accepted risk factors for the development of sporadic AD (although several cardiovascular risk factors, such as diabetes, dyslipidemia, and hypertension, have also been suggested to portray prognostic information).38 Even though serum cystatin C was an independent risk factor for AD in the present study, further studies are needed to evaluate the role of serum cystatin C as a screening tool to assess the risk of AD in the community.
The strengths of the present study include the longitudinal study design, the large population-based study sample, the nearly complete follow-up, the validation of the cases limiting the inclusion of false-positive cases, and the repeated measurements of serum cystatin C at two different baselines. Serum cystatin C is closely associated with GFR. Consequently, creatinine based GFR was included in the multivariable models to investigate whether the association of cystatin C and AD incidence was independent of GFR (table e-3). However, because creatinine-based GFR has been questioned as a marker for GFR, particularly in the elderly, this approach is limited. Ideally, analyses would have been adjusted for the gold standard methods of measuring GFR (isotope clearance measurements). Isotope clearance measurements are seldom used in epidemiologic research because it is a time-consuming and costly procedure. Because these data are not available in the ULSAM cohort, it is not possible to fully elucidate whether the association of cystatin C and AD incidence is confounded by GFR or whether GFR per se is associated with AD incidence in our study sample. Further studies are needed to investigate this important issue. There is a potential risk that results were influenced by unidentified dementia cases. Misclassification is an issue for any clinical study of dementia but is likely to be random with respect to cystatin C levels, and false negatives due to misclassification would most probably drive the results toward the null hypothesis. Other limitations include the limited generalizability of our results to women and other age and ethnic groups. The multiple lines of research reporting a neuroprotective role for cystatin C argue against competing risks as the sole explanation for our findings. Our results should be considered as hypothesis generating, and replication of our associations in another cohort is desirable to validate our findings.
AUTHOR CONTRIBUTIONS
J. Sundelöf conducted the statistical analysis, had full access to all the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Supplementary Material
Address correspondence and reprint requests to Dr. Johan Sundelöf, Uppsala University, Department of Public Health/Geriatrics, Uppsala Science Park, Dag Hammarskölds väg 14B, 751 85 Uppsala, Sweden johan.sundelof@pubcare.uu.se
Supplemental data at www.neurology.org
*These authors contributed equally.
Supported by Wallenberg Consortium North, Hjärnfonden, Bertil Hållstens forskningsstiftelse, Alzheimerfonden, The Swedish Research Council (2003-5546 and 2006-6555), Stiftelsen Gamla Tjänarinnor, Capios Forskningsstiftelse, Gun och Bertil Stohnes Forskningsstiftelse, Swedish Heart-Lung Foundation, Swedish Lions Research Foundation, Erik, Karin och Gösta Selanders Stiftelse, Loo och Hans Ostermans stiftelse, NIH 1T32NS048005-01, AG05134 (Massachusetts AD Research Center), AFAR Beeson Award (to M.C.I.), J.D. French Alzheimer’s Foundation, and Astra Zeneca with an unrestricted grant. The funding sources did not play any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. There are no conflicts of interest for any of the authors.
Disclosure: The authors report no disclosures.
Received January 7, 2008. Accepted in final form June 10, 2008.
REFERENCES
- 1.Mussap M, Plebani M. Biochemistry and clinical role of human cystatin C. Crit Rev Clin Lab Sci 2004;41:467–550. [DOI] [PubMed] [Google Scholar]
- 2.Wang ZZ, Jensson O, Thorsteinsson L, Vinters HV. Microvascular degeneration in hereditary cystatin C amyloid angiopathy of the brain. APMIS 1997;105:41–47. [DOI] [PubMed] [Google Scholar]
- 3.Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N. Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to beta protein and cystatin C. Stroke 1990;21:397–403. [DOI] [PubMed] [Google Scholar]
- 4.Sastre M, Calero M, Pawlik M, et al. Binding of cystatin C to Alzheimer’s amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol Aging 2004;25:1033–1043. [DOI] [PubMed] [Google Scholar]
- 5.Levy E, Carman MD, Fernandez-Madrid IJ, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990;248:1124–1126. [DOI] [PubMed] [Google Scholar]
- 6.Levy E, Sastre M, Kumar A, et al. Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J Neuropathol Exp Neurol 2001;60:94–104. [DOI] [PubMed] [Google Scholar]
- 7.Vinters HV, Nishimura GS, Secor DL, Pardridge WM. Immunoreactive A4 and gamma-trace peptide colocalization in amyloidotic arteriolar lesions in brains of patients with Alzheimer’s disease. Am J Pathol 1990;137:233–240. [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford FC, Freeman MJ, Schinka JA, et al. A polymorphism in the cystatin C gene is a novel risk factor for late-onset Alzheimer’s disease. Neurology 2000;55:763–768. [DOI] [PubMed] [Google Scholar]
- 9.Kaeser SA, Herzig MC, Coomaraswamy J, et al. Cystatin C modulates cerebral beta-amyloidosis. Nat Genet 2007;39:1437–1439. [DOI] [PubMed] [Google Scholar]
- 10.Mi W, Pawlik M, Sastre M, et al. Cystatin C inhibits amyloid-beta deposition in Alzheimer’s disease mouse models. Nat Genet 2007;39:1440–1442. [DOI] [PubMed] [Google Scholar]
- 11.Deng A, Irizarry MC, Nitsch RM, Growdon JH, Rebeck GW. Elevation of cystatin C in susceptible neurons in Alzheimer’s disease. Am J Pathol 2001;159:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuo LJ, Sheu WH, Pai MC, Kuo YM. Genotype and plasma concentration of cystatin C in patients with late-onset Alzheimer disease. Dement Geriatr Cogn Disord 2007;23:251–257. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Amyloid beta protein precursor and the pathogenesis of Alzheimer’s disease. Cell 1989;58:611–612. [DOI] [PubMed] [Google Scholar]
- 14.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 15.Sundelof J, Giedraitis V, Irizarry MC, et al. Plasma beta amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol 2008;65:256–263. [DOI] [PubMed] [Google Scholar]
- 16.Larsson A. Cystatin C: an emerging glomerular filtration rate marker. Scand J Clin Lab Invest 2005;65:89–91. [DOI] [PubMed] [Google Scholar]
- 17.Larsson A, Helmersson J, Hansson LO, Basu S. Increased serum cystatin C is associated with increased mortality in elderly men. Scand J Clin Lab Invest 2005;65:301–305. [DOI] [PubMed] [Google Scholar]
- 18.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 19.Meulen EF, Schmand B, van Campen JP, et al. The seven minute screen: a neurocognitive screening test highly sensitive to various types of dementia. J Neurol Neurosurg Psychiatry 2004;75:700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 21.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
- 22.Lindblad U, Rastam L, Ranstam J, Peterson M. Validity of register data on acute myocardial infarction and acute stroke: the Skaraborg Hypertension Project. Scand J Soc Med 1993;21:3–9. [DOI] [PubMed] [Google Scholar]
- 23.So L, Evans D, Quan H. ICD-10 coding algorithms for defining comorbidities of acute myocardial infarction. BMC Health Serv Res 2006;6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalman J, Marki-Zay J, Juhasz A, Santha A, Dux L, Janka Z. Serum and cerebrospinal fluid cystatin C levels in vascular and Alzheimer’s dementia. Acta Neurol Scand 2000;101:279–282. [DOI] [PubMed] [Google Scholar]
- 25.Straface E, Matarrese P, Gambardella L, et al. Oxidative imbalance and cathepsin D changes as peripheral blood biomarkers of Alzheimer disease: a pilot study. FEBS Lett 2005;579:2759–2766. [DOI] [PubMed] [Google Scholar]
- 26.Nixon RA. A “protease activation cascade” in the pathogenesis of Alzheimer’s disease. Ann NY Acad Sci 2000;924:117–131. [PubMed] [Google Scholar]
- 27.Beyer K, Lao JI, Gomez M, et al. Alzheimer’s disease and the cystatin C gene polymorphism: an association study. Neurosci Lett 2001;315:17–20. [DOI] [PubMed] [Google Scholar]
- 28.Maruyama H, Izumi Y, Oda M, et al. Lack of an association between cystatin C gene polymorphisms in Japanese patients with Alzheimer’s disease. Neurology 2001;57:337–339. [DOI] [PubMed] [Google Scholar]
- 29.Jensson O, Palsdottir A, Thorsteinsson L, Arnason A. The saga of cystatin C gene mutation causing amyloid angiopathy and brain hemorrhage: clinical genetics in Iceland. Clin Genet 1989;36:368–377. [DOI] [PubMed] [Google Scholar]
- 30.Simonsen AH, McGuire J, Hansson O, et al. Novel panel of cerebrospinal fluid biomarkers for the prediction of progression to Alzheimer dementia in patients with mild cognitive impairment. Arch Neurol 2007;64:366–370. [DOI] [PubMed] [Google Scholar]
- 31.Galteau MM, Guyon M, Gueguen R, Siest G. Determination of serum cystatin C: biological variation and reference values. Clin Chem Lab Med 2001;39:850–857. [DOI] [PubMed] [Google Scholar]
- 32.Shlipak MG, Katz R, Sarnak MJ, et al. Cystatin C and prognosis for cardiovascular and kidney outcomes in elderly persons without chronic kidney disease. Ann Intern Med 2006;145:237–246. [DOI] [PubMed] [Google Scholar]
- 33.Wasen E, Isoaho R, Mattila K, Vahlberg T, Kivela SL, Irjala K. Renal impairment associated with diabetes in the elderly. Diabetes Care 2004;27:2648–2653. [DOI] [PubMed] [Google Scholar]
- 34.Peralta CA, Shlipak MG, Fan D, et al. Risks for end-stage renal disease, cardiovascular events, and death in Hispanic versus non-Hispanic white adults with chronic kidney disease. J Am Soc Nephrol 2006;17:2892–2899. [DOI] [PubMed] [Google Scholar]
- 35.Luc G, Bard JM, Lesueur C, et al. Plasma cystatin-C and development of coronary heart disease: the PRIME Study. Atherosclerosis 2006;185:375–380. [DOI] [PubMed] [Google Scholar]
- 36.Ix JH, Shlipak MG, Chertow GM, Whooley MA. Association of cystatin C with mortality, cardiovascular events, and incident heart failure among persons with coronary heart disease: data from the Heart and Soul Study. Circulation 2007;115:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dharnidharka VR, Kwon C, Stevens G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta-analysis. Am J Kidney Dis 2002;40:221–226. [DOI] [PubMed] [Google Scholar]
- 38.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005;65:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.