

Abstract
Background:
A consensus conference on multiple system atrophy (MSA) in 1998 established criteria for diagnosis that have been accepted widely. Since then, clinical, laboratory, neuropathologic, and imaging studies have advanced the field, requiring a fresh evaluation of diagnostic criteria. We held a second consensus conference in 2007 and present the results here.
Methods:
Experts in the clinical, neuropathologic, and imaging aspects of MSA were invited to participate in a 2-day consensus conference. Participants were divided into five groups, consisting of specialists in the parkinsonian, cerebellar, autonomic, neuropathologic, and imaging aspects of the disorder. Each group independently wrote diagnostic criteria for its area of expertise in advance of the meeting. These criteria were discussed and reconciled during the meeting using consensus methodology.
Results:
The new criteria retain the diagnostic categories of MSA with predominant parkinsonism and MSA with predominant cerebellar ataxia to designate the predominant motor features and also retain the designations of definite, probable, and possible MSA. Definite MSA requires neuropathologic demonstration of CNS α-synuclein–positive glial cytoplasmic inclusions with neurodegenerative changes in striatonigral or olivopontocerebellar structures. Probable MSA requires a sporadic, progressive adult-onset disorder including rigorously defined autonomic failure and poorly levodopa-responsive parkinsonism or cerebellar ataxia. Possible MSA requires a sporadic, progressive adult-onset disease including parkinsonism or cerebellar ataxia and at least one feature suggesting autonomic dysfunction plus one other feature that may be a clinical or a neuroimaging abnormality.
Conclusions:
These new criteria have simplified the previous criteria, have incorporated current knowledge, and are expected to enhance future assessments of the disease.
GLOSSARY
- [11C]HED
= [11C]hydroxyephedrine;
- AAN
= American Academy of Neurology;
- AAS
= American Autonomic Society;
- DSM-IV
= Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition;
- DWI
= diffusion-weighted imaging;
- ED
= erectile dysfunction;
- FDG
= [18F]fluorodeoxyglucose;
- FXTAS
= fragile X–associated tremor/ataxia syndrome;
- MCP
= middle cerebellar peduncle;
- MIBG
= [123I]metaiodobenzylguanidine;
- MR
= magnetic resonance;
- MSA
= multiple system atrophy;
- MSA-C
= MSA with predominant cerebellar ataxia;
- MSA-P
= MSA with predominant parkinsonism;
- OH
= orthostatic hypotension;
- PD
= Parkinson disease;
- SCA
= spinocerebellar ataxia;
- SMC
= Safety Monitoring Committee;
- RV
= residual volume;
- UPDRS
= Unified Parkinson's Disease Rating Scale.
Multiple system atrophy (MSA) is an adult-onset, sporadic, progressive neurodegenerative disease characterized by varying severity of parkinsonian features, cerebellar ataxia, autonomic failure, urogenital dysfunction, and corticospinal disorders.1–4 The disease frequently begins with bladder dysfunction, and in males erectile dysfunction (ED) usually precedes this complaint.5 The presenting motor disorder most commonly consists of parkinsonism with bradykinesia, rigidity, gait instability, and at times tremor, but cerebellar ataxia is the initial motor disorder in a substantial percentage of patients.2 The defining neuropathology of MSA consists of degeneration of striatonigral and olivopontocerebellar structures accompanied by profuse numbers of distinctive glial cytoplasmic inclusions formed by fibrillized α-synuclein proteins.6–8 Although the etiology is unknown, this disorder, like Parkinson disease (PD) and dementia with Lewy bodies, seems to result from a disturbance of α-synuclein and is designated as an α-synucleinopathy.
A consensus conference on diagnosis held in 1998 defined two categories, MSA with predominant parkinsonism (MSA-P) and MSA with predominant cerebellar ataxia (MSA-C).3 Three levels of certainty were established, possible, probable, and definite MSA, with the diagnosis of definite MSA requiring autopsy confirmation. These guidelines emphasized the importance of autonomic by requiring this feature for the diagnosis of probable MSA. Validation studies of the consensus criteria demonstrated high predictive accuracy but suboptimal sensitivity, particularly in the early stages of the disease.9,10 For the category of possible MSA, predictive accuracy was relatively lower at the first neurologic visit, but sensitivity was higher at this time point as compared with criteria defining probable MSA.
Widely accepted, the original consensus criteria have served as the gold standard for diagnosis. Nevertheless, the criteria used separate features and criteria for diagnosis that were complex and difficult to keep in mind. Moreover, additional information relevant to diagnostic criteria has accumulated since these criteria were published, including clinical and laboratory studies,4 neuropathologic and biochemical findings,8,11 and neuroimaging studies.12–14 Accordingly, the time had come to initiate a second consensus conference to develop new guidelines for diagnosis. These guidelines are intended for both practicing clinical neurologists and investigators studying the disease.
METHODS
Development of this consensus conference began with a grant application to the NIH for support. The grant was funded, additional support was obtained, and the American Academy of Neurology (AAN) agreed to cosponsor the event. A Steering Committee was selected that included investigators with expertise in MSA-P (G.K.W.), MSA-C (S.G.), and autonomic failure (P.A.L.). The Steering Committee selected members for the consensus group based on the members' expertise in the parkinsonian, cerebellar, and autonomic features; in the neuropathologic and biochemical disturbances; and in the structural and functional imaging characteristics of the disease. Selection of members required evidence that they were active investigators and published in the areas relevant to the diagnostic considerations, and included efforts to involve qualified women and minorities in the group. All of those who were contacted initially agreed to serve, and they were divided into five task groups: MSA-P, MSA-C, Autonomic, Neuropathology, and Neuroimaging. A Chair was appointed for each group based on a long track record of leadership in the discipline of the group. The MSA-P group included Gregor K. Wenning (Chair), Carlo Colosimo, Andrew Lees, Werner Poewe, Niall Quinn, and Marie Vidailhet. Members of the MSA-C group were Nicholas W. Wood (Chair), Alexandra Dürr, Thomas Klockgether, and Sid Gilman. The Autonomic group consisted of Christopher J. Mathias (Chair), Clare J. Fowler, Horacio Kaufmann, Phillip A. Low, David Robertson, and Paola Sandroni. The Neuroimaging group included David J. Brooks (Chair) and Klaus Seppi. The Neuropathology group consisted of John Q. Trojanowski (Chair) and Tamas Revesz.
Each task group received from the Steering Committee a request to develop a position paper regarding consensus criteria for diagnosis limited to their area of expertise. The task group chairs initially wrote the position papers, sent them to group members for comments and criticism, and after an iterative process, sent their draft reports to the Steering Committee. The Committee reviewed them and returned them to the task group chairs with comments as needed. Final drafts of the position papers were circulated to the entire membership of the consensus committee in advance of the meeting. A 2-day meeting was held in Boston, Massachusetts, on April 26 and 27, 2007, immediately preceding the AAN meeting. The meeting involved the use of consensus methodology to arrive at the current criteria for diagnosis. Consensus methodology uses the collected judgment of seasoned investigators closely familiar with the disease from years of experience. The consensus process involves the following principles: all members 1) contribute to the discussion, 2) can state each issue in their own words, 3) have the opportunity and time to express their opinion about each issue, and 4) agree to take responsibility for the implementation of a decision. Members who disagree will agree to support the group decision initially on a trial basis, pending further discussion. Achieving consensus requires that all members 1) listen nonjudgmentally to the opinions of other members and 2) check for understanding by summarizing what they think they hear while building on each other's thoughts and exploring minority opinions. The advantages of this methodology are that the quality of a decision is often excellent because it is based on shared information and opinion; and the level of support for each decision is often great because all members participate in making the decision and there is no minority group whose opinions are discounted. The Steering Committee developed the present article with contributions from the entire membership of the consensus group.
RESULTS
Diagnostic categories.
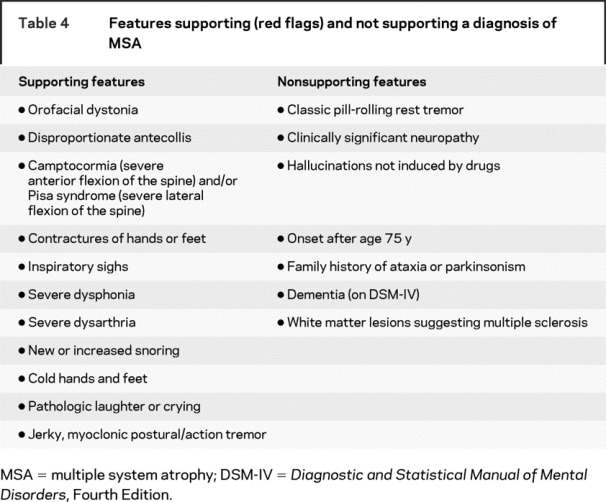
Similar to the first consensus conference, we determined that the diagnosis of MSA should be divided into three groups. The first, definite MSA, requires the neuropathologic findings of widespread and abundant CNS α-synuclein–positive glial cytoplasmic inclusions (Papp–Lantos inclusions) in association with neurodegenerative changes in striatonigral or olivopontocerebellar structures.15 Criteria for probable MSA are listed in table 1 and for possible MSA are listed in tables 2 and 3.3 Patients with predominantly parkinsonian features should continue to be designated MSA-P, and patients with predominantly cerebellar ataxia should be designated MSA-C. We appreciate that the predominant motor feature can change with time; thus, patients who present with cerebellar ataxia can develop increasingly severe parkinsonian features until these latter features dominate the clinical presentation. Hence the designation of MSA-P or MSA-C refers to the predominant feature at the time the patient is evaluated, and the predominant feature can change with time. Also, determining the predominant feature becomes a matter of clinical judgment in subjects with combinations of parkinsonian and cerebellar features. We do not recommend using the term “MSA-mixed” to describe patients with combinations of cerebellar ataxia and parkinsonian features, because this category could be used as a default for any combination of ataxia and parkinsonism, irrespective of the severity of each. Disease onset is defined as the initial presentation of any motor problem, whether parkinsonian or cerebellar, or autonomic features, as defined in the criteria for possible MSA, with the exception of male ED. Although the disease process must start earlier within the CNS, for research purposes, a pragmatic definition of disease onset is required. We excluded ED in men5 and reduced genital sensitivity in women16 because these symptoms have myriad causes in older people. Table 1 provides the criteria for autonomic failure, which is an integral part of MSA, and table 4 gives features supporting and not supporting a diagnosis of MSA for cases with possible MSA.
Table 1 Criteria for the diagnosis of probable MSA

Table 2 Criteria for possible MSA

Table 3 Additional features of possible MSA

Table 4 Features supporting (red flags) and not supporting a diagnosis of MSA
Autonomic failure.
Orthostatic hypotension (OH) may indicate autonomic failure and can be asymptomatic or symptomatic. When symptomatic, it frequently occurs after the onset of ED and urinary symptoms.5 Symptoms of OH result from hypoperfusion, and syncope may occur.17 The clinical diagnosis of probable MSA requires a reduction of systolic blood pressure by at least 30 mm Hg or of diastolic blood pressure by at least 15 mm Hg after 3 minutes of standing from a previous 3-minute interval in the recumbent position. This orthostatic decline is usually accompanied by a compensatory increase in heart rate that is inadequately low for the level of blood pressure decline. We note that this is a more pronounced decrease of blood pressure than recommended previously in the American Autonomic Society (AAS)–AAN consensus statement on the definition of orthostatic hypotension.18 Blood pressure can be decreased additionally by drugs, fluid depletion, food ingestion, an increased temperature, and physical deconditioning. Other disorders known to cause OH, such as diabetes mellitus with autonomic neuropathy, should be excluded or at least taken into account.
Genitourinary dysfunction.
ED is often the earliest symptom of MSA and affects virtually all male patients5; apart from one report of decreased genital sensitivity,16 there is little information about female sexual dysfunction in MSA. Because the prevalence of ED increases with age, the symptom has a low specificity; however, preserved erectile function makes a diagnosis of MSA unlikely. Urinary complaints are common in the aging population, but the recent, unexplained onset of urinary incontinence, especially in men, and incomplete bladder emptying increase the likelihood of a diagnosis of MSA. Constipation often accompanies the other autonomic symptoms.
Laboratory investigations of autonomic failure.
Autonomic failure can be evaluated not only by a history of urinary incontinence and orthostatic blood pressure measurements in the clinic, but also by a comprehensive battery that examines the distribution and severity of cardiovascular, sudomotor, and urinary bladder deficits.19 Cardiovascular and sudomotor autonomic function tests may help to separate MSA from other sporadic cerebellar ataxias and from PD.20 Measurement of urine residual volume (RV) by ultrasound can reveal incomplete bladder emptying of >100 mL. RV tends to increase as MSA progresses. Imaging of cardiac innervation with SPECT and [123I]metaiodobenzylguanidine (MIBG) and with PET and [18F]fluorodopa in many reports have shown preserved sympathetic postganglionic neurons in MSA, in contrast to PD21; however, some MIBG studies have shown denervation in MSA,22 and a recent investigation with PET and [11C]hydroxyephedrine ([11C]HED) revealed severe cardiac denervation in MSA.23
Parkinsonism.
Most MSA patients develop parkinsonism (bradykinesia with rigidity, tremor, or postural instability) at some stage. The tremor is usually irregular and postural/action, often incorporating myoclonus, but a classic pill-rolling rest tremor is uncommon. The parkinsonism can be asymmetric. Postural instability, as defined by item 30 of the Unified Parkinson's Disease Rating Scale (UPDRS) part III (motor examination),24 occurs earlier and progresses more rapidly than in PD. Moreover, the UPDRS part III score typically worsens by less than 10% per annum in PD but by more than 20% in MSA.25
Levodopa responsiveness.
Parkinsonism usually responds poorly to chronic levodopa therapy; however, up to 30% of patients show a clinically significant, but usually waning, response.26 Responsiveness should be tested with escalating doses of levodopa with a peripheral decarboxylase inhibitor over 3 months up to at least 1 g/d (if necessary and tolerated). A positive response is defined as clinically significant motor improvement. This should be demonstrated by objective evidence such as an improvement of 30% or more on part III of the UPDRS or on part II of the Unified Multiple System Atrophy Rating Scale.27
Sleep disorders.
REM sleep behavior disorder and obstructive sleep apnea occur frequently in MSA, but also affect PD patients and are not diagnostically definitive.28
Laboratory investigations.
Structural and functional imaging can assist diagnosis. MRI demonstration of putaminal, pontine, and middle cerebellar peduncle (MCP) atrophy is helpful in both MSA-P and MSA-C.29 T2-signal changes on 1.5-tesla MRI in the basal ganglia and brainstem can be helpful, including posterior putaminal hypointensity, hyperintense lateral putaminal rim, hot cross bun sign, and MCP hyperintensities.
Functional imaging demonstration of striatal or brainstem hypometabolism by PET with [18F]fluorodeoxyglucose can help in the diagnosis of MSA.14 In the absence of clinically evident ataxia in a patient with parkinsonian features, demonstration of cerebellar hypometabolism can point to the diagnosis of MSA-P rather than PD. Conversely, in the absence of parkinsonian features in a patient with cerebellar ataxia, evidence of nigrostriatal dopaminergic denervation from functional imaging (SPECT and PET) may point to the diagnosis of MSA-C.30 As noted above, cardiac sympathetic postganglionic imaging with SPECT shows denervation in PD21 and uncommonly in MSA,22 but PET with [11C]HED may reveal extensive denervation in MSA.23
Other imaging techniques, such as brain parenchymal sonography, magnetic resonance (MR) spectroscopy, MR diffusion-weighted imaging (DWI), MR diffusion tensor imaging, MR magnetization transfer imaging, and MR voxel-based morphometry remain investigational; however, MR DWI has been shown to discriminate MSA-P, even in the early disease stages, from PD and from healthy controls on the basis of increased putaminal and MCP diffusivity measures.31
The CSF neurofilament light chain and tau tests have been reported to differentiate MSA from PD but remain in an exploratory phase of development.32
Cerebellar ataxia.
Ataxia of gait, the most common cerebellar feature of MSA-C, is often accompanied by ataxia of speech (cerebellar dysarthria) and cerebellar oculomotor dysfunction. Limb ataxia may be seen but is generally less prominent than gait or speech disturbances. Although gaze-evoked nystagmus occurs in the majority of later-stage MSA-C patients, earlier oculomotor abnormalities may not involve nystagmus, but include square wave jerks, jerky pursuit, and dysmetric saccades. Limitations of supranuclear gaze and severe slowing of saccadic velocities are not features of MSA.
Differential diagnosis of adult-onset cerebellar ataxia.
The criteria for probable MSA-C shown in table 1 have high predictive value.9,10 MSA-C generally presents clinically as a midline cerebellar disorder that progresses more rapidly than other late-onset sporadic ataxias; typically a patient becomes wheelchair dependent by 5 years after onset.33 The features detailed in tables 2 and 3 for possible MSA help to provide strong indicators of the diagnosis of MSA-C, and table 4 indicates features supporting and not supporting a diagnosis of MSA. Clinicians evaluating patients with progressive ataxia should include a large differential diagnostic list, because many diseases can produce an adult-onset progressive ataxia. The dominantly inherited spinocerebellar ataxias (SCAs) can result in an apparently sporadic disorder, because even with a negative family history, there is a 15% to 20% chance of a mutation in one of the polyglutamine SCAs, notably SCAs 1, 2, 3, 6, and 7. Fragile X–associated tremor/ataxia syndrome (FXTAS) is a neurodegenerative disorder with core features of action tremor and ataxia of gait.34 Frequently this disorder includes parkinsonism, abnormalities of executive function, dementia, neuropathy, and autonomic failure. FXTAS results from a premutation (moderate expansions of 55–200 repeats) of a CGG trinucleotide in the fragile X mental retardation 1 gene, the gene that causes fragile X syndrome when the full mutation develops (over 200 CGG repeats). A search for the fragile X mutation in a large series of possible, probable, and pathologically confirmed cases suggests the frequency is less than 1%.35 FXTAS shows characteristic MRI features, hyperintensity in the middle cerebellar peduncles and supratentorial white matter, that may overlap with those of MSA. A diagnosis of paraneoplastic disease should be considered in patients with an aggressive clinical course with or without general systemic malaise, and a search for appropriate antibodies and for the putative primary should be undertaken. After these diseases have been excluded, and anatomic imaging shows cerebellar and brainstem atrophy, patients usually receive the diagnosis of sporadic adult-onset ataxia, which is also known as idiopathic late-onset cerebellar ataxia or sporadic olivopontocerebellar atrophy.36 The cause of this disorder is unknown, and many patients who develop this disorder do not progress to MSA.
Findings that cast doubt on the diagnosis of MSA-C.
The presence of a family history of a similar disorder makes the diagnosis of MSA-C unlikely; one of the SCAs should be considered. Nevertheless, recent studies of several multiplex families suggest familial MSA may be due to autosomal recessive inheritance,37 and autopsy-confirmed MSA has been reported in association with an abnormal expansion of one allele of the SCA type 3 gene.38 Dementia also makes the diagnosis of MSA-C unlikely. MRI studies showing supratentorial white matter lesions other than those commonly seen in this age group make the diagnosis doubtful and raise the possibility of MS.
Differences between the first and second consensus criteria.
Although the current criteria have the same structure as the earlier criteria, there are differences in each category. In the previous criteria for definite MSA, α-synuclein–positive glial cytoplasmic inclusions were not required. The previous criteria for probable MSA used separate “features” and “criteria,” which have been abandoned in the current criteria. Similarly, the separate “features” and “criteria” in possible MSA have been deleted and, in addition to parkinsonism or a cerebellar syndrome, now there must be one feature involving autonomic dysfunction plus one other finding, and the latter is detected either with clinical examination or with imaging.
DISCUSSION
On clinical presentation, MSA appears with a combination of autonomic failure with parkinsonism or cerebellar ataxia or both, and the previous criteria and the present criteria recognize this fundamental feature of the disease. The current criteria also retain the distinctions between levels of diagnostic certainty, using the term definite MSA for subjects with autopsy demonstration of typical histologic features,15 probable MSA for patients with autonomic failure plus parkinsonism or cerebellar ataxia, and possible MSA for people with clinical findings that do not as yet clearly represent this disease.
The principal differences between the current criteria and the previous criteria concern clinical domains and requirements for the diagnosis of possible MSA. In the previous criteria, we used “features” to describe clinical findings and “criteria” to indicate the features that could be used for diagnosis. This distinction proved to be confusing and difficult to retain; hence it has been discarded. The current criteria include straightforward descriptions of the clinical findings required for the diagnoses of probable and possible MSA. The diagnosis of probable MSA is now considerably simplified, as shown in table 1. The diagnosis of possible MSA has been changed to require at least one feature suggesting autonomic dysfunction in addition to parkinsonism or a cerebellar syndrome. At least one additional feature will be required for this diagnosis and can include findings on history, clinical examination, and results from either structural or functional imaging. This change, particularly the requirement of a feature suggesting autonomic dysfunction, hopefully will decrease the false positives that characterize clinical diagnosis in the early stages of the disorder.
The new criteria were created using consensus methodology, as were the first consensus criteria. This methodology presents the advantage of using the collected experience of active investigators, but the disadvantage that the criteria do not result from an evidence-based approach. We anticipate that validation studies will be performed in the future and that these studies will show the high predictive accuracy of the first set of criteria and hopefully better sensitivity in the early stages of the disease than the previous criteria.9,10
We anticipate that the new criteria will be less cumbersome to apply in patients with possible MSA than the first set of criteria. The principal differences between the first and second set of consensus criteria are in the group with possible MSA, and in this group it is possible now to use both clinical and imaging results to buttress the diagnosis in subjects with parkinsonian features or cerebellar dysfunction plus autonomic symptoms that do not meet the level needed for the diagnosis of probable MSA. In the latter group, we use criteria for autonomic failure that are more rigorous than those used by the AAS and AAN.18 This is to ensure a high level of accuracy in the diagnosis of MSA, because the disease is a grave one and carries the prognosis of a markedly shortened life span.
DISCLOSURE
S.G. serves as Chair of the Safety Monitoring Committee (SMC) for the Elan and Wyeth trials of immunotherapy for Alzheimer disease and of the SMC for the Elan and Transition trials of scyllo-inositol for Alzheimer disease. He has served as a consultant for Wyeth, ReNeuron, Adamas, and Kyowa and has grant support from Cortex Pharmaceuticals. He is a consultant for PPD Development, Longitude Capital, and the Gerson Lehrman Group and a member of the Board of Directors of Balboa Bioscience. C.C. has received grant support and other honoraria from Allergan, Boehringer-Ingelheim, Eli-Lilly, GlaxoSmithKline, and Ipsen. W.P. has served as a consultant for GSK, Novartis, Teva, Boehringer Ingelheim, Schwarz Pharma/UCB, AstraZeneca, and General Electric. N.Q. has served as a consultant for Medtronic, Schwarz-Pharma, UCB, Boehringer & Ingelheim, GlaxoSmithKline, Dainippon Sumitomo, Solvay, Teva, and Novartis. The remaining authors have no disclosures.
Address correspondence and reprint requests to Dr. Sid Gilman, Department of Neurology, University of Michigan, 300 N. Ingalls St., 3D15, Ann Arbor, MI 48109-5489 sgilman@umich.edu
Author's affiliations are listed at the end of the article.
Supported in part by grants from the NIH (National Institute of Neurological Disorders and Stroke grant 1 R13 NS055459), Novartis Pharmaceuticals, and Chelsea Therapeutics.
Disclosure: Author disclosures are provided at the end of the article.
Received January 4, 2008. Accepted in final form May 23, 2008.
REFERENCES
- 1.Quinn N. Multiple system atrophy–the nature of the beast. J Neurol Neurosurg Psychiatry 1989;(suppl):78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133–147. [DOI] [PubMed] [Google Scholar]
- 3.Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94–98. [DOI] [PubMed] [Google Scholar]
- 4.Geser F, Wenning GK, Seppi K, et al. Progression of multiple system atrophy (MSA): a prospective natural history study by the European MSA Study Group (EMSA SG). Mov Disord 2006;21:179–186. [DOI] [PubMed] [Google Scholar]
- 5.Kirchhof K, Apostolidis AN, Mathias CJ, Fowler CJ. Erectile and urinary dysfunction may be the presenting features in patients with multiple system atrophy: a retrospective study. Int J Impot Res 2003;15:293–298. [DOI] [PubMed] [Google Scholar]
- 6.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989;94:79–100. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205–208. [DOI] [PubMed] [Google Scholar]
- 8.Ozawa T, Healy DG, Abou-Sleiman PM, et al. The alpha-synuclein gene in multiple system atrophy. J Neurol Neurosurg Psychiatry 2006;77:464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002;59:1486–1491. [DOI] [PubMed] [Google Scholar]
- 10.Colosimo C, Vanacore N, Bonifati V, et al. Clinical diagnosis of multiple system atrophy: level of agreement between Quinn's criteria and the consensus conference guidelines. Acta Neurol Scand 2001;103:261–264. [PubMed] [Google Scholar]
- 11.Ozawa T, Paviour D, Quinn NP, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 2004;127:2657–2671. [DOI] [PubMed] [Google Scholar]
- 12.Seppi K, Schocke MF, Mair KJ, et al. Progression of putaminal degeneration in multiple system atrophy: a serial diffusion MR study. Neuroimage 2006;31:240–245. [DOI] [PubMed] [Google Scholar]
- 13.Seppi K, Schocke MF, Donnemiller E, et al. Comparison of diffusion-weighted imaging and [123I]IBZM-SPECT for the differentiation of patients with the Parkinson variant of multiple system atrophy from those with Parkinson's disease. Mov Disord 2004;19:1438–1445. [DOI] [PubMed] [Google Scholar]
- 14.Gilman S. Functional imaging with positron emission tomography in multiple system atrophy. J Neural Transm 2005;112:1647–1655. [DOI] [PubMed] [Google Scholar]
- 15.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 2007;33:615–620. [DOI] [PubMed] [Google Scholar]
- 16.Oertel WH, Wachter T, Quinn NP, Ulm G, Brandstadter D. Reduced genital sensitivity in female patients with multiple system atrophy of parkinsonian type. Mov Disord 2003;18:430–432. [DOI] [PubMed] [Google Scholar]
- 17.Allcock LM, Ullyart K, Kenny RA, Burn DJ. Frequency of orthostatic hypotension in a community based cohort of patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 2004;75:1470–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Neurology 1996;46:1470. [DOI] [PubMed] [Google Scholar]
- 19.Suarez GA, Opfer-Gehrking TL, Offord KP, Atkinson EJ, O'Brien PC, Low PA. The Autonomic Symptom Profile: a new instrument to assess autonomic symptoms. Neurology 1999;52:523–528. [DOI] [PubMed] [Google Scholar]
- 20.Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991;1:147–155. [DOI] [PubMed] [Google Scholar]
- 21.Courbon F, Brefel-Courbon C, Thalamas C, et al. Cardiac MIBG scintigraphy is a sensitive tool for detecting cardiac sympathetic denervation in Parkinson's disease. Mov Disord 2003;18:890–897. [DOI] [PubMed] [Google Scholar]
- 22.Nagayama H, Hamamoto M, Ueda M, Nagashima J, Katayama Y. Reliability of MIBG myocardial scintigraphy in the diagnosis of Parkinson's disease. J Neurol Neurosurg Psychiatry 2005;76:249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raffel DM, Koeppe RA, Little R, et al. PET measurement of cardiac and nigrostriatal denervation in parkinsonian syndromes. J Nucl Med 2006;47:1769–1777. [PubMed] [Google Scholar]
- 24.Fahn S, Elton RL, UPDRS Development Committee. Unified Parkinson Disease Rating Scale. Floral Park, NJ: Macmillan, 1987. [Google Scholar]
- 25.Seppi K, Yekhlef F, Diem A, et al. Progression of parkinsonism in multiple system atrophy. J Neurol 2005;252:91–96. [DOI] [PubMed] [Google Scholar]
- 26.Hughes AJ, Colosimo C, Kleedorfer B, Daniel SE, Lees AJ. The dopaminergic response in multiple system atrophy. J Neurol Neurosurg Psychiatry 1992;55:1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenning GK, Tison F, Seppi K, et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19:1391–1402. [DOI] [PubMed] [Google Scholar]
- 28.Iranzo A, Santamaria J, Rye DB, et al. Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology 2005;65:247–252. [DOI] [PubMed] [Google Scholar]
- 29.Seppi K, Schocke MF, Wenning GK, Poewe W. How to diagnose MSA early: the role of magnetic resonance imaging. J Neural Transm 2005;112:1625–1634. [DOI] [PubMed] [Google Scholar]
- 30.Gilman S, Koeppe RA, Junck L, et al. Decreased striatal monoaminergic terminals in multiple system atrophy detected with positron emission tomography. Ann Neurol 1999;45:769–777. [DOI] [PubMed] [Google Scholar]
- 31.Nicoletti G, Lodi R, Condino F, et al. Apparent diffusion coefficient measurements of the middle cerebellar peduncle differentiate the Parkinson variant of MSA from Parkinson's disease and progressive supranuclear palsy. Brain 2006;129:2679–2687. [DOI] [PubMed] [Google Scholar]
- 32.Abdo WF, Bloem BR, Van Geel WJ, Esselink RA, Verbeek MM. CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson's disease. Neurobiol Aging 2007;28:742–747. [DOI] [PubMed] [Google Scholar]
- 33.Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 1998;121(pt 4):589–600. [DOI] [PubMed] [Google Scholar]
- 34.Berry-Kravis E, Abrams L, Coffey SM, et al. Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Mov Disord 2007;22:2018–2030. [DOI] [PubMed] [Google Scholar]
- 35.Kamm C, Healy DG, Quinn NP, et al. The fragile X tremor ataxia syndrome in the differential diagnosis of multiple system atrophy: data from the EMSA Study Group. Brain 2005;128:1855–1860. [DOI] [PubMed] [Google Scholar]
- 36.Abele M, Minnerop M, Urbach H, Specht K, Klockgether T. Sporadic adult onset ataxia of unknown etiology: a clinical, electrophysiological and imaging study. J Neurol 2007;254:1384–1389. [DOI] [PubMed] [Google Scholar]
- 37.Hara K, Momose Y, Tokiguchi S, et al. Multiplex families with multiple system atrophy. Arch Neurol 2007;64:545–551. [DOI] [PubMed] [Google Scholar]
- 38.Nirenberg MJ, Libien J, Vonsattel JP, Fahn S. Multiple system atrophy in a patient with the spinocerebellar ataxia 3 gene mutation. Mov Disord 2007;22:251–254. [DOI] [PubMed] [Google Scholar]