

Abstract
Viruses like particles (VLPs), assembled from capsid structural subunits of several different viruses, have found a number of biomedical applications such as vaccines and novel delivery systems for nucleic acids and small molecules. Production of recombinant proteins in different plant systems has been intensely investigated and improved upon in the last two decades. Plant derived antibodies, vaccines, and microbicides have received great attention and shown immense promise. In the case of mucosal vaccines, orally delivered plant produced VLPs require minimal processing of the plant tissue, thus offering an inexpensive and safe alternative to more conventional live attenuated and killed virus vaccines. For other applications which require higher level of purification, recent progress in expression levels using plant viral vectors have shown that plants can compete with traditional fermentation systems. In this review the different methods used in the production of VLPs in green plants are described. Specific examples of expression, assembly, and immunogenicity of several plant-derived VLPs are presented.
Keywords: VLP, transgenic plants, plant viral systems, vaccines
1. Introduction
The minimal structural features of a typical virus consist of one molecule of nucleic acid encased in a protective capsid that is assembled from several identical proteins. More complex viruses have genomes made up of several nucleic acid molecules. Their capsids may be formed by several different structural proteins, which can be surrounded by a lipid membrane envelope acquired from the cell membranes, or functional proteins involved in virus replication. Unlike viruses with very complex nucleocapsids whose structures are not fully understood, such as members of the Poxviridae and Retroviridae family, most virions follow one of two basic structural symmetries: helical or icosahedral, giving a rod and spherical shape, respectively. The study of virus assembly dates back to 1955 when Fraenckel-Conrat and Williams [1] demonstrated that the simple capsid of the rod-shaped Tobacco Mosaic Virus (TMV) could be reconstituted in vitro from separated RNA and protein components. In fact, 2130 copies of the 158 amino acid coat protein (CP) self-assemble in a helical viral particle 300 nm in length. Since that time, structurally simple viruses have been found to be an excellent model for many aspects of self-assembly of macromolecular structures, because their molecules seem to have all the properties needed for the formation of large particles. This is not the case for more complex viruses such as the phage T4, which must follow an obligate assembly pathway and is mediated by several scaffolding factors and other viral or host components [2–5]
The assembly of the viral structural proteins into organized macromolecular structures (capsids) generates viral “empty shells” known as Virus Like Particles (VLPs). All VLPs lack viral nucleic acid and are noninfectious. In the last ten years methods used to directly visualize particles structure, such as cryo-electron microscopy (cryo-EM) and X-ray crystallography, as well as bio-molecular and biochemical methods like recombinant expression and mutational analysis of the structural subunits, have generated remarkable progress in different areas of structural virology and in the understanding of the assembly, shape, and functional properties of different VLPs [6]. The coat proteins that form a VLP can self-assemble or follow the more intricate assembly pathway. Viral coat proteins and VLPs have been produced in different prokaryotic and eukaryotic heterologous expression systems (Table 1). VLPs can be directly purified and used after the in vivo self-assembly or can be subjected to ex vivo reassembly to improve efficiency, product homogeneity and, if desired, may be packed together with therapeutically active molecules.
Table 1.
VLPs produced in heterologous systems other than plants.
| HOST | Structural subunit | Viral origin | Ref. | |
|---|---|---|---|---|
| Bacteria | E. coli | JGMV CP | Johnson grass mosaic virus | [22] |
| DGNNV CP | Dragon grouper nervous necrosis virus | [98] | ||
| (IPCV) -H CP | Indian peanut clump virus | [99] | ||
| S. thyphimurium | HBc/p re-S2 | Hepatitis B virus | [100] | |
| HPV16 L1 | Human papilloma virus | [101] | ||
|
| ||||
| Yeast | S. cerevisiae | HBsAg | Hepatitis B virus | [11] |
| HBc | Hepatitis B virus | [102] | ||
| S. pombe | HPV 16 L1 | Human papilloma virus | [103] | |
| P. pastoris | HCVc/E1 | Hepatitis C virus | [104] | |
|
| ||||
| Insect cells | S. frugiperda (Sf-9) | SIV | Simian immunodeficiency virus | [105] |
| NV CP | Norwalk virus | [81] | ||
| T. ni (Hi-5) | HPV 18 L1 | Human papilloma virus | [13] | |
|
| ||||
| Xenopus oocytes | X. laevis | HBc | Hepatitis B virus | [106] |
|
| ||||
| Mammalian cells | COS-1 | DENV2 E | Dengue virus 2 | [107] |
| CHO | DENV2 E | Dengue virus 2 | [107] | |
| HepG2 | L-HDAg | Hepatitis delta virus | [108] | |
| HeLa | L-HDAg | Hepatitis delta virus | [108] | |
| BHK | L-HDAg | Hepatitis delta virus | [108] | |
The major application of VLPs is their use in vaccines, especially as a safer alternative to attenuated live or inactivated killed virus based vaccines, including non-cultivable viruses. The fact that VLPs can induce protective antiviral immune responses without having the risk of infection represents a major advantage for the design safer vaccines. VLPs derived from viruses that infect the gastrointestinal system are very well suited to study mucosal immunization and oral delivery of antigens. In particular, extensive data relating to expression, assembly and immunity has been generated using VLPs derived from rotavirus and Norwalk virus (NV), both of which cause acute gastroenteritis in man and animals [7]. VLPs can generate a strong general humoral and mucosal immune response [8]. Like particulate emulsions, microparticles, and liposomes, VLPs can be targeted to antigen presenting cells (APC) indirectly and directly, in the latter case displaying or incorporating specific molecules that interact with dendritic cells (DC). Contact with monocytes and DC induces maturation and triggers a lymphoproliferative response together with production of inflammatory cytokines [9,10]. A very successful use of VLPs is the development of effective vaccines against the hepatitis B virus (HBV), the human papillomavirus (HPV), and NV. The HBV surface antigen (HBsAg-S) can self-assemble in to 22 nm lipoprotein VLPs containing about 100 HBsAg-S molecules without the contribution of nucleocapsid. HBV-like particles produced in Saccharomyces cerevisiae were the first example of a vaccine effective against a human viral infection created in a recombinant system [11]. Currently in the US, Merck & Co. and GlaxoSmithKline (GSK) are marketing FDA approved yeast derived HBV-like particles, known commercially as Recombivax HB and Energix-B respectively.
Papillomavirus VLPs are non-enveloped icosahedral structures of 55 nm in diameter, consisting of a regular array of 72 pentameric oligomers composed of the major coat protein, L1. They are able to induce a strong B-cell response at a very low dose even without the use of adjuvants. Merck and GSK are again playing a major role, having successfully completed Phase II clinical trials and are currently performing Phase III trials. The Merck vaccine, called Gardasil, targets HPV types 16 and 18, which cause about 70% of cervical cancer cases, and types 6 and 11 which cause about 90% of all cases of genital warts. L1 VLPs are synthesized in Saccharomyces cerevisiae [12]. The GSK vaccine called Cervarix is a bivalent vaccine targeting HPV 16 and 18, produced by the baculovirus system in Spodoptera frugiperda Sf-9 and Trichoplusia ni Hi-5 insect cells, respectively [13].
A series of studies, conducted mainly by the group of M. K. Estes (Baylor College of Medicine), revealed that NV-like particles, made from a single recombinant coat protein, are a very promising orally delivered vaccine. These VLPs are composed of 90 dimers of the virus coat protein arranged in a T=3 symmetry; they are stable at low pH and to lyophilization, and easily produced and purified in large quantities using the baculovirus expression system [14]. The VLP vaccine stimulated systemic and mucosal immune response when administered orally in mice [8] and humoral, mucosal and cellular immune response in human volunteers [10,15].
Another possible application of VLPs explored in recent years is their use as vessels for the delivery of small therapeutics and as display systems for biologically active molecules. Particularly attractive is their use in transporting viral vectors for gene therapy and DNA vaccines, exploiting in some cases the natural tropism of the viral particles. It has been reported that supercoiled plasmid DNA up to 17 kb was encapsidated in vitro inside simian virus 40 capsid proteins [15] and that the HPV-16 L1 VLPs are able to package unrelated plasmid DNA in vitro and then to deliver this foreign DNA to eukaryotic cells with the consequent expression of the encoded gene. The gene transfer rate observed was higher than DNA delivered alone or with liposome [16]. HBV nanoparticles have been recently proven to be incredibly successful in specifically delivering genes or drugs to human hepatocytes. HBsAg-L particles, derived from the expression of the L gene encoding preS2 + preS1 + S, are surrounded by a layer of phospholipids derived from the endoplasmic reticulum membrane, like HBV virions. Nucleic acids or small molecules can be introduced in to the yeast-derived VLPs by electroporation, and high targeting specificity was demonstrated using human cell lines and a mouse xenograft model [17]. The incorporation of oligodeoxynucleotides containing CpG motifs, a vaccine adjuvant known to interact with the toll-VLPs was also recently reported [18].
Heterologous epitope presentation is another attractive use of VLPs. Chimeric HBsAg particles displaying a poliovirus neutralization epitope stimulated the production of neutralizing antibodies against poliovirus [19], and another fusion of selected epitopes from HIV-1 and HCV to the HBsAg were immunogenic in rodents and primates [20,21]. A fusion between a 9 amino acid peptide derived from the Japanese encephalitis virus (JEV) E protein and a C-terminal truncated version of the Johnson grass mosaic virus (JGMV) generated chimeric VLPs in E. coli [22].
The immense potential of VLPs technology is just beginning to be fully understood and developed. In this review paper we describe the various methods of production of VLPs in green plants as an alternative expression system for large-scale production. In particular, we focus on vaccines as the major application, including updates on expression and assembly of VLPs in plant as well as their immunogenicity in model animals and in humans.
2. Green plant production
There are three main methods for production of recombinant proteins in plants: stable transformation of the nuclear or chloroplast genomes, and viral transient infection. Only nuclear and viral systems are considered below, as chloroplast transformation has never been reported for VLPs production.
2.1. Stable nuclear transformation
The introduction and expression of foreign DNA in the plant nuclear genome has been used to create commercially important new genetic characteristics such as viral, insect and herbicide resistance. Moreover, stably transformed plants can be used to produce valuable recombinant proteins. Transgenic plants have the advantage of permitting large scale, low-cost biomass production of selected high-expressing genes using agricultural practice, and the potential for crossing transgenic lines to obtain multiple proteins expressed in the same plant [23]. The first transgenic plants were produced in 1983 when the ability of Agrobacterium to transfer DNA plasmids to plants was first discovered [24,25].
DNA can be transferred into plants by either direct or indirect methods, the choice depending on the target species and on the traits being introduced. Direct DNA transfer techniques range from transfection of protoplast by electroporation or chemical methods to laser micropuncture of isolated cells [26,27]. The most common method is particle bombardment, also known as the biolistic method, a combination of biological and ballistic. Microprojectile bombardment is an important tool, especially when plants are recalcitrant to Agrobacterium mediated DNA transfer. Naked DNA is adsorbed on small metal particles, usually gold, tungsten or platinum, and shot inside the plant tissue using either gas or powder discharged systems (gene gun,[28]). Indirect DNA transfer implies the use of Agrobacterium for the delivery of exogenous DNA into plant cells. The natural host range of Agrobacterium includes several species of dicots, gymnosperms and a few monocots. Efficient transformation protocols have been developed for these plants, including several crop species. However, transformation efficiency varies in different species, and some are altogether recalcitrant. Agrobacterium-mediated transfer requires that the gene of interest be inserted in a DNA vector between the natural borders of the transfer DNA (T-DNA) present in the original plasmid of the bacteria, called the tumor inducing (Ti) plasmid. These borders are two sequences of imperfect direct repeats 25 base pairs long and are the only cis-elements required for DNA transfer from the bacteria to the plant cell nucleus [29].
Regardless of the method used to introduce the exogenous DNA, the gene of interest must be cloned into an expression cassette whose minimal requirements are a promoter and a terminator of transcription functional in the plant system. Another critical aspect is the early selection of the transformed cells from the non-transformed tissue, achieved by inclusion of a selectable marker that is often a resistance gene for an antibiotic or herbicide. From the transformed cells a fertile plant can be regenerated (Fig. 1 (A–E)). Unfortunately homologous recombination has not been efficiently achieved for plants. Thus, the transferred DNA integrates randomly in the genome so that its expression is subject to positional effects. Moreover, multiple locus insertions can occur, increasing the probability of unstable gene expression, gene silencing, and complex patterns of transgene inheritance. The analysis of several independently transformed lines to select the one that performs best in a standard agronomic environment is therefore a common practice. This, together with the length of time required to develop the transgenic plants and the often low level of expression, are the major disadvantages of stable genetic transformation.
Fig. 1.

Different stages of plant transformation, nuclear stable (A–F) and viral transient (G–I). (A) Selection of explants after co-cultivation with A. tumefaciens, (B–D) calli formation, (E) regeneration of whole transgenic plants, (F) generation of stable transgenic cell suspension culture (G–H) A. tumefaciens mediated viral infection in contained greenhouse environment, (I) expression of GFP reporter gene after 10 days post infection.
An alternative to whole plants is the use of plant cell cultures. Cells lack the extreme scalability of agricultural production systems, similar to bacterial and mammalian cells. On the other hand, like bacteria, they can be cultivated in simple media and, like mammalian and other eukaryotic cells, are able to synthesized complex multimeric proteins and perform eukaryotic posttranslational processes. They can be cultivated in different vessels according to the required scale of production, from normal shaker flasks and small fermenters up to large certified good manufacturing practice (cGMP) controlled bioreactors. Cell suspension cultures are claimed to be an excellent system for production of recombinant proteins under cGMP and certified good laboratory practice (cGLP), a detail that assumes great importance when the recombinant proteins are intended for clinical use, as are most VLPs based applications. Techniques for plant cell propagation were developed more than 50 years ago mainly for the production of small therapeutic molecules. At present, plant cell culture is regularly used to investigate basic questions in plant science and for the production of recombinant proteins.
Suspension cells are produced from an aggregate of undifferentiated cells (callus) generated by treating tissue explants from leaves, stems and roots with a combination of hormones that induce proliferation (Fig. 1 (A–D)). Calli and the derived cell lines can be created from a genetically modified plant, or unmodified plant cell lines may be transformed. Levels of expression are variable according to the construct used (promoter/enhancers/terminator combination), the kind of protein expressed, and the performance of the specific cell line. Several lines from different plant species such as Arabidopsis thaliana, rice, tomato, tobacco, alfalfa and soybean have been successfully used. The most popular lines, widely used for good efficiency of transformation, ease of propagation and growth rate characteristics, are derived from tobacco: BY2 cells from the Bright Yellow cultivar and NT1 cells from Nicotiana tobacco 1 [30–36].
2.2. Plant virus based expression systems
The use of viral gene vectors has found many applications in several molecular biology laboratories. Bacteriophages, in bacterial systems, are commonly used for gene cloning and gene library screening purposes, as well as recombinant protein display. In animal systems, double-stranded DNA viruses and RNA viruses have found application as expression and gene delivery systems.
Autonomously replicating plant viruses provide an alternative to stable genetic transformation for the expression of recombinant proteins in plants. The use of such vectors offers numerous advantages. Heterologous protein production can reach very high levels in a relatively short time ranging from 3 to 14 days post-infection, depending on the system used. Viral vectors provide the advantage of easily producing large numbers of different constructs that can be quickly tested. Moreover, fully functional and systemic infectious vectors are easily transmissible by mechanical inoculation, making large-scale infections feasible.
Plant viruses from which robust expression systems are being developed are single-stranded DNA geminiviruses, double-stranded non-integrating DNA pararetroviruses, and plus-sense RNA viruses. Within the past three decades, several genetic manipulation techniques have been developed to overcome the limitations in the use of viral vectors. Reverse transcription of viral RNA into cDNA allowed their insertion into plasmid vectors for molecular cloning. In vitro transcription systems facilitated the synthesis of infectious RNA transcripts from full-length cDNA clones [37]. In addition, the ability to clone viral genes into Agrobacterium tumefaciens enabled this bacterium to be used as a vector to deliver viral genomes into the plant cell’s nucleus, allowing the virus to be transcribed, to replicate and move throughout the plant [38]. Thus, viruses that are not mechanically transmissible, such as luteoviruses and many geminiviruses, can be introduced into plants after manipulation in vitro and cloning in E. coli and A. tumefaciens (Fig. 1G). In the case of the fully infectious Potato Virus X (PVX) based vector, the direct injection of a DNA plasmid carrying a cDNA copy of the viral RNA genome could initiate the viral infection [39,40].
The first viral vectors developed were the so-called “gene insertion vectors” or “full virus vectors” which consisted of a viral element that behaved as a wild type virus but also able to express an additional heterologous sequence. These vectors were essentially fully functional viruses that, despite carrying and expressing sequences of the gene of interest, retained infectivity, were relatively stable, had the ability to move systemically within their host, and produced infectious virus particles. The next step, mainly elicited by the instability of large insertions of foreign DNA and by the understanding that not all of the viral functions are necessary in an expression vector, was to develop “gene replacement vectors” substituting existing viral genes with foreign genes. The extreme evolution of this concept lead to deconstructed viral vectors missing several components of the original virus and usually delivered to the plant by independent constructs.
Two full virus vectors, the potato virus X (PVX) and the cowpea mosaic virus (CPMV), and two deconstructed vectors, based upon bean yellow dwarf virus (BeYDV) and magnICON® tobacco mosaic virus based (TMV), have been used to produced heterologous VLPs in plants.
2.2.1. Full virus vectors: PVX and CPMV
Historically both vectors have been extensively used for epitope display. Immunodominant peptides were fused with the viral coat proteins and the resulting chimeric full viral particles were able to assemble and showed different degrees of immunogenicity when delivered in animal studies [39,41–43]. These chimeric viruses differ from VLPs in that they are fully infectious agents containing the viral genome. However, recently the two systems have been used for the production of heterologous VLPs [40].
CPMV belongs to the Comoviridae family and has an icosahedral virion made of two capsid proteins. The viral genome consists of two separately positive-strand RNA molecules of 6 kb (RNA1) and 3.5 kb (RNA2). RNA1 encodes the proteins involved in replication while RNA2 encodes the proteins necessary for cell-to-cell movement and the two capsid proteins. PVX belongs to the Potexviridae family and has a rod-shaped virion made of different copies of the same coat protein. The genome consists of a single stranded positive monopartite RNA.
2.2.2. Deconstructed viral vectors: BeYDV and the ICON expression systems
Geminiviruses, named for their unique geminate capsid morphology, have small single-stranded circular DNA genomes that replicate in the nuclei of the infected cells via a rolling circle, forming a double-stranded DNA intermediate. BeYDV is monopartite and belongs to the Mastreviruses genus adapted to dicotyledonous plants. The system does not involve the use of the viral movement and coat protein and is deconstructed into two vectors, each containing different portions of the viral genome. One vector encodes for the Rep protein, the “trans-factor” which is necessary for rolling circle replication. The second vector incorporates the cis-acting elements of the BeYDV genome, essential for its episomal replication. In the context of a plant expression cassette, the gene of interest is contained between the viral DNA elements. The proof of concept for this system was shown using NT1 cell suspension culture [44] but the system is also functional on whole leaf delivered by agroinfection.
Icon Genetics (Halle, Germany) developed an extraordinary deconstructed system from TMV, a positive single stranded RNA virus. The system is divided into two major cDNA modules, the 5’ of which contains the viral RNA dependent RNA polymerase and the movement protein, and the 3’ module that carries the gene of interest and is depleted of the coat protein. The modules are delivered to the plant cell nucleus by Agrobacterium mediated infection and are assembled together in vivo by the action of a site specific recombinase driven by a constitutive promoter delivered as well by a third Agrobacterium cell line. In addition, different 5’ modules carry different organelle targeting signals, which fuse in frame with the gene of interest after recombination and nuclear processing. This feature allows the easy testing of different cell compartment accumulation with minimal cloning effort [45,46]. Both systems are being extensively used in our laboratory to produce VLPs assembled from Norovirus virus capsid proteins, L1 protein from HPV-16, HBsAg, and HBc (unpublished results).
3. Expression, assembly and immunogenicity of VLPs produced in plants
The production of correctly formed VLPs in different plant systems has been extensively reported (Table 2). Our group has examined the use of plants for expression of vaccine antigens with the aim of convenient delivery of minimally processed plant material by ingestion [47]. Antigens having the ability to assemble VLPs are of particular interest, because: 1) the compact, highly ordered structures of VLPs may provide resistance to degradative enzymes in the gut; 2) the particulate nature of VLPs allows them to be efficiently sampled by the “M” cells of the gut epithelium that transport antigens across the mucosal barrier; and 3) the presence of a structure that mimics the authentic viral particle may present a “danger signal” that can overcome the perception of gut antigens as benign and thus provoke potent immune responses.
Table 2.
VLPs produced in plants
| Antigen | Expression system | Highest Yield | VLP proof | Ref. |
|---|---|---|---|---|
| S-HBsAg | Transgenic tobacco | 0.01% TSP leaf | G, EM | [49] |
| Transgenic potato | 16 μg/g tuber | EM | [52] | |
| Transgenic lupine callus | 150 ng/g FW | ND | [50] | |
| Transgenic lettuce | 5.5 ng/g leaf | ND | [50] | |
| Transgenic tobacco cells | 2 μg/g of FW | G | [54] | |
| 8 μg/g FW | G, EM | [53] | ||
| Transgenic cherry tomatillo | 10 ng/g FW fruit | ND | [51] | |
| Transgenic soybean cells | 74 μg/g FW | G, EM | [53] | |
|
| ||||
| M-HBsAg | Transgenic N. benthamiana | 394 ng/mg TSP leaf | G | [67] |
| Transgenic potato | 15 ng/mg TSP tuber | ND | [68] | |
| 0.09% TSP tuber | ND | [69] | ||
|
| ||||
| VSPá-S-HBsAg | Transgenic tobacco cells | 226 ng/mg TSP | G | [57] |
| GFP-HBsAg | Transient/N. benthamiana | 25 ng/mg TSP leaf | G, EM | [58] |
|
| ||||
| HBcAg | Transgenic tobacco | 24 μg/g FW leaf | EM | [76] |
| PVX/ N. benthamiana | 50 μg/g leaf | EM | [40] | |
| CPMV/cowpea | 10 μg/g leaf | EM | [40] | |
| MagnICON/ N. benthamiana | 2 mg/g FW leaf | G, EM | [78] | |
|
| ||||
| NVCP | Transgenic tobacco | 0.23% of TSP leaf | EM, 38nm | [84] |
| Transgenic potato | 10–20 μg/g tuber | ND | [84] | |
| Transgenic tomato | 20–30 μg/g dry fruit | G | [67] | |
| MagnICON/ N. benthamiana | 0.8 mg/g FW leaf | G, EM | [UR] | |
|
| ||||
| HPV11-L1 | Transgenic potato | 20 ng per g tuber | EM | [95] |
|
| ||||
| HPV16 L1 | Transgenic tobacco | 0.5% of TSP leaf | G | [97] |
| Transgenic potato | 0.2% of TSP tuber | G, EM | [97] | |
| Transgenic tobacco | 2–4 μg/kg FW leaf | EM | [95] | |
| MagnICON/ N. benthamiana | 86 μg/g FW leaf | ND | [UR] | |
Abbreviation used: TSP, total soluble protein; FW, fresh weight; G, gradient; EM, electron microscopy; ND, not determined; UR unpublished result.
3.1. Hepatitis B surface antigen (HBsAg)
Vaccination is the best way to prevent hepatitis B virus (HBV) infection. Plasma-derived 22 nm virus-like particles consisting of HBsAg were used as the first-generation HBV vaccine [48]. Extraction and purification of HBsAg from recombinant yeast resulted in homogeneous HBsAg particles that constituted the second-generation vaccine [11].
Inspired by this first and the most successful example of recombinant subunit vaccine in history, our group first tested the expression of HBsAg in plants [49]. When expressed in transgenic tobacco, HBsAg was correctly folded to form the conformational ‘a’ determinant, at levels up to 66 ng/mg of total soluble protein from leaf. To facilitate oral delivery, HBsAg was expressed in lettuce leaves and cherry tomatillos, however, at very low levels (<0.000001% of fresh weight) [50,51]. The highest HBsAg expression was obtained in transgenic potato at levels up to 16 μg/g tuber [52] by optimizing the regulatory elements and subcellular targeting signals. Production of HBsAg in plant cell culture systems has also been attempted. The yield of HBsAg approached 22 mg/L in soybean culture while it was one tenth of that in tobacco culture [53]. NT1 cell lines were used to express S gene either with or without a C-terminal ER retention signal [54]. Both versions correctly assembled 22 nm VLPs as demonstrated by sedimentation analysis. Interestingly, the version not retained in the ER was secreted in the medium. The possibility of having the recombinant protein in the culture medium would greatly facilitate downstream processing, bypassing problems relating to plant cell-derived contaminants. However, the levels of secreted HBsAg were very poor, at 10 μg per liter of medium [54]. The authors suggested that optimization, in terms of stronger promoters, 5’ UTR translational enhancer as well as the employment of different secretion signals to increase the export of particles in the medium, is required for production purposes.
In mammalian cells, HBsAg is co-translationally inserted into the endoplasmic reticulum (ER) membrane [55] and dimerized via intermolecular disulfide bonds in the ER lumen. Dimers are then transported to the post-ER, pre-Golgi compartment where oligomerization and VLP formation occur prior to secretion [56]. For plant systems, HBsAg dimer formation was demonstrated by Western blot of protein extracts from cultured plant cells [53,57] and from infiltrated leaves [58], indicating that plants can provide a suitable environment for correct processing of HBsAg. The ability of plant-produced HBsAg to assemble VLPs was determined by sucrose gradients and electron microscopy. Mason et al. found that HBsAg purified from extracts of transgenic tobacco leaves was present as spherical VLPs with an average diameter of 22 nm (Fig. 2A), similar to those derived from yeast [49]. Similar results were obtained from N. benthamiana leaves transiently expressing HBsAg [58]. Electron microscopy of sectioned plant samples expressing HBsAg further revealed the structure of HBsAg in planta. In transgenic potato leaves, HBsAg was found inside membrane vesicles as 17 nm particulate structures. In transgenic cell cultures and potato tubers (which were used in the oral immunization of mice [59,60] and human clinical trials [60]), the antigen accumulated within dilated ER membranes as tubular structures [61], similar to those observed in Chinese hamster ovary (CHO) cells expressing HBsAg [62], but with a complex size distribution. Thus, HBsAg expression in plant cells seems to adopt the same mechanism as in mammalian cells for ER insertion and post-translational processing. The resulting VLP formation and ER dilation may interfere with normal cell functions, and most likely explains the low HBsAg expression and toxic effects observed in plant systems.
Fig. 2.
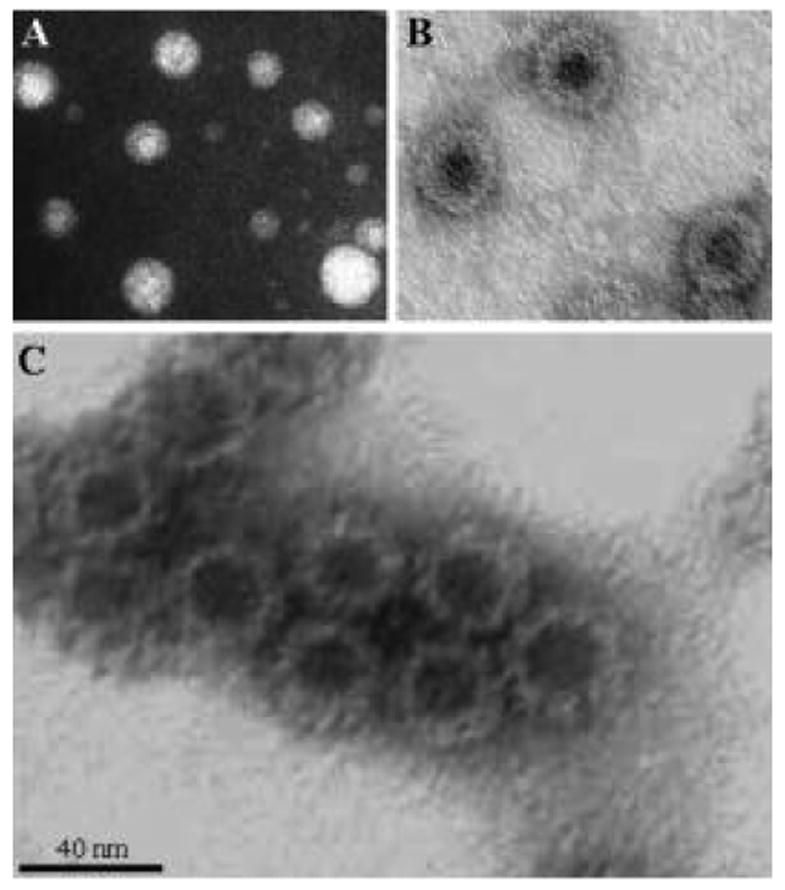
Plant produced VLPs visualized by negative staining and electron microscopy. (A) HBsAg particles from transgenic tobacco, (B) HBcAg particles from N. benthamiana infected with the MagnICON viral vector, (C) NVCP particles from transgenic tobacco.
When delivered by intraperitoneal (i.p.) injection, partially purified HBsAg VLPs from transgenic tobacco leaves stimulated antibody and T cell responses in mice, as did the yeast recombinant vaccine [63]. Feeding mice with 5 grams of raw potato containing ~42 μg of HBsAg primed and boosted high-level anti-HBsAg IgG responses, although 10 μg of cholera toxin (CT) adjuvant was required. It was also found that cooking HBsAg potato significantly reduced its immunogenicity, with the highest anti-HBsAg titer being 135 mIU/ml, in contrast to the peak secondary response of 3,300 mIU/ml in mice fed uncooked tubers [59].
For humans, the protective anti-HBsAg level has been defined as >10 mIU/ml. The ability of HBsAg-expressing plant materials to elicit such antibody responses has been demonstrated in two clinical trials. Kapusta and colleagues showed that ingestion of two doses of transgenic lettuce containing 1μg of HBsAg elicited serum anti-HBsAg IgG at >10 mIU/ml in two out of three naïve human individuals, although antibody levels declined rapidly [50]. In a larger clinical trial, doses of 100 g of uncooked potato tuber containing ~850 μg of HBsAg were administered by ingestion to volunteers who were previously vaccinated against HBV; in 10 of 16 volunteers who ate three doses of potatoes and 9 of 17 who ate 2 doses, serum anti-HBsAg titers increased significantly after oral doses [60].
Most efforts in plant expression of HBsAg have focused on the major surface antigen (also called small surface antigen, S-protein or SHBs). Some groups have also examined plant expression of HBsAg middle protein (M-protein), which has an additional 55 amino acid pre-S2 region at the N-terminus of the S-protein. The yeast-derived recombinant pre-S2 component provides additional T-cell help to render higher titer [64] and earlier appearance [65] of anti-HBsAg response in clinical trials and to overcome non-responsiveness to S protein immunization in mice [66] and humans [65]. Indeed, our group showed that i.p. injection of transgenic N. benthamiana-derived HBsAg M protein generated better serum anti-HBsAg responses in mice than the S protein [67]. However, the levels of M protein expression in N. benthamiana [67] and in potato [68,69] are consistently lower than those of S protein by 3- to 10-fold.
Our group also demonstrated that it is feasible to modify the HBsAg with an N-terminal fusion of up to 239 amino acids without altering its major antigenic properties. We showed that a fusion between the green fluorescent protein (GFP) and the HBsAg S-protein transiently expressed in N. benthamiana leaves retained its abilities to form immunogenic ‘a’ determinants and to assemble VLPs. Interesting, co-expression of the fusion and native HBsAg led to the formation of heterodimers, suggesting the assembly of mosaic VLPs [58]. Furthermore, a plant signal peptide-HBsAg fusion produced in tobacco culture cells accumulates better as ‘a’ determinant, is more stable, and is more immunogenic than the unmodified HBsAg when i.p. injected into mice [57]. In some studies (unpublished), we found that plant-derived HBsAg particles could be used to present foreign T-cell and B-cell epitopes as well as a mucosal-targeting protein, resulting in enhanced or multivalent immune responses.
3.2. Hepatitis B core antigen (HBcAg)
During HBV infection, HBcAg self-assembles into subviral nucleocapsid particles packaging the viral polymerase and pregenomic RNA during HBV infection. Recombinant HBcAg from a variety of non-plant expression systems assembles VLPs composed of 180 or 240 subunits arranged with T=3 or T=4 icosahedral symmetry [70], and stimulates high-titer, long-lasting antibody responses, making it an attractive carrier for high-density presentation of foreign epitopes (reviewed in [71–73]). HBcAg-specific antibody does not provide protection against HBV infection. However, it was found that HBcAg has an immunoenhancing effect on co-delivered HBsAg, and was hence suggested for inclusion in the design of more potent therapeutic and preventive vaccines against HBV infection [74,75].
HBcAg was expressed in transgenic tobacco leaves at levels up to 24 μg per gram of fresh weight [76]. HBcAg was also transiently expressed in plants using two viral vector systems based on PVX and CPMV. In the PVX vector, the HBcAg gene was inserted as an extra ORF downstream of the triple gene block and upstream of the coat protein and a duplication of the coat protein subgenomic promoter was used to drive the transgene expression. N. benthamiana plants were inoculated directly with the DNA from this construct and systemic infection was achieved. In the CPMV vector system, the HBc coding sequence was fused to the CPMV small coat protein via the foot and mouth disease virus (FMDV) 2A peptide, which mediates, by ribosomal skip [77], the discrete production of separate HBc and small coat proteins. Immunosorbent electron microscopy clearly demonstrated that both systems were able to produce correctly assembled HBc VLPs. However, especially in the case of CPMV, the expression levels were very low, 10 μg per g fresh weight, perhaps due to a lack of plant codon optimization. Another disadvantage of full virus based vectors for the production of VLPs is the co-production of wild-type virus particles that can create problems in downstream bioprocessing, purification, and manufacture. The immunogenicity of VLPs produced by both the PVX and CPMV vector systems was not tested.
Recently, our group used a “deconstructed”, TMV-based expression system [45] for rapid production of HBcAg using a plant-optimized gene, and obtained striking results. The antigen produced in N. benthamiana leaves makes up to 7% of total soluble protein (TSP) or 0.2% of fresh weight (FW), and is present as 30 nm VLPs with morphology similar to that of its E.coli-derived counterpart (Fig. 2B). When i.p. injected into mice, the partially purified HBcAg VLPs from plant leaves stimulated serum IgG responses with the same timing and intensity as E. coli-derived VLPs. Furthermore, mucosal immunization with plant-derived VLPs without any adjuvants evoked the serum IgG and mucosal IgA responses [78], suggesting that the plant-derived HBcAg VLPs is an excellent system for epitope presentation and mucosal delivery. Indeed, we produced a fusion between HBcAg and the neutralizing epitope of HPV16 L2 protein [79] in plant leaves, and found that the fusion formed chimeric VLPs (unpublished result).
3.3. NVCP
Norwalk virus (NV) is a member of the Caliciviridae family and cause epidemic acute gastroenteritis in humans [80]. Efforts to develop vaccines against NV infection have been made mainly by the research group led by M. Estes. They discovered that Norwalk virus capsid protein (NVCP) expressed in insect cells yield VLPs with sizes of 38 nm and 23 nm [81,82], react with sera from infected humans, are acid-stable and stimulate serum responses in humans. More significantly, oral immunization with NVLP induced systemic and mucosal responses in mice [8] and in humans [10,83].
NVCP expression in transgenic plants was first demonstrated by our group. The level of NVCP accumulation was up to 0.23% of the total soluble protein in the leaves of tobacco transformants and up to 0.37% in potato tubers (34 μg per g of tuber weight) (Table 2). The presence of 38 nm VLPs purified from transgenic tobacco leaves was demonstrated by negative staining and electron microscopy (Fig. 2C). However, only ~50% of the NVCP subunits were assembled as VLPs in the potato cells. Oral immunization of mice with purified VLPs from tobacco or with raw NVCP potato tubers induced serum IgG and fecal IgA responses, which were slightly enhanced by the supplement of CT adjuvant [84]. NVCP was later produced in tomato at up to 20 μg per gram fruit mass, primarily in the form of the 23 nm VLP. Feeding mice with freeze-dried NVCP-expressing tomato powder stimulated excellent IgG and IgA responses against NVCP [67]. By using a synthetic, plant codon-optimized gene, NVCP accumulation in tomato fruit was increased to 8% of total soluble protein or 160 μg per g dry weight, with the majority of NVCP being 38 nm and 23 nm VLPs (Zhang & Mason, manuscript submitted).
A clinical trial with transgenic potatoes expressing NVCP VLPs was conducted at the Center for Vaccine Development [85]. Twenty adult volunteers ingested either two or three doses each of 150 g raw transgenic potato containing 215–750 μg NVCP (expression was variable). Nineteen of the 20 subjects showed significant increases in the numbers of specific anti-NVCP antibody-secreting cells of the IgA subtype, and six out of 20 developed increases in IgG antibody-secreting cells. Although only six volunteers showed increased anti-NVCP IgA in their stool samples, the 17-fold mean increase among these was substantial. This study proved that orally delivered plant-expressed VLPs could stimulate immune responses. In light of these supporting studies using plant derived material we are currently utilizing the magnICON® and the BeYDV replicon systems to reach higher levels of expression. With both systems VLPs are properly assembled and the levels of accumulation are at least 20 times higher than reported in stably transformed lines (Santi & Mason, unpublished result).
3.4. Human papillomavirus L1 protein
More than 99% of cervical cancers are associated with infection by high-risk types of human papillomavirus (HPV). The HPV major capsid L1 protein produced in insect cells self assembles into VLP structures and induce high titers of antibodies [86] that prevent infection by authentic virions [87]. Several clinical trials with injectable HPV L1 VLP-based vaccines have been carried out and their prophylactic efficacy was demonstrated [88–93]. Oral immunization with HPV VLPs was also found to be successful in mice with the addition of Escherichia coli heat-labile enterotoxin mutant R192G or CpG DNA as adjuvants [94].
Several attempts have been made to produce HPV L1 protein in transgenic plants. Our group expressed a plant codon-optimized gene for HPV11 L1 protein in potato and found that deletion of the nuclear localization signal enhanced mRNA stability and protein accumulation [95]. Two other groups produced HPV16 L1 protein in transgenic tobacco and potato [96,97]. Interestingly, Biemelt et al found that the L1 gene optimized for expression in human cells, resulted in better protein accumulation than the unmodified plant codon-optimized gene, and a translation enhancer derived from the tobacco mosaic virus greatly increased transcript stability and L1 protein accumulation. In general, L1 protein expression level in transgenic plants was from low to modest (Table 2). However, in all three cases, the plant-expressed L1 protein obtained correctly folded conformations and, more importantly, assembled into VLPs with the diameter of 55 nm as evidenced by electron microscopy. By sucrose gradient fractionation it was further shown that the L1 protein is present primarily in a VLP form in transgenic potatoes and with a small portion of capsomeres in transgenic tobacco [97].
Plant-derived HPV16 VLPs were found to be as immunogenic as those from insect cells in mice when administered subcutaneously (s.c.). However, feeding mice four times with 5 g of transgenic potatoes did not evoke detectable serum antibody responses; with the supplement of CpG plasmid DNA or cholera toxin B subunit as adjuvants, weak and transient responses against L1 were observed in 3 out of 16 mice. Similar results were obtained from oral vaccination of mice with HPV11 L1-expressing potatoes. Both groups found that the L1-specific responses could be significantly boosted by a subimmunogenic dose of purified VLPs delivered by s.c. injection or by oral gavage, indicating the priming effect and the establishment of VLP-specific immune memory by the feeding of L1-containing tubers. No clinical trial with transgenic plant materials is being conducted at this point, because the low level of expression (such as ~20 ng per g of fresh tuber) in plants precludes the direct use of these materials in human subjects[95,97]. It is apparent that constitutive high expression of L1 protein is toxic to plants; thus, future efforts should be focused on the use of developmentally regulated or chemically inducible promoters to control its expression. With regard to viral infection, we are currently using the magnICON® system for the expression of a plant optimized HPV16 L1 antigen gene and have achieved expression levels of up to 86 μg per gram of fresh leaf weight (~ 1% TSP).
4. Conclusions
VLP production in plant systems is a very promising technology that has been studied primarily for vaccine production. Although much research has focused on oral vaccine delivery of minimally processed (e.g. freeze-dried) plant materials, extraction and purification of VLPs for parenteral delivery is also a highly feasible strategy. Recent developments in plant viral vectors for transient expression, as well as regulated induction of stably integrated viral replicons, have greatly increased expression levels and indicate that plant systems can compete with microbial fermentation for production of VLPs. Future work will focus on scaling up to commercial production levels in cGMP conditions for selected antigens, and development of chimeric VLPs that display heterologous antigenic determinants.
Acknowledgments
We thank Dr. Michelle Kilcoyne for the careful review of the manuscript. We also thank all the current and former members of the Mason lab involved with the production of VLPs in plant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fraenkel-Conrat H, Williams RC. Proc Natl Acad Sci U S A. 1955;41:690–698. doi: 10.1073/pnas.41.10.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood WB, Edgar RS, King J, Lielausis I, Henninger M. Fed Proc. 1968;27:1160–1166. [PubMed] [Google Scholar]
- 3.Kikuchi Y, King J. J Supramol Struct. 1975;3:24–38. doi: 10.1002/jss.400030104. [DOI] [PubMed] [Google Scholar]
- 4.Dokland T. Cell Mol Life Sci. 1999;56:580–603. doi: 10.1007/s000180050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chromy LR, Pipas JM, Garcea RL. Proc Natl Acad Sci U S A. 2003;100:10477–10482. doi: 10.1073/pnas.1832245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson JE, Chiu W. Curr Opin Struct Biol. 2000;10:229–235. doi: 10.1016/s0959-440x(00)00073-7. [DOI] [PubMed] [Google Scholar]
- 7.Estes MK, Ball JM, Crawford SE, O'Neal C, Opekun AA, Graham DY, Conner ME. Adv Exp Med Biol. 1997;412:387–395. doi: 10.1007/978-1-4899-1828-4_61. [DOI] [PubMed] [Google Scholar]
- 8.Ball JM, Hardy ME, Atmar RL, Conner ME, Estes MK. J Virol. 1998;72:1345–1353. doi: 10.1128/jvi.72.2.1345-1353.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz P, Thompson CD, Day PM, Bacot SM, Lowy DR, Schiller JT. Clin Immunol. 2003;106:231–237. doi: 10.1016/s1521-6616(02)00039-6. [DOI] [PubMed] [Google Scholar]
- 10.Tacket CO, Sztein MB, Losonsky GA, Wasserman SS, Estes MK. Clin Immunol. 2003;108:241–247. doi: 10.1016/s1521-6616(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 11.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Nature. 1984;307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 12.Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, Wheeler CM, Koutsky LA, Malm C, Lehtinen M, Skjeldestad FE, Olsson SE, Steinwall M, Brown DR, Kurman RJ, Ronnett BM, Stoler MH, Ferenczy A, Harper DM, Tamms GM, Yu J, Lupinacci L, Railkar R, Taddeo FJ, Jansen KU, Esser MT, Sings HL, Saah AJ, Barr E. Lancet Oncol. 2005;6:271–278. doi: 10.1016/S1470-2045(05)70101-7. [DOI] [PubMed] [Google Scholar]
- 13.Harper DM, Franco EL, Wheeler C, Ferris DG, Jenkins D, Schuind A, Zahaf T, Innis B, Naud P, De Carvalho NS, Roteli-Martins CM, Teixeira J, Blatter MM, Korn AP, Quint W, Dubin G. Lancet. 2004;364:1757–1765. doi: 10.1016/S0140-6736(04)17398-4. [DOI] [PubMed] [Google Scholar]
- 14.Prasad BV, Hardy ME, Jiang X, Estes MK. Arch Virol. 1996;(Suppl 12):237–242. doi: 10.1007/978-3-7091-6553-9_25. [DOI] [PubMed] [Google Scholar]
- 15.Kimchi-Sarfaty C, Arora M, Sandalon Z, Oppenheim A, Gottesman MM. Human Gene Therapy. 2003;14:167–177. doi: 10.1089/104303403321070865. [DOI] [PubMed] [Google Scholar]
- 16.Touze A, Coursaget P. Nucleic Acids Res. 1998;26:1317–1323. doi: 10.1093/nar/26.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada T, Iwasaki Y, Tada H, Iwabuki H, Chuah MK, VandenDriessche T, Fukuda H, Kondo A, Ueda M, Seno M, Tanizawa K, Kuroda S. Nat Biotechnol. 2003;21:885–890. doi: 10.1038/nbt843. [DOI] [PubMed] [Google Scholar]
- 18.Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF. J Immunol. 2004;172:1777–1785. doi: 10.4049/jimmunol.172.3.1777. [DOI] [PubMed] [Google Scholar]
- 19.Delpeyroux F, Chenciner N, Lim A, Malpiece Y, Blondel B, Crainic R, van der Werf S, Streeck RE. Science. 1986;233:472–475. doi: 10.1126/science.2425433. [DOI] [PubMed] [Google Scholar]
- 20.Lee IH, Kim CH, Ryu WS. J Med Virol. 1996;50:145–151. doi: 10.1002/(SICI)1096-9071(199610)50:2<145::AID-JMV7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 21.Schlienger K, Mancini M, Riviere Y, Dormont D, Tiollais P, Michel ML. J Virol. 1992;66:2570–2576. doi: 10.1128/jvi.66.4.2570-2576.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saini M, Vrati S. Protein Expr Purif. 2003;28:86–92. doi: 10.1016/s1046-5928(02)00647-2. [DOI] [PubMed] [Google Scholar]
- 23.Tacket CO, Mason HS, Losonsky G, Clements JD, Levine MM, Arntzen CJ. Nat Med. 1998;4:607–609. doi: 10.1038/nm0598-607. [DOI] [PubMed] [Google Scholar]
- 24.Herrera-Estrella L, Depicker A, Van Montagu M, Schell J. Biotechnology. 1992;24:377–381. [PubMed] [Google Scholar]
- 25.Fraley RT, Rogers SG, Horsch RB, Sanders PR, Flick JS, Adams SP, Bittner ML, Brand LA, Fink CL, Fry JS, Galluppi GR, Goldberg SB, Hoffmann NL, Woo SC. Proc Natl Acad Sci U S A. 1983;80:4803–4807. doi: 10.1073/pnas.80.15.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davey MR, Rech EL, Mulligan BJ. Plant Mol Biol. 1989;13:273–285. doi: 10.1007/BF00025315. [DOI] [PubMed] [Google Scholar]
- 27.Badr YA, Kereim MA, Yehia MA, Fouad OO, Bahieldin A. Photochem Photobiol Sci. 2005;4:803–807. doi: 10.1039/b503658e. [DOI] [PubMed] [Google Scholar]
- 28.Christou P. Methods Cell Biol. 1995;50:375–382. [PubMed] [Google Scholar]
- 29.Zupan JR, Zambryski P. Plant Physiol. 1995;107:1041–1047. doi: 10.1104/pp.107.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathur J, Szabados L, Schaefer S, Grunenberg B, Lossow a, Jonas-Straube E, Schell J, Koncz C, Koncz-Kalman Z. The Plant Journal. 1998;13:707–716. doi: 10.1046/j.1365-313x.1998.00059.x. [DOI] [PubMed] [Google Scholar]
- 31.Kawasaki T, Henmi K, Ono E, Hatakeyama S, Iwano M, Satoh H, Shimamoto K. Proc Natl Acad Sci U S A. 1999;96:10922–10926. doi: 10.1073/pnas.96.19.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon TH, Kim YS, Lee JH, Yang MS. Biotechnology Letters. 2003;25:1571–1574. doi: 10.1023/a:1025409927790. [DOI] [PubMed] [Google Scholar]
- 33.Chaubet N, Petiard V, Parelleux A. Plant Sci Lett. 1981;22:369–378. [Google Scholar]
- 34.An GH. Plant Physiol. 1985;79:568–570. doi: 10.1104/pp.79.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horn M, Sherrard J, Widholm J. Plant Physiol. 1983:426–429. doi: 10.1104/pp.72.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagata T, Kumagai F. Methods in Cell. Science. 1999;21:123–127. doi: 10.1023/a:1009832822096. [DOI] [PubMed] [Google Scholar]
- 37.Ahlquist P, Janda M. Mol Cell Biol. 1984;4:2876–2882. doi: 10.1128/mcb.4.12.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turpen TH, Turpen AM, Weinzettl N, Kumagai MH, Dawson WO. J Virol Methods. 1993;42:227–239. doi: 10.1016/0166-0934(93)90035-p. [DOI] [PubMed] [Google Scholar]
- 39.Marusic C, Rizza P, Lattanzi L, Mancini C, Spada M, Belardelli F, Benvenuto E, Capone I. J Virol. 2001;75:8434–8439. doi: 10.1128/JVI.75.18.8434-8439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mechtcheriakova IA, Eldarov MA, Nicholson L, Shanks M, Skryabin KG, Lomonossoff GP. JVirol. 2006;131:10–15. doi: 10.1016/j.jviromet.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 41.Usha R, Rohll JB, Spall VE, Shanks M, Maule AJ, Johnson JE, Lomonossoff GP. Virology. 1993;197:366–374. doi: 10.1006/viro.1993.1598. [DOI] [PubMed] [Google Scholar]
- 42.Porta C, Spall VE, Loveland J, Johnson JE, Barker PJ, Lomonossoff GP. Virology. 1994;202:949–955. doi: 10.1006/viro.1994.1417. [DOI] [PubMed] [Google Scholar]
- 43.McLain L, Porta C, Lomonossoff GP, Durrani Z, Dimmock NJ. AIDS Res Hum Retroviruses. 1995;11:327–334. doi: 10.1089/aid.1995.11.327. [DOI] [PubMed] [Google Scholar]
- 44.Mor TS, Moon YS, Palmer KE, Mason HS. Biotechnol Bioeng. 2003;81:430–437. doi: 10.1002/bit.10483. [DOI] [PubMed] [Google Scholar]
- 45.Marillonnet S, Giritch A, Gils M, Kandzia R, Klimyuk V, Gleba Y. Proc Natl Acad Sci U S A. 2004 doi: 10.1073/pnas.0400149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marillonnet S, Thoeringer C, Kandzia R, Klimyuk V, Gleba Y. Nat Biotechnol. 2005;23:718–723. doi: 10.1038/nbt1094. [DOI] [PubMed] [Google Scholar]
- 47.Mason HS, Warzecha H, Mor T, Arntzen CJ. Trends Mol Med. 2002;8:324–329. doi: 10.1016/s1471-4914(02)02360-2. [DOI] [PubMed] [Google Scholar]
- 48.Krugman S, Overby LR, Mushahwar IK, Ling CM, Frosner GG, Deinhardt F. N Engl J Med. 1979;300:101–106. doi: 10.1056/NEJM197901183000301. [DOI] [PubMed] [Google Scholar]
- 49.Mason HS, Lam DM, Arntzen CJ. Proc Natl Acad Sci U S A. 1992;89:11745–11749. doi: 10.1073/pnas.89.24.11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kapusta J, Modelska A, Figlerowicz M, Pniewski T, Letellier M, Lisowa O, Yusibov V, Koprowski H, Plucienniczak A, Legocki AB. FASEB J. 1999;13:1796–1799. doi: 10.1096/fasebj.13.13.1796. [DOI] [PubMed] [Google Scholar]
- 51.Gao Y, Ma Y, Li M, Cheng T, Li SW, Zhang J, Xia NS. World J Gastroenterol. 2003;9:996–1002. doi: 10.3748/wjg.v9.i5.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter LJ, Thanavala Y, Arntzen CJ, Mason HS. Nat Biotechnol. 2000;18:1167–1171. doi: 10.1038/81153. [DOI] [PubMed] [Google Scholar]
- 53.Smith ML, Mason HS, Shuler ML. Biotechnol Bioeng. 2002;80:812–822. doi: 10.1002/bit.10444. [DOI] [PubMed] [Google Scholar]
- 54.Sunil Kumar GB, Ganapathi TR, Revathi CJ, Prasad KS, Bapat VA. Protein Expr Purif. 2003;32:10–17. doi: 10.1016/j.pep.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 55.Eble BE, Lingappa VR, Ganem D. Mol Cell Biol. 1986;6:1454–1463. doi: 10.1128/mcb.6.5.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huovila AP, Eder AM, Fuller SD. J Cell Biol. 1992;118:1305–1320. doi: 10.1083/jcb.118.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sojikul P, Buehner N, Mason HS. Proc Natl Acad Sci U S A. 2003;100:2209–2214. doi: 10.1073/pnas.0438037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang Z, Mason HS. Plant Biotechnology Journal. 2004;2:241–249. doi: 10.1111/j.1467-7652.2004.00068.x. [DOI] [PubMed] [Google Scholar]
- 59.Kong Q, Richter L, Yang YF, Arntzen CJ, Mason HS, Thanavala Y. Proc Natl Acad Sci U S A. 2001;98:11539–11544. doi: 10.1073/pnas.191617598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thanavala Y, Mahoney M, Pal S, Scott A, Richter L, Natarajan N, Goodwin P, Arntzen CJ, Mason HS. Proc Natl Acad Sci U S A. 2005;102:3378–3382. doi: 10.1073/pnas.0409899102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith ML, Richter L, Arntzen CJ, Shuler ML, Mason HS. Vaccine. 2003;21:4011–4021. doi: 10.1016/s0264-410x(03)00268-8. [DOI] [PubMed] [Google Scholar]
- 62.Patzer EJ, Nakamura GR, Yaffe A. J Virol. 1984;51:346–353. doi: 10.1128/jvi.51.2.346-353.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thanavala Y, Yang YF, Lyons P, Mason HS, Arntzen C. Proc Natl Acad Sci U S A. 1995;92:3358–3361. doi: 10.1073/pnas.92.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miskovsky E, Gershman K, Clements ML, Cupps T, Calandra G, Hesley T, Ioli V, Ellis R, Kniskern P, Miller W. Vaccine. 1991;9:346–350. doi: 10.1016/0264-410x(91)90062-b. [DOI] [PubMed] [Google Scholar]
- 65.Suzuki H, Iino S, Shiraki K, Akahane Y, Okamoto H, Domoto K, Mishiro S. Vaccine. 1994;12:1090–1096. doi: 10.1016/0264-410x(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 66.Milich DR, Thornton GB, Neurath AR, Kent SB, Michel ML, Tiollais P, Chisari FV. Science. 1985;228:1195–1199. doi: 10.1126/science.2408336. [DOI] [PubMed] [Google Scholar]
- 67.Huang Z, Elkin G, Maloney BJ, Beuhner N, Arntzen CJ, Thanavala Y, Mason H. Vaccine. 2005;23:1851–1858. doi: 10.1016/j.vaccine.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 68.Ehsani P, Khabiri A, Domansky NN. Gene. 1997;190:107–111. doi: 10.1016/s0378-1119(96)00647-6. [DOI] [PubMed] [Google Scholar]
- 69.Joung YH, Youm JW, Jeon JH, Lee BC, Ryu CJ, Hong HJ, Kim HC, Joung H, Kim HS. Plant Cell Rep. 2004;22:925–930. doi: 10.1007/s00299-004-0775-1. [DOI] [PubMed] [Google Scholar]
- 70.Wynne SA, Crowther RA, Leslie AG. Mol Cell. 1999;3:771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 71.Pumpens P, Borisova GP, Crowther RA, Grens E. Intervirology. 1995;38:63–74. doi: 10.1159/000150415. [DOI] [PubMed] [Google Scholar]
- 72.Pumpens P, Grens E. FEBS Lett. 1999;442:1–6. doi: 10.1016/s0014-5793(98)01599-3. [DOI] [PubMed] [Google Scholar]
- 73.Schodel F, Peterson D, Hughes J, Wirtz R, Milich D. J Biotechnol. 1996;44:91–96. doi: 10.1016/0168-1656(95)00118-2. [DOI] [PubMed] [Google Scholar]
- 74.Lobaina Y, Palenzuela D, Pichardo D, Muzio V, Guillen G, Aguilar JC. Mol Immunol. 2005;42:289–294. doi: 10.1016/j.molimm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 75.Aguilar JC, Lobaina Y, Muzio V, Garcia D, Penton E, Iglesias E, Pichardo D, Urquiza D, Rodriguez D, Silva D, Petrovsky N, Guillen G. Immunol Cell Biol. 2004;82:539–546. doi: 10.1111/j.0818-9641.2004.01278.x. [DOI] [PubMed] [Google Scholar]
- 76.Tsuda S, Yoshioka K, Tanaka T, Iwata A, Yoshikawa A, Watanabe Y, Okada Y. Vox Sang. 1998;74:148–155. [PubMed] [Google Scholar]
- 77.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. J Gen Virol. 2001;82:1013–1025. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- 78.Huang Z, Santi L, LePore K, Kilbourne J, Arntzen CJ, Mason HS. Vaccine. 2006;24:2506–2513. doi: 10.1016/j.vaccine.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 79.Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. J Virol. 1999;73:6188–6190. doi: 10.1128/jvi.73.7.6188-6190.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xi JN, Graham DY, Wang KN, Estes MK. Science. 1990;250:1580–1583. doi: 10.1126/science.2177224. [DOI] [PubMed] [Google Scholar]
- 81.Jiang X, Wang M, Graham DY, Estes MK. J Virol. 1992;66:6527–6532. doi: 10.1128/jvi.66.11.6527-6532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.White LJ, Hardy ME, Estes MK. J Virol. 1997;71:8066–8072. doi: 10.1128/jvi.71.10.8066-8072.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ball JM, Graham DY, Opekun AR, Gilger MA, Guerrero RA, Estes MK. Gastroenterology. 1999;117:40–48. doi: 10.1016/s0016-5085(99)70548-2. [DOI] [PubMed] [Google Scholar]
- 84.Mason HS, Ball JM, Shi JJ, Jiang X, Estes MK, Arntzen CJ. Proc Natl Acad Sci U S A. 1996;93:5335–5340. doi: 10.1073/pnas.93.11.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tacket CO, Mason HS, Losonsky G, Estes MK, Levine MM, Arntzen CJ. J Infect Dis. 2000;182:302–305. doi: 10.1086/315653. [DOI] [PubMed] [Google Scholar]
- 86.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Proc Natl Acad Sci U S A. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzich JA, Ghim SJ, Palmer-Hill FJ, White WI, Tamura JK, Bell JA, Newsome JA, Jenson AB, Schlegel R. Proc Natl Acad Sci U S A. 1995;92:11553–11557. doi: 10.1073/pnas.92.25.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang LF, Zhou J, Chen S, Cai LL, Bao QY, Zheng FY, Lu JQ, Padmanabha J, Hengst K, Malcolm K, Frazer IH. Vaccine. 2000;18:1051–1058. doi: 10.1016/s0264-410x(99)00351-5. [DOI] [PubMed] [Google Scholar]
- 89.Harro CD, Pang YY, Roden RB, Hildesheim A, Wang Z, Reynolds MJ, Mast TC, Robinson R, Murphy BR, Karron RA, Dillner J, Schiller JT, Lowy DR. J Natl Cancer Inst. 2001;93:284–292. doi: 10.1093/jnci/93.4.284. [DOI] [PubMed] [Google Scholar]
- 90.Evans TG, Bonnez W, Rose RC, Koenig S, Demeter L, Suzich JA, O'Brien D, Campbell M, White WI, Balsley J, Reichman RC. J Infect Dis. 2001;183:1485–1493. doi: 10.1086/320190. [DOI] [PubMed] [Google Scholar]
- 91.Brown DR, Bryan JT, Schroeder JM, Robinson TS, Fife KH, Wheeler CM, Barr E, Smith PR, Chiacchierini L, DiCello A, Jansen KU. J Infect Dis. 2001;184:1183–1186. doi: 10.1086/323645. [DOI] [PubMed] [Google Scholar]
- 92.Emeny RT, Wheeler CM, Jansen KU, Hunt WC, Fu TM, Smith JF, MacMullen S, Esser MT, Paliard X. J Virol. 2002;76:7832–7842. doi: 10.1128/JVI.76.15.7832-7842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kahn TW, Beachy RN, Falk MM. Curr Biol. 1997;7:R207–208. doi: 10.1016/s0960-9822(06)00100-x. [DOI] [PubMed] [Google Scholar]
- 94.Gerber S, Lane C, Brown DM, Lord E, DiLorenzo M, Clements JD, Rybicki E, Williamson AL, Rose RC. J Virol. 2001;75:4752–4760. doi: 10.1128/JVI.75.10.4752-4760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Warzecha H, Mason HS, Lane C, Tryggvesson A, Rybicki E, Williamson AL, Clements JD, Rose RC. J Virol. 2003;77:8702–8711. doi: 10.1128/JVI.77.16.8702-8711.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Varsani A, Williamson AL, Rose RC, Jaffer M, Rybicki EP. Arch Virol. 2003;148:1771–1786. doi: 10.1007/s00705-003-0119-4. [DOI] [PubMed] [Google Scholar]
- 97.Biemelt S, Sonnewald U, Galmbacher P, Willmitzer L, Muller M. J Virol. 2003;77:9211–9220. doi: 10.1128/JVI.77.17.9211-9220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu MW, Lin CS. Arch Virol. 2003;148:345–355. doi: 10.1007/s00705-002-0901-8. [DOI] [PubMed] [Google Scholar]
- 99.Bragard C, Duncan GH, Wesley SV, Naidu RA, Mayo MA. J Gen Virol. 2000;81:267–272. doi: 10.1099/0022-1317-81-1-267. [DOI] [PubMed] [Google Scholar]
- 100.Schodel F, Peterson D, Hughes J, Milich DR. Vaccine. 1993;11:143–148. doi: 10.1016/0264-410x(93)90010-u. [DOI] [PubMed] [Google Scholar]
- 101.Nardelli-Haefliger D, Roden R, Benyacoub J, Sahli R, Kraehenbuhl J, Schiller J, Lachat P, Potts A, De Grandi P. Infect Immun. 1997;65:3328–3336. doi: 10.1128/iai.65.8.3328-3336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Clarke BE, Newton SE, Carroll AR, Francis MJ, Appleyard G, Syred AD, Highfield PE, Rowlands DJ, Brown F. Nature. 1987;330:381–384. doi: 10.1038/330381a0. [DOI] [PubMed] [Google Scholar]
- 103.Sasagawa T, Tani M, Basha W, Rose RC, Tohda H, Giga-Hama Y, Azar KK, Yasuda H, Sakai A, Inoue M. Virus Res. 2005;110:81–90. doi: 10.1016/j.virusres.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Falcon V, Garcia C, de la Rosa MC, Menendez I, Seoane J, Grillo JM. Tissue Cell. 1999;31:117–125. doi: 10.1054/tice.1999.0032. [DOI] [PubMed] [Google Scholar]
- 105.Yao Q, Vuong V, Li M, Compans RW. Vaccine. 2002;20:2537–2545. doi: 10.1016/s0264-410x(02)00160-3. [DOI] [PubMed] [Google Scholar]
- 106.Zhou S, Standring D. Proc Natl Acad Sci U S A. 1992;89:10046–10050. doi: 10.1073/pnas.89.21.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crill WD, Chang GJ. J Virol. 2004;78:13975–13986. doi: 10.1128/JVI.78.24.13975-13986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuei-Chun Wang J-CW, Yao-Chi Chung, Yi-Chen Ho, Margaret Dah-Tsyr Chang, Yu-Chen Hu. Biotechnol Bioeng. 2005;89:464–473. doi: 10.1002/bit.20385. [DOI] [PubMed] [Google Scholar]