

Abstract
CD56+ T cells are abundant in liver and play an important role in defense against viral infections. However, the role of CD56+ T cells in control of HCV infection remains to be determined. We investigated the noncytolytic anti-HCV activity of primary CD56+ T cells in human hepatocytes. When HCV JFH-1-infected hepatocytes were co-cultured with CD56+ T cells or incubated in media conditioned with CD56+ T cell culture supernatants (SN), HCV infectivity and replication were significantly inhibited. The antibodies to interferon (IFN)-γ or IFN-γ receptor could largely block CD56+ T cell-mediated anti-HCV activity. Investigation of mechanism(s) responsible for CD56+ T cell-mediated noncytolytic anti-HCV activity showed that CD56+ T SN activated the multiple elements of janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and enhanced the expression of IFN regulatory factors (IRFs) 1, 3, 7, 8 and 9, resulting in the induction of endogenous IFN-α/β expression in hepatocytes. Moreover, CD56+ T SN treatment inhibited the expression of HCV-supportive miRNA-122 and enhanced the levels of anti-HCV miRNA-196a in human hepatocytes. Conclusion: These findings provide direct in vitro evidence at cellular and molecular levels that CD56+ T cells may have an essential role in innate immune cell-mediated defense against HCV infection.
Keywords: Interferon (IFN)-γ, JAK/STAT, IFN regulatory factor, Type I IFNs, microRNA
CD56+ T cells are a subset of human T lymphocytes expresses the cell-surface molecular, CD56, which is typically expressed by natural killer cells.1, 2 CD56+ T cells express both NK and T cells markers, therefore they functionally display properties of both NK cells and T cells. Unlike classical T cells, CD56+ T cells possess the ability to rapidly produce large quantities of both Th1 and Th2 cytokines, particularly interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukin-2 (IL-2), IL-4, and IL-13 without need for priming or clonal expansion, which promote Th1 or Th2 adaptive immune responses.2-7 This ability of CD56+ T cells permits them to play a key role in the communications between the innate and adaptive immune cells. The dual innate and adaptive immune functions place CD56+ T cells alongside natural killer T cells as frontline innate immune effectors and potential regulators of adaptive immune responses against microorganisms.8 The properties of CD56+ T cells make them important innate immune cells as the first line defense mechanism against pathogens including viruses. In support of this notion, numerical and functional deficiencies and phenotypic alterations of CD56+ T cells have been reported in patients with various infectious and autoimmune diseases and cancer.7, 9-13
The intrahepatic immune system is characterized by a unique repertoire of lymphocytes. A normal human liver as the primary site of HCV infection contains lymphocytes that usually enriched for CD56+ T cells.6, 14 CD56+ T cells comprise approximately 5 to 15% of the peripheral T-cell pool, and up to 50% of T lymphocytes within the liver environment.14 This innate lymphocyte population can recognize conserved structures that signal viral invasion, thus providing an important first line of defense against viral infections.14, 15 More importantly, recent studies have highlighted that CD56+ T cells play an important role in determining outcome of acute hepatitis C virus infection,16 and are known to be depleted either in the livers or in peripheral bloods of patients with chronic HCV infection.10, 16-18 In the present study, we examined the ability of CD56+ T cells isolated from peripheral blood of healthy donors to inhibit full cycle HCV infection and replication in human hepatocytes. We also explored the cellular and molecular mechanisms underlying the CD56+ T cell-mediated action on HCV infection.
Materials and Methods
Reagents
Recombinant human IFN-γ and mouse anti-IFN-γ, goat anti-IFN-γ receptor (anti-IFN-γR1 and IFN-γR2) antibodies, and IL-12 were purchased from R&D Systems Inc. (Minneapolis, MN). Goat anti-IFN-α/β-receptor 1 (anti-IFN-α/β R1) was purchased from Sigma-Aldrich Inc. (St Louis, MO). Rabbit anti-HCV NS5A antibody was obtained from Chiron Company (Emeryville, CA). Mouse anti-HCV NS3 antibody was generated by Dr. Guangxiang Luo (University of Kentucky College of Medicine, Lexington, Kentucky). Rabbit anti-signal transducer and activator of transcription-1 (STAT-1), IFN regulatory factor-1 (IRF-1), IRF-9 antibodies, mouse anti-STAT-2 antibody, and goat anti-IRF-3, IRF-7, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Goat anti-IRF-5 antibody was purchased from Abcam Inc. (Cambridge, MA). Rabbit anti-IRF-8 antibody was obtained from Aviva Systems Biology (San Diego, CA). The secondary antibodies (horseradish peroxidase-conjugated goat-anti-rabbit IgG, goat-anti-mouse IgG, and donkey anti-goat IgG) used for Western blot were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Primary cells and cell lines
Primary CD56+ T cells were enriched and purified from PBMCs of three health donors (two males and one female) with CD3+CD56+ T Cell Isolation Kit (Miltenyi Biotec, Auburn, CA). The purity (% of CD3+CD56+) of CD56+ T cells measured by flow cytometry was greater than 95%. For the purpose of control cell population, CD3+CD56- T cells were isolated by using CD3 and CD56 microbeads (Miltenyi Biotec, Auburn, CA) and the purity was also great then 95%. Purified CD56+ T cells or CD56- T cells were cultured in RPMI 1640 containing 10% fetal bovine serum (HyClone, Logan, UT) and activated by a standard T Cell Activation/Expansion Kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. Briefly, CD56+ T cells or CD56- T cells were stimulated with MACSiBead particles loaded with CD2, CD3 and CD28 antibodies. The activation of CD56+ T cells or CD56- T cells was accomplished by using one loaded Anti-Biotin MACSiBead particle per two cells (bead-to-cell ratio 1:2). The cells were cultured at 37°C with 5% CO2 for 48h. Cell-free supernatants (SN) collected from the activated CD56+ T cell or CD56- T cell cultures were used as CD56+ T or CD56- T SN. Primary human hepatocytes were isolated from independent healthy donors by Lonza Walkersville Inc. (Walkersville, MD). Cells were cultured on collagen-coated plates and maintained with Hepatocyte Culture Medium (Lonza Walkersville Inc.) according to the manufacturer’s protocol. A hepatoma cell line (Huh7.5.1) is a gift from Dr. Charles Rice (The Rockefeller University, New York, NY).
HCV JFH-1 infection of human hepatocytes
The generation of infectious HCV JFH-1 and infection of hepatocytes were carried out as described.19 For detection of HCV positive- and negative-strand RNAs, a strand-specific RT-PCR assay was used as previously described.20, 21
CD56+ T SN treatment and Co-culture of human hepatocytes with CD56+ T cells
For the co-culture experiments, the purified CD56+ T cells were first activated by MACSiBead particles with CD2, CD3 and CD28 antibodies for 48h, then collected and transferred to the co-culture system. To maintain the activation status of CD56+ T cells in the co-culture system, the cultures were supplemented with IL-12 (20ng/mL). HCV JFH-1-infected Huh7.5.1 cells were co-cultured with CD56+ T cells at different ratios (CD56+ T: Huh7.5.1, 0.1:1; 1:1; 10:1) in 0.4-μm-pore-transwell tissue culture plates (Costar, Cambridge, MA). Huh7.5.1 cells were in the lower compartment and CD56+ T cells were placed in the upper compartment. The hepatocytes were collected for RNA extraction and HCV RNA real-time RT-PCR at different time points after co-culture. For the experiments using CD56+ T SN, heaptocytes were cultured in media conditioned with or without CD56+ T SN (1%, 5%, 10%, 25%, and 50%, v/v). CD56+ T SN was added to the hepatocyte cultures at either 24h prior to HCV infection or day 3 postinfection. In addition, CD56- T cells or SN were used as a control in the experiments above.
Quantitative Real-Time RT-PCR
Total cellular RNA extraction and the reverse transcription were performed as described.22 The real-time RT-PCR for the quantification of HCV, IFN-α, IFN-β, IRF-1, IRF-3, IRF-4, IRF-5, IRF-7, IRF-8, IRF-9, and GAPDH mRNA was performed with the iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA). The levels of GAPDH mRNA were used as an endogenous reference to normalize the quantities of target mRNA. The special oligonucleotide primers used in this study were listed in Table 1. The oligonucleotide primers were synthesized by Integrated DNA Technologies Inc. (Coralville, IA).
Table 1.
Primers for Real-Time RT-PCR
| Primer | Orientation | Sequence |
|---|---|---|
| GAPDH | Sense: | 5′-GGTGGTCTCCTCTGACTTCAACA-3′ |
| Antisense: | 5′-GTTGCTGTAGCCAAATTCGTTGT-3′ | |
| HCV | Sense: | 5′-RAYCACTCCCCTGTGAGGAAC -3′ |
| Antisense: | 5′-TGRTGCACGGTCTACGAGACCTC -3′ | |
| IFN-α | Sense: | 5′-TTTCTCCTGCCTGAAGGACAG-3′ |
| Antisense: | 5′-GCTCATGATTTCTGCTCTGACA-3′ | |
| IFN-β | Sense: | 5′-AAAGAAGCAGCAATTTTCAGC-3′ |
| Antisense: | 5′-CCTTGGCCTTCAGGTAATGCA-3′ | |
| IFN-γ | Sense: | 5′-AGAAAAATAATGCAGAGCCAAATT-3′ |
| Antisense: | 5′-TGACTCCTTTTTCGCTTCCCTGTT-3′ | |
| CXCL-9 | Sense: | 5′-TGCAATGAACCCCAGTAGTGA-3′ |
| Antisense: | 5′-GGTGGATAGTCCCTTGGTTGG-3′ | |
| IRF-1 | Sense: | 5′-TGAAGCTACAACAGATGAGG-3′ |
| Antisense: | 5′-AGTAGGTACCCCTTCCCATC-3′ | |
| IRF-3 | Sense: | 5′-ACCAGCCGTGGACCAAGAG-3′ |
| Antisense: | 5′-TACCAAGGCCCTGAGGCAC-3′ | |
| IRF-5 | Sense: | 5′-AAGCCGATCCGGCCAA-3′ |
| Antisense: | 5′-GGAAGTCCCGGCTCTTGTTAA-3′ | |
| IRF-7 | Sense: | 5′-TGGTCCTGGTGAAGCTGGAA-3′ |
| Antisense: | 5′-GATGTCGTCATAGAGGCTGTTGG-3′ | |
| IRF-8 | Sense: | 5′-GGAGTGCGGTCGCTCTGAAA-3′ |
| Antisense: | 5′-GTCGTAGGTGGTGTACCCCGTCA-3′ | |
| IRF-9 | Sense: | 5′-GCATCAGGCAGGGCACGCTGCACC-3′ |
| Antisense: | 5′-GCCTGCATGTTTCCAGGGAATCCG-3′ |
PCR-Based Gene Expression Array
The pathway-focused (Janus Kinase/signal transducer and activator of transcription, JAK/STAT) gene expression array was carried out according to the protocol provided by SuperArray Bioscience Corporation (Frederick, MD). Briefly, total cellular RNA was extracted from human hepatocytes treated with or without CD56+ T SN for 6h using SuperArray RT2 qPCR-Grade RNA Isolation Kit. The elimination of genomic DNA contamination and first strand cDNA synthesis were carried out using RT2 First Strand Kit. Pathway-focused (JAK/STAT) gene expression array was performed using RT2 Profiler TM PCR Array System. Statistical analysis of data was performed with the Data-Analysis-Template provided by the SuperArray Bioscience Corporation.
Quantitative Real-Time RT-PCR of MicroRNA (miRNA)
Total cellular RNA, including miRNA, was extracted from cells using miRNeasy Mini Kit from Qiagen (Valencia, CA). Total RNA (1μg) was reverse-transcribed with miScript Reverse Transcription Kit from Qiagen. The real-time RT-PCR for the quantification of a subset of miRNAs (miR-122, miR-196a, miR-196b, miR-296, miR-351, miR-431, and miR-448) was carried out with miScript Primer Assays and miScript SYBR Green PCR Kit from Qiagen.
Immunoassays
The ELSIA kits for IFN-α, IFN-β, and IFN-γ were purchased from PBL Biomedical Laboratories (Piscataway, NJ) and ELISA kits for TNF-α, IL-2, IL-4, and IL-10 were from BD Biosciences (San Jose, CA). Immunofluorescent evaluation of HCV NS5A protein in HCV-infected hepatocytes and Western blot assays were carried out as previous described.22
Statistical analysis
Where appropriate, data were expressed as mean ±SD of triplicate cultures. For comparison of the mean of two groups, statistical significance was assessed by student’s t test. If there were more than 2 groups, one-way repeated measures of ANOVA were used. Statistical analyses were performed with Graphpad Instat Statistical Software. Statistical significance was defined as P<0.05.
Results
CD56+ T cells inhibit HCV infection and replication
We first examined whether CD56+ T cells or CD56+ T SN have cytotoxicity effect on human hepatocytes. No cytotoxic effect was observed in the hepatocytes directly contacted with CD56+ T cells or treated with CD56+ T SN (data not shown). We then examined whether CD56+ T cells release soluble factor(s) that suppresses HCV replication in human hepatocytes. We demonstrated that HCV replication in the hepatocytes co-cultured with CD56+ T cells was significantly inhibited, and the degree of suppression was correlated with the number of CD56+ T cells added to the co-culture system (Fig. 1A). In contract, CD56- T cells and SN, when added to HCV JFH-1-infected cells, had little effect on HCV replication (Fig. 1A). We also determined whether SN from CD56+ T cell cultures has anti-HCV activity. CD56+ T SN, when added to HCV-infected Huh7.5.1 cells, inhibited HCV RNA expression in a dose-dependence fashion (Fig. 1B). The CD56+ T SN-mediated inhibition of HCV replication was also confirmed by the observation that the percentage of NS5A-positive cells in HCV-infected and CD56+ T SN-treated Huh7.5.1 cells decreased significantly compared with that in untreated cells (Fig. 2). The anti-HCV activity of CD56+ T SN was also determined in HCV JFH-1-infected primary human hepatocytes. CD56+ T-SN-treated primary hepatocytes had undetectable negative-strand HCV RNA and lower levels of HCV positive-strand RNA than untreated cells (Fig. 3). In order to determine whether CD56+ T SN can protect hepatocytes from HCV infection, we pretreated Huh7.5.1 cells or primary hepatocytes with CD56+ T SN for 24h. The pretreatment of hepatocytes resulted in a 10-100 fold decrease in HCV RNA expression (Fig. 4). Moreover, HCV RNA expression in primary hepatocytes became undetectable at day 4 and afterwards postinfection (Fig. 4B).
Fig. 1. CD56+ T cells or supernatant (SN) suppress HCV replication in Huh 7.5.1 cells.

(A) Co-culture of HCV infected-Huh7.5.1 cells with CD56+ T cells or CD56- T cells via a trans-well system. HCV JFH-1-infected Huh7.5.1 cells (day 3 post infection) were plated in the low compartment of a 24-well plate (105cells/well), while the different numbers of activated CD56+ T cells or CD56- T cells were added in the top compartment as indicated. The cultures were supplemented with IL-12 (20ng/mL) to maintain the activated status of T cells. After 48h co-culture, total cellular RNA extracted from Huh7.5.1 cells was subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. (B) Effect of CD56+ T or CD56- T SN on HCV replication in Huh7.5.1 cells. HCV JFH-1-infected Huh7.5.1 cells (day 3 post infection) were cultured in the presence or absence of CD56+ T or CD56- T SN at indicated concentrations for 48h. Total cellular RNA extracted from Huh7.5.1 cells was subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as HCV RNA levels relative (%) to control (without T cells or SN treatment, which is defined as 100). The results are mean ±SD of triplicate cultures, representative of three experiments (* p<0.05, ** p<0.01).
Fig. 2. Effect of CD56+ T supernatant (SN) on HCV NS5A protein expression in Huh7.5.1 cells.

HCV JFH-1-infected Huh7.5.1 cells (day 3 post infection) were cultured in the presence or absence of CD56+ T SN (50%, v/v) for 48h. HCV NS5A protein expression was determined by immunofluoresence staining with antibody against NS5A (green). The nuclei were stained with Hoechst 33258 (blue). One representative experiment is shown (200x).
Fig. 3. CD56+ T supernatant (SN) inhibition of HCV replication in primary human hepatocytes.

(A) Effect of CD56+ T SN on HCV RNA expression. CD56+ T SN was added to HCV JFH-1-infected primary human hepatocyte cultures (day 3 post infection). Total cellular RNA extracted from the cell cultures at 48h posttreatment was subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as HCV RNA levels relative (%) to control (without CD56+ T SN treatment, which is defined as 100). The results are mean ±SD of triplicate cultures, representative of three experiments (** p<0.01). (B) Effect of CD56+ T SN on the expression of both HCV positive (+) and negative (-) strand RNAs. Total cellular RNA extracted from CD56+ T SN-treated or untreated HCV JFH-1-infected primary human hepatocytes was subjected to a strand-specific RT-PCR. Amplified RT-PCR products were run on a 1.5% agarose gel.
Fig. 4. Effect of CD56+ T supernatant (SN) on susceptibility of hepatocytes to HCV infection.

Huh 7.5.1 cells (A) or primary human hepatocytes (B) were cultured in the presence or absence of CD56+ T SN (50%, v/v) for 24h prior to HCV JFH-1 infection. Total cellular RNA was extracted from the cell cultures at the indicated time points and subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as log HCV RNA copies/μg RNA. The results shown are mean ±SD of triplicate cultures, representative of three experiments. UD: undetectable.
IFN-γ is a major factor responsible for the CD56+ T SN-mediated anti-HCV activity
Upon activation, CD56+ T cells can release the antiviral cytokines, such as IFN-γ. Thus, we examined whether IFN-γ is responsible for CD56+ T SN-mediated anti-HCV activity. Preincubation of CD56+ T SN with antibody to IFN-γ largely reversed CD56+ T SN-mediated anti-HCV activity (Fig. 5A). Furthermore, Pretreatment of HCV-infected hepatocytes with antibodies to IFN-γ receptor (anti-IFN-γR1 and/or anti-IFN-γR2) also blocked the most of CD56+ T SN-mediated anti-HCV activity (Fig. 5B).
Fig. 5. IFN-γ is the major factor responsible for CD56+ T supernatant (SN)-mediated anti-HCV activity.

(A) Anti-IFN-γ antibody blocks CD56+ T SN-mediated anti-HCV activity. HCV JFH-1-infected Huh7.5.1 cells were cultured in the presence or absence of CD56+ T SN and/or antibody against IFN-γ (10μg/mL). For the cultures in the presence of CD56+ T SN and the antibody against IFN-γ, the CD56+ T SN was preincubated with the antibody against IFN-γ for 30 min prior to the addition to Huh7.5.1 cultures. IFN-γ (200U/mL) alone was added to the cell cultures as a positive control to determine the neutralization ability of the antibody to IFN-γ. Mouse IgG2A was used as a control antibody to determine the specificity of the antibody to IFN-γ. (B) Antibodies to IFN-γR1 and IFN-γR2 block anti-HCV activity of CD56+ T SN. HCV JFH-1-infected Huh7.5.1 cells were incubated with or without antibodies to IFN-γR1 (10μg/mL) and/or IFN-γR2 (10μg/mL) for 1 h prior to the addition of the CD56+ T SN. Goat IgG was used as a control IgG to determine the specificity of the antibodies to IFN-γR. HCV RNA levels in Huh7.5.1 cells were determined at 48 h post exposure to CD56+ T SN. Total cellular RNA extracted from Huh7.5.1 cells was subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as HCV RNA levels relative (%) to control cultures (no antibody treatment and no CD56+ T SN added, which is defined as 100). The results shown are mean ±SD of triplicate cultures, representative of three separate experiments (* p<0.05, ** p<0.01).
The impact of CD56+ T SN on JAK/STAT pathway
We hypothesized that the activation of IFN-γ-mediated signaling pathway is a potential mechanism involved in CD56+ T cell-mediated noncytolytic anti-HCV action. Therefore, we used gene expression array to examine the profile of CD56+ T SN-mediated regulation of genes in JAK/STAT pathway. When omitted genes that displayed less than 2 fold up- or down-regulation relative to that of the control group, we showed that CD56+ T SN was able to up-regulate 24 genes (Table 2) and down-regulate 10 genes (Table 3) in JAK/STAT pathway. On the top of the list of upregulated genes by CD56+ T SN treatment was CXCL-9, an IFN-γ-inducible chemokine (Table 2). In addition, the expression of the key elements in JAK/STAT pathway, including JAK-2, JAK-3, STAT-1, STAT-2, and STAT-5A, were all significantly enhanced by CD56+ T SN treatment (Table 2). Furthermore, the key regulators in JAK/STAT pathway, such as IRF-1 and ISGF3G (IRF-9), were also induced by CD56+ T SN treatment (Table 2). Notably, CD56+ T SN treatment resulted in the increased expression of antiviral genes, including IFN-γ, NOS2A, GBP1, OSA1 and ISG15 (Table 2), and the decreased expression of the inhibitor of JAK/STAT pathway, such as PIAS1(Table 3).
Table 2. Up-Regulated Genes in JAK/STAT Pathway by CD56+ T Supernatant.
Gene array analysis of CD56+ T SN-regulated genes in JAK/STAT pathway in primary human hepatocytes Primary human hepatocytes were incubated with or without CD56+ T SN (50%, v/v) for 6h. Total RNA was harvested and subjected to the gene array using RT2 ProfilerTM PCR Array System (SuperArray Bioscience Corporation). Data analysis was performed using Data-Analysis-Template from Superarray Bioscience Corporation. The genes which displayed less than 2-fold up- or down-regulation relative to that of the control group were omitted. The CD56+ T SN up-regulated about 24 genes (Table 2) and down-regulated 10 genes (Table 3) over 2-fold in JAK/STAT pathway.
| Gene | Description | Fold |
|---|---|---|
| CXCL9 | Chemokine (C-X-C motif) ligand 9 | 112.99 |
| NOS2A | Nitric oxide synthase 2A (inducible, hepatocytes) | 73.01 |
| IFNG | Interferon, gamma | 43.41 |
| IL2RA | Interleukin 2 receptor, alpha | 31.34 |
| GBP1 | Guanylate binding protein 1, interferon-inducible, 67kDa | 22.78 |
| CSF2RB | Colony stimulating factor 2 receptor, beta, low-affinity (granulocyte-macrophage) |
12.21 |
| IRF-1 | Interferon regulatory factor 1 | 11.08 |
| IL20 | Interleukin 20 | 8.11 |
| JAK3 | Janus kinase 3 (a protein tyrosine kinase, leukocyte) | 4.53 |
| JAK2 | Janus kinase 2 (a protein tyrosine kinase) | 4.41 |
| ISG15 | ISG15 ubiquitin-like modifier | 4.29 |
| STAT1 | Signal transducer and activator of transcription 1, 91kDa | 4.00 |
| FCGR1A | Fc fragment of IgG, high affinity Ia, receptor (CD64) | 3.86 |
| STAT5A | Signal transducer and activator of transcription 5A | 3.27 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 2.55 |
| MMP3 | Matrix metallopeptidase 3 (stromelysin 1, progelatinase) | 2.51 |
| OSA1 | 2′,5′-oligoadenylate synthetase 1, 40/46kDa | 2.50 |
| IL10 RA | Interleukin 10 receptor, alpha | 2.33 |
| ISGF3G | Interferon-stimulated transcription factor 3, gamma 48kDa | 2.27 |
| OSM | Oncostatin M | 2.27 |
| STAT2 | Signal transducer and activator of transcription 2, 113kDa | 2.20 |
| JUN | Jun oncogene | 2.17 |
| SH2B2 | SH2B adaptor protein 2 | 2.07 |
| JUNB | Jun B proto-oncogene | 2.04 |
Table 3. Down-Regulated Genes in JAK/STAT Pathway by CD56+ T Supernatant.
Gene array analysis of CD56+ T SN-regulated genes in JAK/STAT pathway in primary human hepatocytes Primary human hepatocytes were incubated with or without CD56+ T SN (50%, v/v) for 6h. Total RNA was harvested and subjected to the gene array using RT2 ProfilerTM PCR Array System (SuperArray Bioscience Corporation). Data analysis was performed using Data-Analysis-Template from Superarray Bioscience Corporation. The genes which displayed less than 2-fold up- or down-regulation relative to that of the control group were omitted. The CD56+ T SN up-regulated about 24 genes (Table 2) and down-regulated 10 genes (Table 3) over 2-fold in JAK/STAT pathway.
| Gene | Description | Fold |
|---|---|---|
| PRLR | Prolactin receptor | -5.62 |
| F2R | Coagulation factor II (thrombin) receptor | -2.89 |
| SH2B1 | SH2B adaptor protein 1 | -2.81 |
| BCL2L1 | BCL2-like 1 | -2.55 |
| PIAS1 | Protein inhibitor of activated STAT, 1 | -2.53 |
| GHR | Growth hormone receptor | -2.43 |
| SIT1 | Signaling threshold regulating transmembrane adaptor 1 | -2.41 |
| INSR | Insulin receptor | -2.39 |
| CSF1R | Colony stimulating factor 1 receptor, formerly McDonough feline sarcoma viral (v-fms) oncogene homolog |
-2.14 |
| SRC | V-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (avian) |
-2.04 |
CD56+ T SN enhances the expression of IRFs and STATs
To further determine the mechanism(s) involved in the CD56+ T cell-mediated anti-HCV action, we investigated whether CD56+ T SN has the ability to induce the expression of IRF members and STAT-1/ 2 in Huh7.5.1 cells. Among the IRF family members tested, IRFs 1, 3, 7, 8 and 9 were induced by CD56+ T SN treatment (Fig. 6). This CD56+ T SN-mediated induction of IRF family members was demonstrated at both mRNA (Fig. 6A) and protein (Fig. 6B) levels. We also examined the impact of CD56+ T SN on the expression of STATs. CD56+ T SN treatment enhanced STAT-1 and STAT-2 expression in human hepatocytes as determined by Western blot assay (Fig. 6B). Our additional analysis of the dynamic effect of CD56+ T SN on expression of STATs and IRFs during the course of CD56+ T SN treatment demonstrated that there was significant and stable increase of SATA-1/2 and IRFs (1, 3, 7, 8, and 9) at different time points during the course of suppression of HCV replication in Huh7.5.1 cells by CD56+ T SN treatment (Fig. 6B).
Fig. 6. The effect of CD56+ T supernatant (SN) on the expression of STATs and IRFs in human hepatocytes.

(A) IRF mRNA expression in human hepatocytes. Huh7.5.1 cells were cultured in the presence or absence of CD56+ T SN (50%, v/v) for 24h. Total cellular RNA extracted from cell cultures was subjected to the real-time RT-PCR for IRF and GAPDH RNA quantification. The data are expressed as IRF mRNA levels relative (fold) to the control (without CD56+ T SN treatment, which is defined as 1). The results shown are mean ± SD of triplicate cultures, representative of three experiments (*p<0.05, ** p<0.01). (B) STAT/IRF protein expression in human hepatocytes. HCV JFH-1-infected cells at day 3 postinfection were cultured in the presence or absence of CD56+ T SN (50%, v/v) for 48h. Total proteins extracted from cell cultures at indicated timepoints were subjected to Western blot assay using the antibodies against HCV NS3, STAT-1, STAT-2, IRF-1, IRF-3, IRF-5, IRF-7, IRF-8, IRF-9, and GAPDH.
CD56+ T SN or IFN-γ modulates the expression of intracellular type I IFN and miRNAs
Since IRF-3 and IRF-7, the key regulators of type I IFNs, were induced in human hepatocytes by CD56+ T SN treatment, we examined whether CD56+ T SN or IFN-γ has the ability to induce intracellular IFN-α/β expression in hepatocytes. Huh7.5.1 cells or primary human hepatocytes treated with CD56+ T SN or IFN-γ expressed higher levels of endogenous IFN-α and IFN-β than the untreated control cells (Fig. 7A and 7B). Preincubation of HCV JFH-1-infected Huh7.5.1 cells with antibody to IFN-α/β receptor 1 could significantly reverse CD56+ T SN-mediated anti-HCV activity (Fig. 7C). In addition, we examined the ability of CD56+ T SN to regulate the expression of miRNAs in hepatocytes, as type I IFNs have been shown to regulate several cellular miRNAs that involved in HCV infection and replication.23 Among miRNAs (miRNA-122, miRNA-196a, miRNA-196b, miRNA-296, miRNA-351, miRNA-431, and miRNA-448) tested, the levels of two key miRNAs (miRNA-122 and miRNA-196a) that are involved in HCV replication were significantly altered by CD56+ T SN treatment. While HCV-supportive miRNA-122 was inhibited by CD56+ T SN treatment, anti-HCV miRNA-196a was induced by CD56+ T SN treatment (Fig. 8).
Fig. 7. CD56+ T supernatant (SN) or IFN-γ induces intracellular expression of IFN-α/β.

Huh7.5.1 cells or primary human hepatocytes were cultured in the presence or absence of CD56+ T SN (50%, v/v) or IFN-γ (200U/mL) for 24h. Total cellular RNA extracted from cell cultures was subjected to the real-time RT-PCR for IFN-α, IFN-β and GAPDH RNA quantification. The data are expressed as IFN-α (A) or IFN-β (B) mRNA levels relative (fold) to the control (without CD56+ T SN and IFN-γ treatment, which is defined as 1). The results shown are mean ±SD of triplicate cultures, representative of three experiments (*p<0.05, ** p<0.01). (C) Antibody to IFN-α/β R1 blocks CD56+ T cell-mediated anti-HCV activity in Huh7.5.1 cells. HCV JFH-1-infectd Huh7.5.1 cells were incubated with or without goat anti-IFN-α/β R1 (10μg/mL) for 1 h prior to the treatment with CD56+ T SN. IFN-γ (200U/ml) or IFN-α (20U/mL) treatment of human hepatocytes was used as the controls. Goat IgG was used as a control IgG to determine the specificity of the antibody to IFN-α/β R1. HCV RNA levels in Huh7.5.1 cells were determined at 48h post exposure to CD56+ T SN. Total cellular RNA extracted from Huh7.5.1 cells was subjected to the real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as HCV RNA levels relative (%) to control (no antibody and CD56+ T SN treatment, which is defined as 100). The results shown are mean ±SD of triplicate cultures, representative of three separate experiments (* p<0.05, ** p<0.01).
Fig. 8. The effect of CD56+ T supernatant (SN) on the expression of miRNAs.

Huh7.5.1 cells or primary human hepatocytes were cultured in the presence or absence of CD56+ T SN (50%, v/v) for 6h. Total cellular RNA extracted from cell cultures was subjected to the real-time RT-PCR for miRNA-122 (A) and miRNA-196a (B) quantification. The data are expressed as miRNA levels relative (fold) to the control (without CD56+ T SN treatment, which is defined as 1). The results shown are mean ±SD of triplicate cultures, representative of three experiments (*p<0.05, ** p<0.01).
Discussion
In this study, we examined the noncytolytic anti-HCV activity of primary CD56+ T cells in the newly developed infectious HCV (JFH-1) system.19 We demonstrated that CD56+ T cells isolated from peripheral blood of healthy subjects release soluble factors that inhibit HCV replication in both hepatoma cell line (Huh7.5.1) and primary human hepatocytes. This CD56+ T cell-mediated anti-HCV activity was highly significant, since as much as ∼90% of HCV RNA expression was inhibited in hepatocytes either co-cultured with CD56+ T cells or exposed to CD56+ T SN (Figs. 1-3). In agreement with studies by others,24-26 we showed that the replication of HCV JFH-1 in the primary hepatocytes was lower than that of Huh7.5.1 cells. One of possibilities is that when primary hepatocytes grow as a monolayer culture, they rapidly de-differentiate and have a shorter life span in culture, which limits their support for HCV replication. Nevertheless, the anti-HCV activity of CD56+ T SN on JFH-1-infected primary human hepatocytes is rather strong, because CD56+ T-SN-treated primary human hepatocytes had undetectable negative-strand HCV RNA and lower levels of HCV positive-strand RNA than untreated cells (Fig. 3).
We subsequently identified that IFN-γ is the major player in this CD56+ T cell-mediated anti-HCV activity, as evidenced by the observation that the preincubation CD56+ T SN with antibody to IFN-γ or pretreatment of hepatocytes with antibodies to IFN-γ receptor (IFN-γR1 and/or IFN-γR2) largely blocked CD56+ T SN-mediated anti-HCV activity in hepatocytes (Fig. 5). The role of IFN-γ in CD56+ T cell-mediated anti-HCV activity is also supported by our observation that CD56- T that produced significantly lower levels of IFN-γ (data not shown) had little effect on HCV replication in human hepatocytes (Fig.1). Our finding is in agreement with the reports by others,2, 3, 27 showing that CD56+ T cells produce much quicker and more IFN-γ than CD56- T cells poststimulation. In order to determine whether other antiviral cytokines also play a role in the CD56+ T SN action against HCV, we measured the levels of IFN-α/β and the primary Th1/Th2 cytokines in CD56+ T SN. ELISA revealed that although CD56+ T SN contained undetectable IFN-α/β, TNF-α, IL-2 and IL-10 were still detected. However, these cytokines had little effect on HCV replication since the antibodies to them did not block the CD56+ T-SN-mediated anti-HCV activity in the hepatocytes (data not shown). Therefore, these data demonstrated that CD56+ T cell-produced IFN-γ in the presence of other cytokines is still effective and potent in HCV inhibition.
While it is known that IFN-γ has the antiviral property, the mechanisms for IFN-γ-mediated antiviral activity is still largely unknown. IFN-γ primarily signals through the JAK/STAT pathway that is used by over 50 cytokines, growth factors and hormones to affect gene regulation.28, 29 We demonstrated that the treatment of primary human hepatocytes with CD56+ T SN not only activated the multiple elements of JAK/STAT pathway (JAK-2, JAK-3, STAT-1, STAT-2, and STAT-5A), which is critical for the activation of type I IFNs, but also induced the expression of the multiple antiviral factors, including NOS2A, GBP1, OSA1 and ISG15 (Table 2). Thus, it is likely that the activation of these antiviral genes contributes to the anti-HCV action of CD56+ T cells. Moreover, CD56+ T SN induced high level expression of CXCL-9, an IFN-γ inducible chemokine gene.30, 31 This finding may be significant, as control of HCV infection depends on at least in part on chemokine-mediated recruitment of specific T cells to the liver. It is likely that CD56+ T cell-mediated upregulation of CXCL-9 contributes to host immunity against HCV infection in liver.
Both type I IFNs (IFN-α/β) and type II IFN (IFN-γ) have potent anti-HCV activity in the in vitro systems.32-35 These two types of IFNs, however, act through different cell surface receptors and are structurally unrelated, suggesting that they carry out their activities through distinct but related pathways.36 In our experimental system, IFN-γ plays a major role in inhibition of HCV replication, because the antibodies to IFN-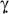 or IFN-γ receptors could largely block CD56+ T cell-mediated anti-HCV activity. Investigation of mechanism(s) responsible for the CD56+ T action showed that CD56+ T SN induced the expression of IRF-1, 3, 7, 8 and 9 in human hepatocytes (Fig. 6). These cellular factors have been shown as the positive regulators of the transcription of type I IFN genes.30, 37, 38 Among them, IRF-3 and IRF-7 are the two key regulators of type I IFN gene expression elicited by viruses. IRF-3 is mainly responsible for the initial induction of the IFN-β gene, whereas IRF-7, the upregulation of which is mediated by type I IFNs themselves, is involved in the late phase of type I IFN gene induction.30, 39 In addition to IRF-3 and IRF-7, IRF-1 is also a strong activator for type I IFN gene expression and able to rapidly mediate the induction of IFN-β.30, 37 Thus, the induction of these important regulators by CD56+ T SN provides a sound mechanism for CD56+ T cell-mediated enhancement of intracellular type I IFN expression in human hepatocytes. Given the vital role of intracellular IFN-α/β in the control of HCV replication in hepatocytes, it is likely that CD56+ T SN, through IFN-γ-mediated upregulation of intracellular IFN-α/β expression, inhibits HCV replication in hepatocytes. Our findings support the notion that there is a cross-talk between type I and type II IFN pathways. This concept was supported by our observations (Fig. 6B) that CD56+ T SN induced the expression of STAT-1 and STAT-2, the nuclear factors that are essential for the activation of type I IFN-mediated antiviral pathways. It is well known that STAT-1 is crucial in initial signaling by IFN-γ, whereas STAT-1 and STAT-2 are both necessary for the signaling by IFN-α/β.30, 40 The activation of intracellular type I IFNs in hepatocytes is of importance, as a recent study23 showed that IFN-α/β regulated the expression of several cellular miRNAs that are involved in HCV infection and replication.23,41 For example, miRNA-122, a liver-specific miRNA, has been shown to be essential for HCV replication.41 This HCV-supportive miRNA, however, could be inhibited by IFN-β.23 Another important miRNA is miRNA-196 that has the anti-HCV property and could be upregulated by type I IFNs.23 Our results that CD56+ T SN treatment of hepatocytes inhibited the expression of miRNA-122 and enhanced the expression of miRNA-196a provide a novel mechanism for the CD56+ T cell-mediated action against HCV.
or IFN-γ receptors could largely block CD56+ T cell-mediated anti-HCV activity. Investigation of mechanism(s) responsible for the CD56+ T action showed that CD56+ T SN induced the expression of IRF-1, 3, 7, 8 and 9 in human hepatocytes (Fig. 6). These cellular factors have been shown as the positive regulators of the transcription of type I IFN genes.30, 37, 38 Among them, IRF-3 and IRF-7 are the two key regulators of type I IFN gene expression elicited by viruses. IRF-3 is mainly responsible for the initial induction of the IFN-β gene, whereas IRF-7, the upregulation of which is mediated by type I IFNs themselves, is involved in the late phase of type I IFN gene induction.30, 39 In addition to IRF-3 and IRF-7, IRF-1 is also a strong activator for type I IFN gene expression and able to rapidly mediate the induction of IFN-β.30, 37 Thus, the induction of these important regulators by CD56+ T SN provides a sound mechanism for CD56+ T cell-mediated enhancement of intracellular type I IFN expression in human hepatocytes. Given the vital role of intracellular IFN-α/β in the control of HCV replication in hepatocytes, it is likely that CD56+ T SN, through IFN-γ-mediated upregulation of intracellular IFN-α/β expression, inhibits HCV replication in hepatocytes. Our findings support the notion that there is a cross-talk between type I and type II IFN pathways. This concept was supported by our observations (Fig. 6B) that CD56+ T SN induced the expression of STAT-1 and STAT-2, the nuclear factors that are essential for the activation of type I IFN-mediated antiviral pathways. It is well known that STAT-1 is crucial in initial signaling by IFN-γ, whereas STAT-1 and STAT-2 are both necessary for the signaling by IFN-α/β.30, 40 The activation of intracellular type I IFNs in hepatocytes is of importance, as a recent study23 showed that IFN-α/β regulated the expression of several cellular miRNAs that are involved in HCV infection and replication.23,41 For example, miRNA-122, a liver-specific miRNA, has been shown to be essential for HCV replication.41 This HCV-supportive miRNA, however, could be inhibited by IFN-β.23 Another important miRNA is miRNA-196 that has the anti-HCV property and could be upregulated by type I IFNs.23 Our results that CD56+ T SN treatment of hepatocytes inhibited the expression of miRNA-122 and enhanced the expression of miRNA-196a provide a novel mechanism for the CD56+ T cell-mediated action against HCV.
In conclusion, the present study using primary CD56+ T cells and the clinical relevant hepatocyte system that produces infectious HCV has for the first time provided direct experimental evidence at cellular and molecular levels, showing that CD56+ T cells have the ability to inhibit HCV infection and replication in human hepatocytes. The finding is particularly important, since CD56+ T cells are abundant in liver and are a key component of host innate immune cell-mediated defense mechanisms. More importantly, we showed that CD56+ T cells, through the secretion of IFN-γ, activate JAK/STAT pathway and enhance the expression of IRFs and STATs, resulting in the induction of intracellular IFN-α/β and anti-HCV miRNA expression. These findings provide a novel mechanism for CD56+ T cell-mediated anti-HCV activity in human hepatocytes, which would be of importance for the design and development of CD56+ T cell-based intervention strategies for intracellularly control and elimination of HCV in hepatocytes.
Acknowledgements
The authors thank Dr. Charles Rice (The Rockefeller University, New York, NY) for generously providing Huh7.5.1 cell line, Dr. Takaji Wakita (Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan) for providing HCV JFH-1 molecular clone, and Dr. Guangxiang Luo (University of Kentucky College of Medicine, Lexington, Kentucky) for providing mouse anti-HCV NS3 antibody.
Financial Support: This work was supported by the National Institute of Health grants DA 12815 and DA 22177 (to Wenzhe Ho) and Foerderer Murray Award (to Wenzhe Ho).
Abbreviations
- IFN
interferon
- HCV
hepatitis C virus
- TNF-α
tumor necrosis factor-α
- IL
interleukin
- IFN-γR
IFN-γ receptor
- IFN-α/β R
IFN-α/β receptor
- STAT
signal transducer and activator of transcription
- IRF
IFN regulatory factor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- SN
supernatant
- JAK
Janus Kinase
- miRNA
microRNA
References
- 1.Schmidt RE, Murray C, Daley JF, Schlossman SF, Ritz J. A subset of natural killer cells in peripheral blood displays a mature T cell phenotype. J Exp Med. 1986;164:351–356. doi: 10.1084/jem.164.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly-Rogers J, Madrigal-Estebas L, O’Connor T, Doherty DG. Activation-induced expression of CD56 by T cells is associated with a reprogramming of cytolytic activity and cytokine secretion profile in vitro. Hum Immunol. 2006;67:863–873. doi: 10.1016/j.humimm.2006.08.292. [DOI] [PubMed] [Google Scholar]
- 3.Ohkawa T, Seki S, Dobashi H, Koike Y, Habu Y, Ami K, Hiraide H, et al. Systematic characterization of human CD8+ T cells with natural killer cell markers in comparison with natural killer cells and normal CD8+ T cells. Immunology. 2001;103:281–290. doi: 10.1046/j.1365-2567.2001.01248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loza MJ, Metelitsa LS, Perussia B. NKT and T cells: coordinate regulation of NK-like phenotype and cytokine production. Eur J Immunol. 2002;32:3453–3462. doi: 10.1002/1521-4141(200212)32:12<3453::AID-IMMU3453>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 5.Jin Y, Fuller L, Carreno M, Esquenazi V, Tzakis AG, Miller J. The regulation of phenotype and function of human liver CD3+/CD56+ lymphocytes, and cells that also co-express CD8 by IL-2, IL-12 and anti-CD3 monoclonal antibody. Hum Immunol. 1998;59:352–362. doi: 10.1016/s0198-8859(98)00030-5. [DOI] [PubMed] [Google Scholar]
- 6.Doherty DG, Norris S, Madrigal-Estebas L, McEntee G, Traynor O, Hegarty JE, O’Farrelly C. The human liver contains multiple populations of NK cells, T cells, and CD3+CD56+ natural T cells with distinct cytotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J Immunol. 1999;163:2314–2321. [PubMed] [Google Scholar]
- 7.Lu PH, Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994;153:1687–1696. [PubMed] [Google Scholar]
- 8.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 9.Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, Lee L, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med. 2006;203:1343–1355. doi: 10.1084/jem.20060028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deignan T, Curry MP, Doherty DG, Golden-Mason L, Volkov Y, Norris S, Nolan N, et al. Decrease in hepatic CD56(+) T cells and V alpha 24(+) natural killer T cells in chronic hepatitis C viral infection. J Hepatol. 2002;37:101–108. doi: 10.1016/s0168-8278(02)00072-7. [DOI] [PubMed] [Google Scholar]
- 11.Norris S, Doherty DG, Curry M, McEntee G, Traynor O, Hegarty JE, O’Farrelly C. Selective reduction of natural killer cells and T cells expressing inhibitory receptors for MHC class I in the livers of patients with hepatic malignancy. Cancer Immunol Immunother. 2003;52:53–58. doi: 10.1007/s00262-002-0331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koreck A, Suranyi A, Szony BJ, Farkas A, Bata-Csorgo Z, Kemeny L, Dobozy A. CD3+CD56+ NK T cells are significantly decreased in the peripheral blood of patients with psoriasis. Clin Exp Immunol. 2002;127:176–182. doi: 10.1046/j.1365-2249.2002.01721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bose A, Chakraborty T, Chakraborty K, Pal S, Baral R. Dysregulation in immune functions is reflected in tumor cell cytotoxicity by peripheral blood mononuclear cells from head and neck squamous cell carcinoma patients. Cancer Immun. 2008;8:10. [PMC free article] [PubMed] [Google Scholar]
- 14.Doherty DG, O’Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunol Rev. 2000;174:5–20. doi: 10.1034/j.1600-0528.2002.017416.x. [DOI] [PubMed] [Google Scholar]
- 15.Vitale M, Caruso A, Licenziati S, Rodella L, Fiorentini S, Zauli G, Castelli F, et al. Differential production of IFN-gamma, analyzed at the single-cell level, by specific subsets of human NK and T cells from healthy and HIV(+) subjects. Cytometry. 2000;39:189–194. doi: 10.1002/(sici)1097-0320(20000301)39:3<189::aid-cyto3>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 16.Golden-Mason L, Castelblanco N, O’Farrelly C, Rosen HR. Phenotypic and functional changes of cytotoxic CD56pos natural T cells determine outcome of acute hepatitis C virus infection. J Virol. 2007;81:9292–9298. doi: 10.1128/JVI.00834-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawarabayashi N, Seki S, Hatsuse K, Ohkawa T, Koike Y, Aihara T, Habu Y, et al. Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology. 2000;32:962–969. doi: 10.1053/jhep.2000.19362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okumura A, Ishikawa T, Maeno T, Sato K, Ayada M, Hotta N, Yamauchi T, et al. Changes in natural killer T cells subsets during therapy in type C hepatitis and hepatocellular carcinoma. Hepatol Res. 2005;32:213–217. doi: 10.1016/j.hepres.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Wang X, Douglas SD, Metzger DS, Woody G, Zhang T, Song L, et al. CD8+ T cell depletion amplifies hepatitis C virus replication in peripheral blood mononuclear cells. J Infect Dis. 2005;192:1093–1101. doi: 10.1086/432957. [DOI] [PubMed] [Google Scholar]
- 21.Laskus T, Radkowski M, Wang LF, Vargas H, Rakela J. The presence of active hepatitis C virus replication in lymphoid tissue in patients coinfected with human immunodeficiency virus type 1. J Infect Dis. 1998;178:1189–1192. doi: 10.1086/515682. [DOI] [PubMed] [Google Scholar]
- 22.Zhang T, Lin RT, Li Y, Douglas SD, Maxcey C, Ho C, Lai JP, et al. Hepatitis C virus inhibits intracellular interferon alpha expression in human hepatic cell lines. Hepatology. 2005;42:819–827. doi: 10.1002/hep.20854. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castet V, Fournier C, Soulier A, Brillet R, Coste J, Larrey D, Dhumeaux D, et al. Alpha interferon inhibits hepatitis C virus replication in primary human hepatocytes infected in vitro. J Virol. 2002;76:8189–8199. doi: 10.1128/JVI.76.16.8189-8199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong TW, Smith RL, Hughes MG, Camden J, Rudy CK, Evans HL, Sawyer RG, et al. Primary human hepatocytes in spheroid formation to study hepatitis C infection. J Surg Res. 2006;130:52–57. doi: 10.1016/j.jss.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 26.Lazaro CA, Chang M, Tang W, Campbell J, Sullivan DG, Gretch DR, Corey L, et al. Hepatitis C virus replication in transfected and serum-infected cultured human fetal hepatocytes. Am J Pathol. 2007;170:478–489. doi: 10.2353/ajpath.2007.060789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ami K, Ohkawa T, Koike Y, Sato K, Habu Y, Iwai T, Seki S, et al. Activation of human T cells with NK cell markers by staphylococcal enterotoxin A via IL-12 but not via IL-18. Clin Exp Immunol. 2002;128:453–459. doi: 10.1046/j.1365-2249.2002.01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 29.Subramaniam PS, Green MM, Larkin J, 3rd, Torres BA, Johnson HM. Nuclear translocation of IFN-gamma is an intrinsic requirement for its biologic activity and can be driven by a heterologous nuclear localization sequence. J Interferon Cytokine Res. 2001;21:951–959. doi: 10.1089/107999001753289569. [DOI] [PubMed] [Google Scholar]
- 30.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, et al. The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J Immunol. 2002;169:1433–1443. doi: 10.4049/jimmunol.169.3.1433. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Zhang T, Ho C, Orange JS, Douglas SD, Ho WZ. Natural killer cells inhibit hepatitis C virus expression. J Leukoc Biol. 2004;76:1171–1179. doi: 10.1189/jlb.0604372. [DOI] [PubMed] [Google Scholar]
- 33.Frese M, Schwarzle V, Barth K, Krieger N, Lohmann V, Mihm S, Haller O, et al. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology. 2002;35:694–703. doi: 10.1053/jhep.2002.31770. [DOI] [PubMed] [Google Scholar]
- 34.Cheney IW, Lai VC, Zhong W, Brodhag T, Dempsey S, Lim C, Hong Z, et al. Comparative analysis of anti-hepatitis C virus activity and gene expression mediated by alpha, beta, and gamma interferons. J Virol. 2002;76:11148–11154. doi: 10.1128/JVI.76.21.11148-11154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo JT, Bichko VV, Seeger C. Effect of alpha interferon on the hepatitis C virus replicon. J Virol. 2001;75:8516–8523. doi: 10.1128/JVI.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 37.Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, Takayanagi H, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem. 2007;282:20065–20069. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- 39.Hengel H, Koszinowski UH, Conzelmann KK. Viruses know it all: new insights into IFN networks. Trends Immunol. 2005;26:396–401. doi: 10.1016/j.it.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 40.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 41.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]