

Abstract
Infection with attenuated Listeria monocytogenes (Lm) is a robust in vivo model for examining how antigen-specific T cells are primed, and subsequent challenge with virulent Lm allows for the protective effects of T cell priming to be quantified. Herein, we investigated the role of PDL-1 in T cell priming and immunity conferred after primary infection with Lm ΔactA followed by virulent Lm challenge. In striking contrast to the inhibitory role of PDL-1 on T cell immunity in other infection models, marked reductions in the magnitude of T cell expansion and the kinetics of T cell proliferation were observed with PDL-1 blockade after primary Lm ΔactA infection. More importantly PDL-1 blockade beginning before primary infection and maintained throughout the experiment resulted in delayed bacterial clearance and T cell expansion after secondary challenge with virulent Lm. These results indicate that for immunity to intracellular bacterial infection, PDL-1 plays an important stimulatory role for priming and expansion of protective T cells.
Keywords: T cell, bacterial infection, costimulation, vaccination
Introduction
PDL-1 (B7-H1) belongs to the B7 family of co-stimulatory molecules that also includes B7-1 (CD80), B7-2 (CD86), and B7-DC (CD273 or PDL-2) (1, 2). T cell engagement with each of these co-stimulatory receptors can confer both stimulatory or inhibitory signals, and the overall magnitude of the antigen-specific T cell response after immunization or infection is controlled by multiple T cell activation and suppression signals. Therefore, understanding how these various opposing signals together control antigen-specific T cell activation is required for the more rational design of vaccines that aim to target T cell-mediated immunity. Although PDL-1 can provide both T cell inhibitory and activation signals examined with both in vitro models of T cell activation or in vivo models of autoimmune disease, functional studies with in vivo infection models have uniformly demonstrated that T cell stimulation by PDL-1 suppresses T cell proliferation and effector function (3-13). For example during chronic LCMV infection, in vivo PDL-1 blockade restores proliferation and cytolytic function to virus-specific “exhausted” CD8 T cells and results in viral clearance (6). Similarly, PDL-1 blockade restores activation and proliferation to virus-specific T cells during both chronic hepatitis B and acute herpes simplex virus in mouse infection models (7, 9), and proliferation and cytolytic function for HIV-specific or hepatitis C virus-specific CD8 T cells from human patients with these chronic infections (10-12). Accordingly, reinvigorating viral T cells through PDL-1 blockade has been proposed as a novel therapeutic intervention for treatment of chronic viral infection.
Listeria monocytogenes (Lm) is an intracellular Gram-positive bacterium that primarily causes localized infections in the gastrointestinal tract in immune-competent individuals, and more severe systemic infections in immune-compromised individuals. During infection, Lm primes a robust antigen-specific CD8 and CD4 T cell response, and accordingly Lm infection is a widely used experimental model whereby priming and activation of antigen-specific T cells, and the protective effects these T cells are examined (14). Furthermore because of the relative ease by which recombinant Lm strains can be generated, and the existence of many highly attenuated and immunogenic Lm mutant strains, recombinant Lm expressing protective antigen from other pathogens are being explored as a new class of live attenuated vaccine vectors (15-17). In this study we examined the effects of PDL-1 blockade on T cell priming and expansion after primary infection with Lm ΔactA, and protective immunity following secondary challenge with virulent Lm. The highly attenuated nature of Lm ΔactA compared with WT Lm normalizes antigen load after primary infection thereby bypassing potential difference in innate susceptibility between groups of mice allowing for a more accurate comparison of the resulting T cell response (18-21). In this report, we first demonstrate that PDL-1 expression is markedly upregulated during Lm ΔactA primary infection, and PDL-1 blockade does not alter antigen load or the kinetics of bacterial clearance following infection with this attenuated Lm strain. Additional studies using Lm ΔactA primary infection to normalize initial antigen load between anti-PDL-1 antibody and control antibody treated mice demonstrate that PDL-1 blockade delays the kinetics of T cell priming and reduces the magnitude of T cell expansion after primary Lm ΔactA infection. Importantly, PDL-1 blockade beginning prior to T cell priming and maintained throughout secondary challenge reveals an important role for PDL-1 in optimal bacterial clearance and secondary T cell expansion after challenge with virulent Lm.
Materials and Methods
Mice
C57B6 (H-2Kb) female mice were purchased from the National Cancer Institute and used at 6-8 weeks of age. OT-1 TCR transgenic mice have been described and were intercrossed with CD90.1 mice and maintained on a RAG-1-deficient background (22). All experiments were performed under University of Minnesota IACUC approved protocols.
Listeria monocytogenes
The recombinant Lm strain Lm-OVA and Lm-OVA ΔactA derived through targeted deletion in the actA gene were used allowing for an analysis of the immune response to the surrogate Lm-specific H-2Kb OVA257-264 antigen as described (20, 22). For infections, Lm were grown to early log phase (OD600 0.1) in brain heart infusion media at 37°C, washed, and diluted with saline to 200 μl final volume and injected IV. At the indicated time points after infection, the number of recoverable bacteria in the organs of infected mice were quantified by homogenization in saline containing Triton X (0.05%), and plating serial dilutions of the homogenate on brain heart infusion plates as described (21, 23).
Reagents, in vitro cultures, cell staining, and adoptive transfer
For in vivo PDL-1 blockade, anti-mouse PDL-1 (clone 10F.9G2) or rat IgG2b isotype control (clone LTF-2) antibodies (6, 9) were purchased from Bio Express, and injected IP in the following manner: one day prior to primary infection (500 μg per mouse), days 4 and 8 after primary infection and day 1 prior to rechallenge (250 μg per mouse). The CD8 T cell response to OVA257-264 was examined with H-2Kb dimer X loaded with OVA257-264 peptide according to the manufacturer's instructions (BD Biosciences). Antibodies for cell surface staining, reagents for intracellular cytokine and Annexin V staining were purchased from BD Biosciences and used according to manufacturer's recommendations. For in vitro culture, splenocytes were plated into 96-well round bottom plates (5 × 106 cells/ml), and stimulated with the indicated peptides (10-6 M) with Brefeldin-A (BD GolgiPlug reagent) for 5 hours as described (20). For adoptive transfer, 105 CD8 T cells from OT-1 (CD90.1) mice were CFSE labeled (5 μM final concentration) and transferred intravenously into recipient mice one day prior to Lm infection.
Statistics
The differences in number and percentage of antigen-specific cells, and geometric mean CFUs between groups of mice were evaluated by using the Student's t test with P < 0.05 taken as statistically significant (Graph Pad, Prism software).
Results
PDL-1 expression after primary Lm-ΔactA infection
Although expressed constitutively on most lymphoid cells, PDL-1 expression is upregulated on virtually all splenocytes during chronic viral infection (6). Functionally, PDL-1 expression during chronic viral infection actively suppresses virus-specific CD8 T cells since PDL-1 blockade restores the function of virus-specific “exhausted” T cells into “effector” cells capable of viral clearance (6). To gain insight into how and when PDL-1 may alter T cell priming during acute bacterial infection, we examined the kinetics of PDL-1 expression on splenocytes after primary Lm infection. Remarkably, beginning 24 hours after infection with 106 CFUs of Lm ΔactA, PDL-1 was upregulated on the majority of all splenocytes (Fig 1). At this time point, the level of PDL-1 expression was highest among the CD11c+ splenocyte population consistent with the important role this subset of antigen presenting cells plays in T cell priming after Lm infection (24). PDL-1 was also dramatically upregulated on other splenocyte subpopulations including CD11b+, B220+, CD4+ and CD8+ cells. PDL-1 upregulation in response to Lm ΔactA infection was maintained through day 3, dramatically reduced by day 6, and returned to levels present in naive mice day 14 after infection. The kinetics for PDL-1 upregulation and return to baseline on CD11c+ cells was similar to the expression kinetics of other molecules important for T cell priming such as CD80, CD86, MHC class I (H-2Kb) and class II (I-Ab) (Fig 1 and data not shown).
Figure 1.
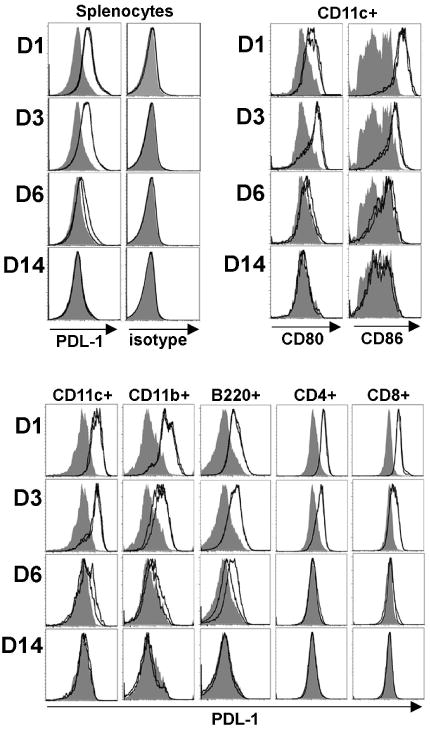
PDL-1, CD80, and CD86 expression among all splenocytes or specific splenocyte cell subsets at the indicated time points after infection with 106 Lm ΔactA (open histograms) compared with no infection mice (shaded histograms). These data are from 2 mice per experimental group per time point, and representative of three independent experiments with similar results.
PDL-1 blockade reduces antigen-specific T cell expansion after primary Lm-ΔactA infection
To evaluate the overall role of PDL-1 in T cell priming accommodating the brisk nature whereby PDL-1 is upregulated during Lm infection, we pretreated mice with either anti-PDL-1 blocking or isotype control antibody beginning one day prior to infection. Remarkably, in vivo PDL-1 blockade effectively blocked access to both constitutive levels of cell-surface PDL-1 and the increased expression levels present after infection since only background levels of PDL-1 staining could be detected in anti-PDL-1 treated mice (Fig 2A). These reductions were specific to PDL-1 since anti-PDL-1 blockade did not alter the cell-surface expression of other costimulation molecules in response to Lm infection (Fig 2A). Other experiments indicate that PDL-1 blockade does not alter the antigen load or kinetics of bacterial clearance after Lm-OVA ΔactA infection since the numbers of recoverable CFUs is virtually identical at early time points (24, 48, and 72 hours) after infection between anti-PDL-1 treated and control mice (Fig 2B). In this experimental model beginning day 5 after Lm ΔactA infection, a ∼70% reduction in the antigen-specific CD8 T cell response enumerated by OVA257-264 dimer staining was present for anti-PDL-1 treated compared with control mice (P < 0.001) (Fig 3A). This reduced magnitude antigen-specific CD8 T cell response was maintained through the peak (day 8) and contraction phase (days 15 to 30) of the T cell response. Similarly during the peak T cell response, there was a ∼70% reduction in number of antigen-specific CD8 T cells in splenocytes from anti-PDL-1 treated compared with control mice (Fig 3B). Moreover, 50 to 70% reductions in both the percentage and total number of cytokine producing CD8 and CD4 T cells among splenocytes were present after stimulation with the MHC class I peptide OVA257-264 or class II peptide LLO189-201, respectively (Fig 3C,D). These results indicate that PDL-1 blockade impedes antigen-specific T cell expansion after primary Lm ΔactA infection.
Figure 2.
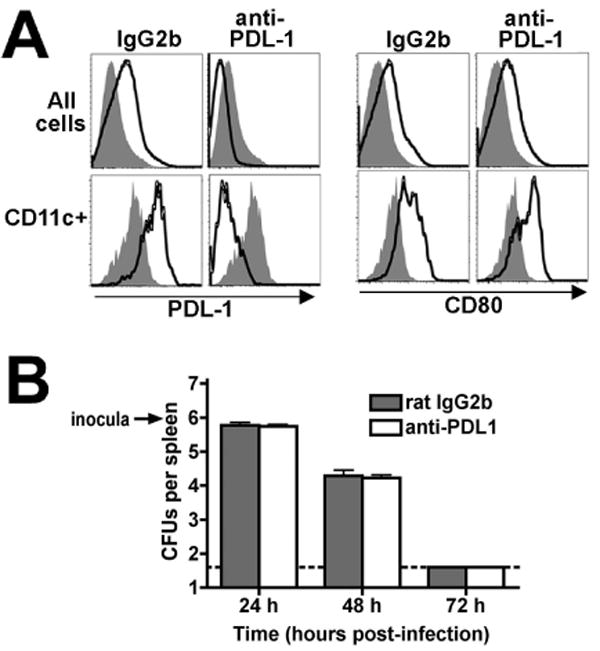
A. PDL-1 and CD80 expression among all splenocytes (top) or CD11c+ splenocyte cells (bottom) from mice 3 days after infection with 106 Lm ΔactA (open histograms) or no infection (shaded histograms) treated with either rat IgG2b isotype control or anti-PDL-1 antibody prior to infection. These data are from 2 mice per experimental group per time point, and representative of three independent experiments. B. Number of recoverable Lm CFUs per spleen in rat IgG2b control (shaded bars) or anti-PDL-1 antibody (open bars) treated mice at the indicated time points after infection. These data are from 4 to 5 mice per group, reflective of two independent experiments. Bar, one standard error.
Figure 3.

A. Percent antigen-specific CD8 T cells among PBMCs in anti-PDL-1 antibody treated and control mice at the indicated time points after infection with 106 Lm ΔactA quantified by H-2Kb OVA257-264 dimer staining. B. Number of H-2Kb OVA257-264 dimer+ CD8 T cells among splenocytes day 8 post-infection. C. Percent and number of IFN-γ producing CD8 T cells among splenocytes after stimulation with OVA257-264 peptide or no stimulation day 8 post-infection. D. Percent and number of IFN-γ producing CD4 T cells among splenocytes after LLO189-201 peptide stimulation day 8 post-infection. These data represent 7 to 12 mice per experimental group and is representative of three independent determinations. Bar, standard error. * P < 0.05.
PDL-1 blockade impedes antigen-specific T cell priming
Early priming events after Lm infection play critical roles regulating both the kinetics and magnitude of the antigen-specific T cell response (19). To determine if reductions in T cell expansion resulting from PDL-1 blockade are due to defects in early T cell priming, we examined the degree of proliferation and magnitude of expansion in adoptively transferred antigen-specific T cells in the first few days following Lm ΔactA infection. Beginning at 48 hours after infection adoptively transferred antigen-specific OT-1 CD8 (CD90.1) T cells could be readily detected, and at this early time point consistent reductions in both cell number and degree of CFSE dilution among antigen-specific T cells adoptively transferred into anti-PDL-1 compared with control mice were present (Fig 4A). Reduced levels of CFSE dilution and expansion among antigen-specific T cells in anti-PDL-1 antibody treated compared with control mice became more pronounced by day 3 after infection (Fig 4A). By day 4 after infection when CFSE was fully dilute among transferred cells in both groups of mice, significant reductions in both percentage and total numbers of antigen-specific OT-1 cells in mice treated with anti-PDL-1 compared with control antibody were still present (Fig 4B). In similar experiments we evaluated the possibility that reduced numbers of antigen-specific T cells present in anti-PDL-1 treated compared with control mice were the result of increased apoptotic cell death since in other systems, direct stimulation of activated human T cells with immobilized anti-PDL-1 antibody in vitro can lead to increased rates of apoptotic cell death (25). These experiments revealed no difference in annexin V staining among antigen-specific OT-1 cells recovered from anti-PDL-1 treated and control mice (Fig 4C), and indicate that potential differences in the rate of apoptotic cell death attributable to anti-PDL-1 antibody in other systems does not contribute significantly to observed reductions in number of antigen specific T cells primed after Lm infection. Together, these results indicate that after primary Lm-ΔactA infection, PDL-1 blockade interferes with early events in T cell priming resulting in reduced proliferation and expansion of antigen-specific T cells.
Figure 4.
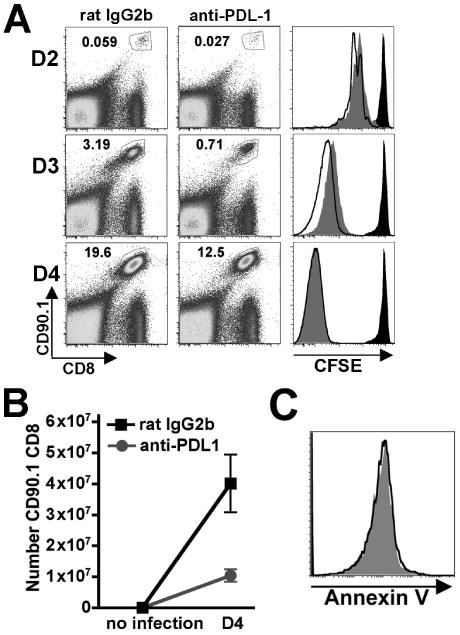
A. Expansion and CFSE dilution in adoptively transferred OT-I (CD90.1+CD8+) cells at the indicated time points after infection with 106 Lm ΔactA in anti-PDL-1 antibody treated (gray histograms), control rat IgG2b treated (open histograms), or no infection control mice (black histograms). The numbers in the upper left quadrant indicate percentage of gated cells among total splenocytes. B. Number of OT-I (CD90.1+CD8+) per mouse spleen prior to infection and day 4 after infection in anti-PDL-1 antibody (circle) or rat IgG2b antibody (square) treated mice. C. Annexin V staining among OT-1 (CD90.1+CD8+) cells day 3 after infection in either anti-PDL-1 treated (gray histograms) or control IgG2b treated (open histograms) mice. These data represent 5-7 mice for each experimental group combined from two independent experiments. Bar, standard error.
PDL-1 blockade delays bacterial clearance after challenge with virulent Lm
To evaluate the overall impact PDL-1 plays in protective immunity primed by attenuated Lm infection we examined the susceptibility of anti-PDL-1 antibody treated and control mice to challenge with an inocula of virulent Lm lethal for naïve mice (1 LD50). Consistent with other studies demonstrating the remarkable efficiency whereby prior infection with Lm-ΔactA primes protective T cell immunity allowing virulent Lm to be rapidly cleared after challenge (18), there were ∼ 4 log-fold reductions in recoverable Lm CFUs in the livers by day 3 after challenge for Lm ΔactA primed compared with naive control mice each treated with isotype control antibody (Fig 5A). However the degree of protection primed with Lm ΔactA was significantly reduced by PDL-1 blockade since ∼2 log-fold increased numbers of Lm were present for anti-PDL-1 antibody treated compared with control mice (P = 0.001). Moreover, this is likely an under-representation of the true difference between these groups since 4 of 7 control mice (rat IgG2b treated) had Lm CFUs below the limits of detection, while Lm CFUs were present for all anti-PDL-1 treated mice (7 total). To evaluate if the increased susceptibility to virulent Lm challenge for anti-PDL-1 compared with control mice reflects either a failure or delay in bacterial clearance, Lm CFUs were also examined at additional later time points. By day 5 after challenge, the number of recoverable Lm for both anti-PDL-1 antibody treated and control mice were below the limits of detection (Fig 5A). These results indicate that PDL-1 blockade delays bacterial clearance after secondary challenge with virulent Lm.
Figure 5.

A. Numbers of recoverable Lm CFUs in the liver days 3 and 5 after challenge with 105 virulent Lm for the indicated groups of naive mice (solid bar), or mice previously inoculated with Lm ΔactA 30 days prior to challenge (shaded and striped bars). For these experiments, mice were treated with either anti-PDL-1 or control antibody prior to primary infection, and throughout the experiment as described in the Materials and Methods. B. Number of IFN-γ producing CD8 T cells after stimulation with OVA257-264 peptide among splenocytes prior to challenge (day 30 after Lm ΔactA infection), and days 3 and 5 after challenge with virulent Lm. These data represent 7 mice for each experimental group combined from two independent experiments. Bar, standard error. * P < 0.05.
Lastly we examined the role of PDL-1 in antigen-specific T cell expansion after secondary infection by enumerating the antigen-specific T cell response to OVA257-264 prior to challenge (day 30 after Lm ΔactA primary infection), and days 3 and 5 after secondary challenge with virulent Lm (Fig 5B). Consistent with the marked reductions in T cell expansion after primary infection with Lm ΔactA attributable to PDL-1 blockade, significantly delayed kinetics and reductions in overall magnitude of T cell expansion after secondary challenge with virulent Lm was observed for anti-PDL-1 antibody treated compared with control mice. Taken together, these results indicate that PDL-1 blockade impedes antigen-specific T cell priming and protective immunity during Lm infection.
Discussion
PDL-1 T stimulation uniformly results in suppression of pathogen-specific T cell proliferation and effector function after evaluation in various in vivo infection models (6-12). However, whether PDL-1 mediated suppression of antigen-specific T cell proliferation is beneficial or detrimental for the host appears to be pathogen and context dependent. For LCMV clone 13 and other viruses that cause chronic infection, active T cell inhibition through PDL-1 results in antigen-specific T cells that are phenotypcially “exhausted” and cannot eradicate virus resulting in chronic infection (6, 8-12). On the other hand for acute HSV-1 infection causing stromal keratitis, PDL-1 mediated suppression of virus-specific T cell priming and apoptosis limits the severity of disease caused by activated T cells (7). Herein we report experimental data demonstrating a stimulatory role for PDL-1 in T cell proliferation and expansion, and a protective role for PDL-1 using an in vivo model of priming model of protective immunity to Lm infection.
First we demonstrate the marked upregulation of PDL-1 expression from baseline levels on virtually all immune cells after Lm infection, with CD11c+ having the highest levels of expression. The role of PDL-1 expression in priming T cell immunity after Lm infection was examined treating mice with either anti-PDL-1 blocking or control antibody prior to and during infection. To bypass potential roles PDL-1 may play in innate resistance to virulent Lm infection leading to differences in Lm antigen load, infection with the ΔactA Lm mutant was used. This highly attenuated Lm lacks the bacterial virulence protein necessary for actin recruitment and intracellular and intercellular spread and therefore is rapidly cleared even in mice lacking key components of innate resistance (18, 20, 21). Similarly we now demonstrate that infection with Lm ΔactA normalize antigen load in anti-PDL-1 treated and control mice at early time points when potential differences can critically impact the immune response magnitude (19). Using this in vivo model to prime Lm specific T cells, we demonstrate consistent ∼70% reductions in the antigen-specific T cell response for anti-PDL-1 antibody treated compared with control mice quantified using both MHC multimer and intracellular cytokine staining techniques. Additional experiments using adoptively transferred antigen-specific T cells from TCR transgenic mice indicate that PDL-1 plays an important role in early T cell priming since cells recovered from anti-PDL-1 compared with control mice had delayed CFSE dilution and decreased overall expansion. Importantly after challenge with virulent Lm in mice primed with Lm ΔactA, PDL-1 blockade throughout the experiment lead to delayed bacterial clearance and secondary expansion of antigen-specific T cells.
Therefore during Lm infection, unlike the other previously described in vivo infection models, PDL-1 is required for optimal T cell proliferation and expansion. These results are consistent with the stimulatory role for PDL-1 previously described in other non-infectious in vivo and in vitro models of T cell activation (3-5). Our results demonstrating PDL-1 stimulates T cell immunity after primary infection with Lm-ΔactA are also consistent with a recent study describing a defect in Lm-specific CD8 T cell expansion after infection with WT Lm using a different anti-PDL-1 blocking antibody and a different mouse strain (26). In this study, the increased susceptibility conferred by PDL-1 blockade to primary WT Lm infection was also demonstrated underscoring the importance of using attenuated Lm for primary infection to normalize antigen load at early time points when the resulting adaptive T cell response is being evaluated. This critical difference or potentially other intrinsic differences between the two experimental systems lead these authors to miss the important functional consequence of PDL-1 blockade resulting in delayed bacterial clearance after re-challenge with virulent Lm that we now report. These results collectively challenge the dogma that T cell stimulation through PDL-1 during infection results in decreased proliferation and suppression of effector function, and instead demonstrates that the role of PDL-1 in T cell immunity is context dependent and varies with the type of infection. Determining the molecular basis for how T cell stimulation through PDL-1 and other co-stimulation signals can result in such drastic differences in proliferation and functional capacity during specific infections is an important area for future investigation.
Acknowledgments
The authors thank Drs Stephen McSorley, David Masopust, and Vaiva Vezys; and members of their respective laboratories for helpful discussions.
The authors gratefully acknowledge funding support from the following sources: NICHD/NIH-K08HD51584, Infectious Disease Society of America, March of Dimes Foundation, and Vikings Children's Fund.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org
References
- 1.Keir ME, Francisco LM, Sharpe AH. PaD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007;19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 3.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 4.Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, Machida U, Iwai H, Azuma M, Chen L, Watanabe M. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171:4156–4163. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- 5.Subudhi SK, Zhou P, Yerian LM, Chin RK, Lo JC, Anders RA, Sun Y, Chen L, Wang Y, Alegre ML, Fu YX. Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest. 2004;113:694–700. doi: 10.1172/JCI19210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 7.Jun H, Seo SK, Jeong HY, Seo HM, Zhu G, Chen L, Choi IH. B7-H1 (CD274) inhibits the development of herpetic stromal keratitis (HSK) FEBS Lett. 2005;579:6259–6264. doi: 10.1016/j.febslet.2005.09.098. [DOI] [PubMed] [Google Scholar]
- 8.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol. 2007;178:2714–2720. doi: 10.4049/jimmunol.178.5.2714. [DOI] [PubMed] [Google Scholar]
- 10.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 11.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 13.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 15.Frankel FR, Hegde S, Lieberman J, Paterson Y. Induction of cell-mediated immune responses to human immunodeficiency virus type 1 Gag protein by using Listeria monocytogenes as a live vaccine vector. J Immunol. 1995;155:4775–4782. [PubMed] [Google Scholar]
- 16.Zhao X, Zhang M, Li Z, Frankel FR. Vaginal protection and immunity after oral immunization of mice with a novel vaccine strain of Listeria monocytogenes expressing human immunodeficiency virus type 1 gag. J Virol. 2006;80:8880–8890. doi: 10.1128/JVI.00894-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orr MT, Orgun NN, Wilson CB, Way SS. Cutting Edge: Recombinant Listeria monocytogenes expressing a single immune-dominant peptide confers protective immunity to herpes simplex virus-1 infection. J Immunol. 2007;178:4731–4735. doi: 10.4049/jimmunol.178.8.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 19.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 20.Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–4505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol. 2003;171:533–537. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 22.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 23.Edelson BT, Unanue ER. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol. 2002;169:3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- 24.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong H, Strome SE, Matteson EL, Moder KG, Flies DB, Zhu G, Tamura H, Driscoll CL, Chen L. Costimulating aberrant T cell responses by B7-H1 autoantibodies in rheumatoid arthritis. J Clin Invest. 2003;111:363–370. doi: 10.1172/JCI16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo SK, Jeong HY, Park SG, Lee SW, Choi IW, Chen L, Choi I. Blockade of endogenous B7-H1 suppresses antibacterial protection after primary Listeria monocytogenes infection. Immunology. 2007;123:90–99. doi: 10.1111/j.1365-2567.2007.02708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]