

Abstract
The Foxp3-expressing subset of regulatory CD4+ T cells have defined antigen-specificity and play essential roles in maintaining peripheral tolerance by suppressing the activation of self-reactive T cells. Similarly during chronic infection, pathogen-specific Foxp3-expressing CD4+ T cells expand and actively suppress pathogen-specific effector T cells. Herein, we used MHC class II tetramers and Foxp3gfp knock-in mice to track the kinetics and magnitude whereby pathogen-specific Foxp3+CD4+ and Foxp3-CD4+ cells are primed and expand after acute infection with recombinant Listeria monocytogenes (Lm) expressing the non-"self' antigen 2W1S52-68. We demonstrate that Lm infection selectively primes proliferation, expansion and subsequent contraction of Lm-specific Foxp3-negative effector CD4+ cells, while the numbers of Lm-specific Foxp3+CD4+ regulatory cells remain essentially unchanged. In sharp contrast, purified 2W1S52-68 peptide primes coordinated expansion of both Foxp3+ regulatory and Foxp3-negative effector T cells with the same antigen specificity. Together, these results indicate selective priming and expansion of Foxp3-negative CD4 T cells is a distinguishing feature for acute bacterial infection.
Keywords: T cells, bacterial infections, cell proliferation and differentiation
INTRODUCTION
The adaptive immune system is intricately regulated allowing for potentially detrimental self-reactive responses to be actively suppressed, while simultaneously allowing protective responses to be rapidly generated and mobilized during infection. The naturally occurring Foxp3+ subset of CD4 T cells, regulatory T cells (Tregs), play important roles in maintaining peripheral tolerance and immune homeostasis. Mice and humans with spontaneous or targeted defects in foxp3 invariably develop fatal multi-organ systemic autoimmunity (1, 2). Tregs, like other T cell subsets, have diverse antigen-specificity and their suppressive function requires cell-intrinsic TCR stimulation (3, 4). However, the degree of overlap in the repertoire of antigens recognized by Tregs and non-Tregs remains uncertain. After adoptive transfer into naïve recipients, CD4+ T cells transduced with TCR sequences from CD25+CD4+ Tregs proliferate to a larger extent compared with CD4+ T cells transduced with TCR from CD25-CD4+ T cells (5). Moreover in transgenic mice that express hemagglutinin (HA) as a neo-“self” antigen, substantial increases in the number of HA-specific CD4+Foxp3+ Tregs are present (6). Collectively, these results suggest high-affinity interactions between developing thymocytes and “self” antigens program their differentiation into self-reactive Tregs. However, larger scale studies comparing the TCR repertoires between Tregs and effectors among human peripheral CD4+ cells indicate these two cell subsets recognize a similar and overlapping array of antigens (7, 8). Moreover, the ability of exogenous antigens to prime naïve CD4 T cells to differentiate in vivo into antigen-specific Foxp3-expressing Tregs with specificity to non-“self” antigens indicates the potential for a high degree of plasticity in the repertoire of antigens recognized by Foxp3+CD4+ Tregs (9-11).
Interrelated with their role in suppressing the activation of self-reactive T cells, Tregs also play important roles in controlling the pathogen-specific adaptive T cell response (12). For each infection, a delicate balance between expansion of pathogen-specific effector T cells that mediate pathogen clearance, and Tregs that limit collateral host damage by suppressing pathogen-specific effector T cells appears to be established. For example during pulmonary Mycobacterium tuberculosis infection where both Tregs and effector T cells are recruited into infected tissue, Treg depletion accelerates the kinetics of bacterial clearance (13). Similarly Tregs potently eliminate the reductions in Mycobacterium tuberculosis CFUs conferred by effector T cells after T cell reconstitution in RAG-deficient mice (14). Accordingly for Mycobacterium tuberculosis infection, Tregs delay pathogen clearance. Interestingly however Tregs through suppression of pathogen specific T cells also play important protective roles during other infections. For example after corneal HSV-1 inoculation, Treg depletion results in more severe viral immunopathological lesions and exacerbates stromal keratitis (15). Thus, for most infections, manipulation of bulk Tregs in a non-antigen specific fashion clearly has the potential to alter both the kinetics of pathogen clearance and infection outcome. Curiously however, the antigen-specificity for infection primed Tregs has not been characterized after most infections, and has been described most extensively in models of Leishmania major or Schistosoma mansoni infection. In both these chronic parasite infection models, Tregs recovered from the infection site proliferate and produce IL-10 in an antigen-specific fashion after stimulation with parasite infected APCs (16-19). More importantly these parasite-specific Tregs through suppression of effector T cells cause local antigen persistence required for immunity to re-infection at secondary sites (20, 21). Therefore coordinated expansion of both pathogen-specific Tregs and effectors cells result in local antigen persistence during chronic infection that appear to offer survival advantages for both host and pathogen. These observations led us to explore potential differences in how pathogen-specific Tregs and effectors CD4 T cells are primed after acute infection when antigen persistence does not occur.
In this study, infection with the intracellular bacterium Listeria monocytogenes (Lm) was used to characterize the magnitude and kinetics whereby acute infection primes the expansion of pathogen-specific Foxp3+CD4+ Tregs and Foxp3-CD4+ effector T cells. Since the number of CD4+ T cells specific for each defined antigen is highly consistent among individual naïve mice and the precursor frequency of CD4+ cells specific for the 2W1S variant of peptides 52-68 from I-Eα is the highest among defined I-Ab antigens in naïve B6 mice (22), we engineered recombinant Lm to express this surrogate Lm-specific antigen so that a detailed comparison of 2W1S-specific CD4+ T cells before and after Lm infection can be made using MHC class II tetramer staining. Recent studies indicate that 2W1S-tetramer staining followed by magnetic bead enrichment is a highly sensitive and specific method allowing for the enumeration and characterization of rare 2W1S-specifc CD4+ cells present in naïve B6 mice (22). Using this technique, similar percentages of Foxp3+CD25+ Tregs were identified among 2W1S-tetramer+CD4+ cells and the remaining tetramer-negative CD4+ cell population in naïve mice. In response to either Lm-2W1S52-68 infection or 2W1S52-68 peptide inoculation, 2W1S-specific cells readily expand allowing characterization for how antigen-specific effector and Treg CD4+ cells are primed under each condition.
MATERIALS AND METHODS
Bacteria
WT Lm (10403s), the isogenic Lm ΔactA mutant (DPL1942) and recombinant Lm-OVA, along with methods for Lm transformation have been described (23-25). For infection, Lm was grown and sub-cultured in BHI media to early log-phase (OD600 0.1), washed and diluted in saline and inoculated IV as described (24). Recombinant Lm-2W1S was grown in media supplemented with chloramphenicol (20μg/ml).
Expression constructs
The low copy episomal pAM401 plasmid based Lm-expression construct allowing insertion of recombinant antigens in-frame with the Lm hly promoter, signal sequence, and HA-tag has been described (24). The following primers were annealed together, and ligated into the Pst1 and Stu1 sites of this vector: 2W1S coding 5'-gaagcatggggtgcactagcaaactgggcagtagactcagcagg-3', 2W1S non-coding 5'-cctgctgagtctactgcccagtttgctagtgcaccccatgcttctgca-3'. Relevant portions of this construct were verified by DNA sequencing.
Western blotting
Lm supernatant protein preparation, SDS gel electrophoresis and protein blotting using anti-HA antibody (clone HA-11, Covance, Princeton, NJ) were performed as described (24).
Mice
C57Bl/6 (B6) mice were purchased from the National Cancer Institute and Foxp3gfp knock-in mice (26) backcrossed for ≥ 10 generations to B6 mice were a gift from Dr. Alexander Y. Rudensky. For BrdU incorporation, mice were administered 500μg of BrdU IP daily for seven days after primary Lm infection. For priming antigen-specific CD4+ cells in non-infection conditions, the indicated quantity of 2W1S52-68 peptide (≥ 95% purity, United Biochemical Research, Inc. Seattle, WA) was dissolved in saline and injected IV. All experiments were performed under University of Minnesota IACUC approved protocols.
Tetramer and cell surface staining
Techniques for MHC class II tetramer production, cell staining, and magnetic bead enrichment with fluorochrome-conjugated 2W1S52-68-tetramer have been described (22). Other reagents for cell staining and quantifying BrdU incorporation (BD Bioscience) were used according to the manufacture's instructions.
RESULTS
Construction of Lm-expressing Eα-2W1S
The absolute number of CD4+ T cells specific for any specific defined antigen is highly consistent among individual mice. However, within an individual mouse, there is significant variation in the absolute numbers of CD4+ T cells with specificity to each defined antigen (22). For example, using MHC class II tetramer staining and magnetic bead enrichment, naïve B6 mice are estimated to contain ~200 CD4 T cells specific for the non-“self” 2W1S52-68 peptide representing the highest frequency of CD4+ T cells among defined I-Ab peptide antigens (22). Accordingly, we sought to exploit the relatively high precursor frequency of 2W1S52-68-specific CD4 T cell in naïve B6 mice to examine pathogen-specific CD4 T cells before and after experimental Lm infection. For these experiments, we engineered recombinant Lm to express the 2W1S52-68 peptide by placing the promoter and signal sequence coding residues from the Lm hly gene in-frame with the coding residues for 2W1S52-68 peptide using the previously described “open” Lm expression construct (24) (Fig. 1A). After transformation, both WT Lm and Lm ΔactA mutant strains each secreted a protein of predicted size (~19kDa) that was readily detected using standard blotting techniques (Fig. 1B).
FIGURE 1.

A. Construct map indicating placement of coding sequences for 2W1S52-68 peptides expressed and secreted behind the hly promoter and signal sequence (SS) within the pAM401 parent vector (cat, chloroamphenicol acetyltransferase). B. Western blot of supernatant protein from WT Lm (WT Lm-2W1S, lane 1) or Lm ΔactA (Lm ΔactA-2W1S, lane 2) each transformed with this construct, and Lm-OVA (lane 3) used as a positive control (25).
Lm-specific CD4 T cells tracked with Class II tetramer
To track the Lm-2W1S-specific CD4+ T cell response, mice were initially infected with recombinant WT Lm-expressing 2W1S (WT Lm-2W1S). However, at relatively low WT Lm-2W1S inocula (103 to 104 CFUs), the expansion of 2W1S-specific CD4 T cells above levels present in naïve mice was not reproducibly detected (Fig. 2). After infection with higher Lm-2W1S inocula (≥ 105 CFUs), the majority of infected mice became moribund within 72 hours after infection consistent with the previously reported lethal dose of 105 CFUs for WT Lm in naïve B6 mice (27). Therefore, the virulence of WT Lm precluded infection with higher WT Lm-2W1S inocula potentially required for more optimal priming and robust expansion of 2W1S-specific CD4+ T cells. To bypass this limitation, we examined the 2W1S-specific response primed by infection with higher inocula of attenuated Lm containing targeted deletion the actA locus (Lm ΔactA-2W1S). Lm ΔactA cannot spread from infected into adjacent non-infected cells, and is rapidly cleared after infection. Therefore the highly attenuated nature of this mutant allows characterization of the adaptive T cell response after infection with relatively high inocula of Lm ΔactA even in neonatal and other mice with targeted immune defects normally highly susceptible to virulent Lm infection (27-30).
FIGURE 2.

Expansion of 2W1S-specific CD4 cells after WT Lm-2W1S or Lm ΔactA-2W1S infection. FACS plots indicating the percent 2W1S-TET+ cells among CD4+ cells before or seven days after infection with the indicated inocula of WT Lm 2W1S and Lm ΔactA-2W1S (top). Histogram plots (bottom) indicating CD62L expression among 2W1S-TET+CD4+ cells (solid line) and 2W1S-TET+CD4+ cells (filled shaded). These data are representative of four independent experiments each with similar results.
During the peak T cell response (day 7 after infection), dose-dependent expansion of 2W1S tetramer-positive (2W1S-TET+) CD4+ cells was readily detected in mice infected with 105 to 108 CFUs of Lm ΔactA-2W1S (Fig. 2). For mice infected with 106 to 108 CFUs of Lm ΔactA-2W1S, 2W1S-TET+ cells were uniformly CD62Llo indicating recent activation (31, 32). By contrast the majority of 2W1S-TET+ cells in naïve mice or mice infected with lower inocula of either WT Lm-2W1S (103 and 104 CFUs) or Lm ΔactA-2W1S (105 CFUs) were not activated and CD62Lhi (Fig. 2). Thus Lm ΔactA-2W1S primes expansion of 2W1S-specific CD4 T cells in a dose dependent manner and 106 CFUs of Lm ΔactA-2W1S is the minimum inocula required for priming a homogenous population of activated 2W1S-specific CD4 T cells. Accordingly, in subsequent studies, 106 CFUs Lm ΔactA-2W1S was used to further characterize the Lm-2W1S-specific CD4+ T cell response.
Selective priming and expansion of Foxp3- antigen-specific CD4 T cells
During chronic infection, expansion of both pathogen-specific Foxp3- “effector” CD4+ and Foxp3+ “regulatory” CD4+ cells occurs and together both these subsets of pathogen-specific CD4+ cells play important roles in pathogen clearance and immunity (12, 16-19, 21). However, it is uncertain whether priming and expansion of both pathogen-specific Treg and effector CD4+ cells is unique to chronic infection conditions where antigen-persistence occurs or is more broadly applicable to acute infection conditions where antigen persistence does not occur. Accordingly, we used Lm ΔactA-2W1S infection to examine the magnitude and kinetics whereby pathogen-specific CD4 effectors and Tregs are primed after acute infection. For these experiments, we used Foxp3gfp reporter mice where the coding sequences for GFP are inserted in-frame with Foxp3 coding sequences allowing Foxp3-expressing cells to be readily identified by FACS (26). Similar to naïve B6 mice, naive Foxp3gfp mice contained a small but defined population of 2W1S-TET+ CD4+ cells (0.002 to 0.003% of total CD4+ cells and between 200 - 400 cells per mouse) (Fig. 3A). Among this relatively small population of 2W1S-TET+CD4+ cells present in naïve Foxp3gfp mice, between 6-12% were Foxp3+CD25+ Tregs quantified using either magnetic bead enrichment or direct tetramer staining, and this percentage of Tregs among 2W1S-TET+CD4+ cells was similar to the percentage of Foxp3+CD25+ Tregs cells among all CD4+ cells (Fig. 3A). By contrast, during the peak T cell response (day 7) after Lm ΔactA-2W1S infection when the percentage of 2W1S-speific CD4 T cells had expanded ~100-fold, the percentage of Foxp3+CD25+ Tregs among 2W1S-TET+CD4+ cells was reciprocally reduced 100-fold from ~10% to ~0.1% (Fig. 3A). Importantly, this reduction in percent 2W1S-TET+ Tregs corresponding to the expansion of 2W1S-specific effector cells was specific to 2W1S-TET+ cells because the overall percentage of Foxp3+ cells among tetramer negative CD4+ cells did not change significantly after Lm ΔactA-2W1S infection. Furthermore, selective priming and expansion of 2W1S-TET+Foxp3-CD4+ T cells could not be attributed to non-specific features related to Lm ΔactA infection because 2W1S-specific CD4 T cells did not expand, and among these few 2W1S-TET+CD4+ cells ~8-10% remained Foxp3+ after infection with Lm ΔactA expressing an irrelevant antigen (Fig. 3A).
FIGURE 3.

Lm-2W1S does not prime expansion of 2W1S-specific Foxp3+CD4+ cells. A. Representative FACS plots indicating the percent 2W1S-TET+CD4+ cells before or seven days after infection with either 106 CFUs of Lm ΔactA-2W1S (Lm-2W1S) or Lm ΔactA expressing an irrelevant antigen (Lm-CONTROL) (top). Percent Foxp3+ cells among 2W1S-TET+ cells (middle) and 2W1S-TET- (bottom) cells for the indicated mice. B. BrdU incorporation for Foxp3+ (line histogram) and Foxp3- cells (filled histogram) among 2W1S-TET+CD4+ cells (top) or tetramer negative CD4+ cells (bottom) in mice treated with either daily BrdU or no BrdU. These data are representative of three independent experiments each with similar results.
To more precisely characterize the priming of antigen-specific Foxp3- and Foxp3+ CD4 cells, we measured the degree of BrdU incorporation among both Foxp3- and Foxp3+ 2W1S-TET+ or 2W1S TET-negative CD4+ cells after Lm ΔactA-2W1S infection. By day 7, the majority of 2W1S-TET+ Foxp3- cells were BrdU+ indicating extensive cell division consistent with their level of expansion. By contrast, 2W1S-TET+ Foxp3+ cells had levels of BrdU similar to background levels found in 2W1S-TET-negative CD4+ cells or in mice that did not receive BrdU (Fig. 3B). Additionally, neither Foxp3+ nor Foxp3- 2W1S-TET+ cells had levels of BrdU above background in mice infected with Lm ΔactA expressing an irrelevant antigen. Together, these results indicate primary Lm-2W1S infection selectively primes the proliferation of antigen-specific Foxp3- CD4+ T cells within week one after infection.
Since systemic administration of peptide antigen without adjuvant primes antigen-specific Treg expansion (10, 11), we next examined the 2W1S-specific CD4 T cell response primed by purified 2W1S52-68 peptide to determine if the small but defined population of 2W1S-TET+Foxp3+ cells present in naïve Foxp3gfp mice could expand in non-infection conditions that favor Treg differentiation. Similar to Lm-2W1S infection, intravenous 2W1S52-68-peptide inoculation selectively primed dose dependent expansion and BrdU incorporation among antigen-specific CD4 T cells identified using 2W1S-tetramer (Fig. 4A). Importantly and in sharp contrast to Lm-2W1S infection, there were no significant differences in the percentage of Foxp3+ cells found among 2W1S52-68 peptide primed antigen-specific CD4 T cells and the remaining tetramer negative CD4 T cell population (Fig. 4B, D). Moreover, among these 2W1S52-68 peptide primed 2W1S-TET+ cells, Foxp3+ and Foxp3- cells each incorporated BrdU to the same extent and to increased levels compared with tetramer negative cells (Fig. 4C). Together, these results indicate 2W1S52-68 peptide readily primes expansion of both Foxp3+ and Foxp3- 2W1S-specific cells, while Lm-2W1S infection selectively primes expansion of Foxp3- 2W1S-specific CD4 T cells.
FIGURE 4.
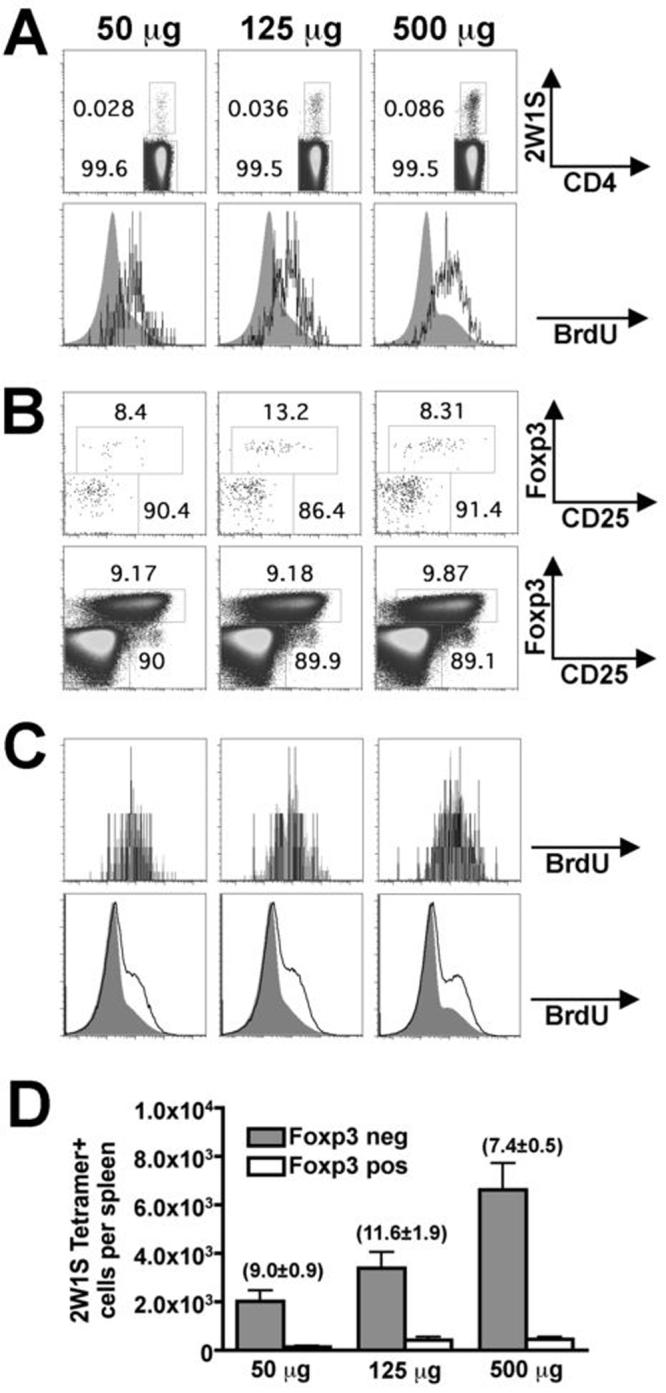
Expansion of 2W1S-specific Foxp3+ and Foxp3-CD4+ cells day seven after 2W1S52-68 peptide inoculation. A. Representative FACS plots indicating percent 2W1S-TET+CD4+ cells (top), and BrdU incorporation among 2W1S-TET+CD4+ cells (line histogram) or tetramer-negative CD4+ T cells (filled histogram) (bottom). B. FACS plots indicating percent Foxp3+ cells among 2W1S-TET+ (top) and tetramer-negative CD4+ cells (bottom). C. BrdU incorporation among Foxp3+ (line histogram) or Foxp3- cells (filled histogram) among 2W1S-TET+CD4+ cells (top) and tetramer-negative CD4+ T cells (bottom). D. Total numbers of 2W1S-TET+CD4+ Foxp3- and Foxp3+ cells among splenocytes at each peptide dose. Numbers indicate the mean (± standard error) percentage of Foxp3+ cells among 2W1S-TET+CD4+ cells. These data are combined from three independent experiments each with similar results containing 6-8 mice per group. Bar, one standared error.
To begin characterizing differences between how purified peptide compared with recombinant Lm infection primes antigen-specific CD4 T cells, we examined the impact co-administration of non-recombinant Lm on the CD4+ T cell response primed by 2W1S52-68 peptide. Both the percentage and absolute numbers of 2W1S-TET+ CD4+ cells were elevated ~10-fold in mice primed with 125 μg of 2W1S52-68 peptide + Lm ΔactA (106 CFUs) compared with mice primed with the same quantity of 2W1S52-68 peptide alone (Fig. 5A). While the percentage of Foxp3+ cells among 2W1S+TET+ cells primed by 2W1S52-68 peptide alone remained consistent with the percentage of Foxp3+ cells among tetramer-negative cells (each ~10%), the percentage of Foxp3+ cells among 2W1S+TET+ cells primed by 2W1S52-68 peptide + Lm was reduced to ~5% (Fig. 5B, C). Notably, despite this modest reduction, the percent Foxp3+ cells among 2W1S-TET+ cells in mice primed with 2W1S52-68 peptide + Lm was dramatically increased compared with mice primed with the same inocula of recombinant LmΔactA-2W1S (5% compared with 0.1%, Fig. 5B and Fig. 3A). Furthermore, while the absolute numbers of 2W1S-TET+Foxp3+ remained constant after recombinant Lm-2W1S infection, significant expansion in absolute numbers of 2W1S-TET+ Foxp3+ cells occurred after co-administration of Lm with 2W1S52-68 peptide (Fig. 5D). Together, these results indicate selective priming and expansion of pathogen-specific Foxp3-negative CD4 T cells observed after recombinant Lm-2W1S infection are not solely due to the inflammatory milieu triggered by acute Lm infection.
FIGURE 5.

Expansion of 2W1S-specific CD4+ cells after inoculation with 2W1S52-68 peptide (125 μg) alone (Peptide) or peptide co-administered with non-recombinant Lm (LmΔactA 106 CFUs) (Peptide + Lm). A. Representative FACS plots indicating percent 2W1S-TET+CD4+ cells among all CD4+ splenocytes for each condition day 7 after inoculation. B. FACS plots indicating percent Foxp3+ cells among 2W1S-TET+ (top) and tetramer-negative CD4+ cells (bottom) after inoculation with either peptide alone or peptide + Lm. C. Percent Foxp3+ cells among either 2W1S-tetramer positive or 2W1S-tetramer negative CD4+ cells for each condition. D. Total numbers of 2W1S-TET+CD4+ Foxp3+ cells per mouse spleen day 7 after inoculation with 2W1S52-68 peptide alone or peptide co-administered with non-recombinant Lm. These data are combined from three independent experiments each with similar results containing 5-6 mice per group. Bar, one standared error.
In related experiments, we extended these studies by characterizing the 2W1S-specific CD4 T cell response at later time points after Lm-2W1S infection. Compared with the day 7 response, the percent and total number 2W1S-TET+ CD4 T cells had contracted ~60% and 90% by days 14 and 30 after infection, respectively. Despite the contraction in absolute numbers of 2W1S-TET+CD4+ cells between days 7 and 30 after infection, the number of 2W1S-specific Foxp3+CD4+ cells remained essentially unchanged (Fig. 6A). Accordingly, the ~90% decline in numbers of 2W1S-TET+ cells between the second and fourth weeks after infection are exclusively among 2W1S-TET+ Foxp3- CD4+ cells. This reduction in absolute numbers of 2W1S-specific Foxp3- CD4+ cells resulted in reciprocal increased percentages of Foxp3+ cells among 2W1S-TET+CD4+ cells from ~0.1% day 7 after infection to ~1% and ~5% by days 14 and day 30, respectively (Fig. 6A). Thus, primary Lm infection selective primes the expansion and subsequent contraction of antigen-specific Foxp3-CD4+ cells, while the numbers of Foxp3+CD4+ cells with the same antigen specificity do not change significantly.
FIGURE 6.

Kinetics of 2W1S-specific CD4 T cell expansion after primary (A) and secondary (B) Lm-2W1S infection. Total numbers of 2W1S-TET+CD4+ cells (top), total numbers of 2W1S-TET+Foxp3+ or 2W1S-TET+Foxp3- cells (middle), and percent Foxp3+ cells among 2W1S-TET+ or all CD4 cells (bottom), at the indicated time points after primary infection (A) with either Lm ΔactA-2W1S (Lm-2W1S) or Lm ΔactA expressing an irrelevant antigen (Lm-CONTROL) or secondary challenge with WT Lm-2W1S (B). The percentage of Foxp3+ cells among all CD4 T cells in the bottom panels reflect data for mice after Lm-2W1S infection, although similar percentages were obtained after Lm-CONTROL infection. These data are combined from four independent experiments each with similar results that together contain 8 - 12 mice per group per time point.
Lastly, we sought to examine the dynamics of antigen-specific CD4 T cell priming after secondary Lm infection by re-challenging with WT Lm-2W1S in mice primed 30 days previously with Lm ΔactA. For both immune-competent, and mice with targeted defects in various cytokines required for T cell activation, a single 106 CFU dose of Lm ΔactA efficiently primes protective immunity to subsequent re-challenge with inocula of virulent Lm normally lethal for naïve mice (27, 28, 30). Protective immunity after secondary challenge is associated with more rapid expansion of both Lm-specific CD8 and CD4 T cells. Interestingly, CD25+CD4+ Tregs under some conditions may limit secondary expansion of protective CD8 T cells during Lm infection, however the antigen-specificity of these Tregs were not characterized (33). Within the first three days after secondary challenge with WT Lm-2W1S, the numbers of 2W1S-TET+CD4+ cells increased ~10-fold in mice primed initially with Lm ΔactA-2W1S and increased an additional ~2-fold by day 5 reflecting the accelerated kinetics of antigen-specific T cell expansion after secondary infection (Fig. 6B). By contrast, for mice that were initially primed with Lm ΔactA expressing an irrelevant antigen, the numbers of 2W1S-TET+ cells did not increase significantly within the first three days but increased dramatically between days 3 and 5 after WT Lm-2W1S challenge reflecting primary expansion kinetics for 2W1S-specific CD4 T cells (Fig. 6B). In these mice, the susceptibility to this normally lethal inocula of WT Lm-2W1S for naïve mice is bypassed by prior infection with Lm ΔactA allowing characterization for how WT Lm-2W1S primes antigen-specific Tregs and effector CD4+ cells. Consistent with the selective priming and expansion of Lm-specific Foxp3- CD4+ cells observed after primary Lm ΔactA infection, both secondary expansion of 2W1S-specific CD4 T cells after WT Lm 2W1S challenge in Lm ΔactA-2W1S primed mice and primary expansion of 2W1S-specific CD4 T cells after WT Lm 2W1S challenge in mice primed initially with Lm ΔactA expressing an irrelevant antigen occurred selectively among Foxp3- CD4+ cells, while the absolute numbers of Foxp3+2W1S-TET+ cells remained unchanged from numbers present in naïve mice (Fig. 5B). Taken together, these data demonstrate that primary and secondary Lm infection selectively primes antigen-specific Foxp3-CD4+, and not Foxp3+CD4+ cells.
DISCUSSION
In this report, we used MHC class II tetramer staining to track the antigen-specific CD4 T cell response after Lm infection. For these studies, we engineered recombinant Lm to express the non-“self” 2W1S antigen and measured the expansion of Lm-2W1S-specific Foxp3-CD4+ and Foxp3+CD4+ T cells after infection using 2W1S-specific tetramer (22). Using this approach, we demonstrate that recombinant Lm-2W1S infection selectively primes expansion and subsequent contraction of 2W1S-specific Foxp3-CD4+ cells in a dose-dependent fashion, while the numbers of 2W1S-specific Foxp3+CD4+ cells remain essentially unchanged throughout the course of infection. Importantly in naïve mice prior to Lm infection, a small but defined population of 2W1S-specific CD4+ cells was consistently identified; and similar percentages of Foxp3+CD25+ cells (~ 6 to 12%) were found among this 2W1S-specific CD4+ cell population compared with all CD4+ T cells. Thus, in naïve B6 mice, specificity for the non-“self” 2W1S52-68 peptide antigen is present in the repertoire of both Foxp3+ and Foxp3- CD4+ T cells. These findings suggest that overlap is present for Tregs and effector CD4 T cells at least for some TCR specificities, and these cells have the potential to respond to the same antigen. Accordingly and in sharp contrast to Lm-2W1S infection, the percentage of Foxp3+ cells among 2W1S tetramer positive cells remained at ~10% after systemic administration of purified 2W1S52-68 peptide. These results, together with the ability of purified peptide and not Lm-2W1S infection to prime BrdU incorporation among 2W1S tetramer+ Foxp3+ cells, suggest that Lm infection selectively inhibits the clonal expansion of antigen-specific Tregs. However, using our currently available tools, we cannot distinguish whether expansion of 2W1S-specific Foxp3+CD4+ cells after peptide administration results from the clonal expansion of pre-existing 2W1S-specific Foxp3+ cells or de-novo differentiation of antigen-specific cells into Foxp3+CD4+ Tregs. Both possibilities are consistent with other reports demonstrating that purified antigen administered without adjuvant primes antigen-specific Treg expansion (10, 11). Nevertheless, these results collectively demonstrate that although expansion of both 2W1S-specific Foxp3- and 2W1S-specific Foxp3+ CD4+ T cells can occur, features related to Lm-2W1S infection primes the selective expansion of only antigen-specific Foxp3-CD4+ T cells.
Therefore, features related to Lm infection distinguish it from non-infection and other infection conditions where both antigen-specific Tregs and effector CD4 T cells are primed (9-11, 16-19). For example, after infection with other pathogens such as Schistosoma or Leishmania that normally cause chronic infection, parallel expansion of both pathogen-specific Foxp3+ and Foxp3-CD4 T cells occurs. Moreover, these pathogen-specific Tregs indirectly confer protective immunity to secondary infection by maintaining local antigen persistence required for sustaining pathogen-specific effector CD4 T cells (17, 20, 21). Interestingly, despite the lack of antigen persistence and detectable priming of pathogen-specific Tregs, Lm-ΔactA readily primes protective immunity to secondary challenge with virulent Lm that is mediated by pathogen-specific CD8 T cells (27, 28, 30). We are currently exploring whether these differential requirements for pathogen-specific Tregs in protective immunity are specific to these infections, relate to unique features of pathogen-specific CD4 versus CD8 T cell immunity, or reflect more general features related to the immune response primed by pathogens that primarily cause acute versus chronic infection. The inability of non-recombinant Lm co-administered with 2W1S52-68 peptide to reproduce the selective expansion of antigen-specific Foxp3- cells observed after recombinant Lm-2W1S infection indicates that these differences are not caused solely by acute inflammatory cytokines produced in response to Lm infection. Therefore, studies that dissect key features related to acute Lm infection that allows selective priming and expansion of antigen-specific effector CD4 cells are important areas for future investigation.
ACKNOWLEDGEMENTS
We thank Drs. Marc Jenkins, Stephen McSorley, and Kevin Urdahl for helpful discussions, and Dr. Alexandar Rudensky for generously providing Foxp3gfp mice.
The authors gratefully acknowledge funding support from the following sources: NICHD/NIH-K08HD51584, March of Dimes Basil O'Conner research award, Minnesota Vikings Children's Fund, and Minnesota Medical Foundation.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org
REFERENCES
- 1.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 2.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 3.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 4.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Rankin AL, Picca CC, Riley MP, Jenks SA, Sant AJ, Caton AJ. CD4+CD25+ regulatory T cell repertoire formation shaped by differential presentation of peptides from a self-antigen. J Immunol. 2008;180:2149–2157. doi: 10.4049/jimmunol.180.4.2149. [DOI] [PubMed] [Google Scholar]
- 7.Kasow KA, Chen X, Knowles J, Wichlan D, Handgretinger R, Riberdy JM. Human CD4+CD25+ regulatory T cells share equally complex and comparable repertoires with CD4+CD25- counterparts. J Immunol. 2004;172:6123–6128. doi: 10.4049/jimmunol.172.10.6123. [DOI] [PubMed] [Google Scholar]
- 8.Fazilleau N, Bachelez H, Gougeon ML, Viguier M. Cutting edge: size and diversity of CD4+CD25high Foxp3+ regulatory T cell repertoire in humans: evidence for similarities and partial overlapping with CD4+CD25- T cells. J Immunol. 2007;179:3412–3416. doi: 10.4049/jimmunol.179.6.3412. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi Y, Tsukumo S, Shiota H, Kishihara K, Yasutomo K. Antigen-specific T cell repertoire modification of CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5240–5248. doi: 10.4049/jimmunol.172.9.5240. [DOI] [PubMed] [Google Scholar]
- 10.Thorstenson KM, Khoruts A. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J Immunol. 2001;167:188–195. doi: 10.4049/jimmunol.167.1.188. [DOI] [PubMed] [Google Scholar]
- 11.Verginis P, McLaughlin KA, Wucherpfennig KW, von Boehmer H, Apostolou I. Induction of antigen-specific regulatory T cells in wild-type mice: visualization and targets of suppression. Proc Natl Acad Sci U S A. 2008;105:3479–3484. doi: 10.1073/pnas.0800149105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 13.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. 2007;204:2159–2169. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kursar M, Koch M, Mittrucker HW, Nouailles G, Bonhagen K, Kamradt T, Kaufmann SH. Cutting Edge: Regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol. 2007;178:2661–2665. doi: 10.4049/jimmunol.178.5.2661. [DOI] [PubMed] [Google Scholar]
- 15.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172:4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 16.Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol. 2006;176:5839–5847. doi: 10.4049/jimmunol.176.10.5839. [DOI] [PubMed] [Google Scholar]
- 17.Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med. 2006;203:777–788. doi: 10.1084/jem.20052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hesse M, Piccirillo CA, Belkaid Y, Prufer J, Mentink-Kane M, Leusink M, Cheever AW, Shevach EM, Wynn TA. The pathogenesis of schistosomiasis is controlled by cooperating IL-10-producing innate effector and regulatory T cells. J Immunol. 2004;172:3157–3166. doi: 10.4049/jimmunol.172.5.3157. [DOI] [PubMed] [Google Scholar]
- 19.McKee AS, Pearce EJ. CD25+CD4+ cells contribute to Th2 polarization during helminth infection by suppressing Th1 response development. J Immunol. 2004;173:1224–1231. doi: 10.4049/jimmunol.173.2.1224. [DOI] [PubMed] [Google Scholar]
- 20.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 21.Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med. 2004;200:201–210. doi: 10.1084/jem.20040298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orr MT, Orgun NN, Wilson CB, Way SS. Cutting Edge: Recombinant Listeria monocytogenes expressing a single immune-dominant peptide confers protective immunity to herpes simplex virus-1 infection. J Immunol. 2007;178:4731–4735. doi: 10.4049/jimmunol.178.8.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 26.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol. 2003;171:533–537. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 28.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 29.Kollmann TR, Reikie B, Blimkie D, Way SS, Hajjar AM, Arispe K, Shaulov A, Wilson CB. Induction of protective immunity to Listeria monocytogenes in neonates. J Immunol. 2007;178:3695–3701. doi: 10.4049/jimmunol.178.6.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orgun NN, Mathis MA, Wilson CB, Way SS. Deviation from a strong Th1-dominated to a modest Th17-dominated CD4 T cell response in the absence of IL-12p40 and type I IFNs sustains protective CD8 T cells. J Immunol. 2008;180:4109–4115. doi: 10.4049/jimmunol.180.6.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung TM, Gallatin WM, Weissman IL, Dailey MO. Down-regulation of homing receptors after T cell activation. J Immunol. 1988;141:4110–4117. [PubMed] [Google Scholar]
- 32.Bradley LM, Atkins GG, Swain SL. Long-term CD4+ memory T cells from the spleen lack MEL-14, the lymph node homing receptor. J Immunol. 1992;148:324–331. [PubMed] [Google Scholar]
- 33.Kursar M, Bonhagen K, Fensterle J, Kohler A, Hurwitz R, Kamradt T, Kaufmann SH, Mittrucker HW. Regulatory CD4+CD25+ T cells restrict memory CD8+ T cell responses. J Exp Med. 2002;196:1585–1592. doi: 10.1084/jem.20011347. [DOI] [PMC free article] [PubMed] [Google Scholar]