

Abstract
Calcium-calmodulin-dependent protein kinase II (CaMKII) is a key mediator of synaptic plasticity and learning. Global pyramidal cell glutamate stimulation induces translocation of CaMKII from dendritic shafts to spines. Here we show that local dendritic stimulation by puffing glutamate onto a region containing 7-32 synapses induces translocation of CaMKII to synapses initially at the puff site, but that translocation subsequently spreads within dendrites to the distal dendrite arbor, resulting in a persistent, widespread synaptic accumulation. This locally-induced propagating synaptic (L-IPS) accumulation of CaMKII requires activation of NMDA receptors and L-type Ca++ channels and is preceded by a Ca++ spike. L-IPS translocation of CaMKII alters biochemical signaling and is associated with an increase in AMPA receptor GluR1 at both stimulated and non-stimulated synapses and thus provides a molecular mechanism for heterosynaptic plasticity.
INTRODUCTION
Its unique holoenzyme structure, autoregulatory properties, binding partners and substrates, and regulated mobility render CaMKII a key player in numerous forms of synaptic plasticity (Hudmon and Schulman, 2002; Soderling et al., 2000). The complex properties of the CaMKII holoenzyme result in different patterns of activation depending on the subunit composition, the nature of the Ca++ signal, and regulatory phosphatases. Activation of CaMKII by Ca++ entry through synaptic NMDA receptors is essential for long term potentiation (LTP), and a point mutation that blocks autophosphorylation of CaMKIIα at T286 inhibits LTP in pyramidal neurons and inhibits memory formation (Lisman et al., 2002). GABAergic interneurons lack CaMKIIα (Sik et al., 1998) but some can still exhibit CaMKII-dependent plasticity (Lamsa et al., 2007), likely dependent on CaMKIIβ.
CaMKII activity is also determined by its subcellular localization. Synaptically targeted CaMKII is maximally activated by Ca++ entry through NMDA receptors and voltage-gated calcium channels (VGCCs) and is in prime position to phosphorylate synaptic targets including AMPA and NMDA receptors (Soderling et al., 2000). Global cell stimulation of NMDA receptors induces translocation of CaMKII from dendrite shafts to spines, increasing its synaptic levels (Shen and Meyer, 1999). Synaptic translocation requires Ca++/CaM binding to CaMKII and is prolonged by autophosphorylation at T286. Postsynaptic accumulation of CaMKII is mediated by its binding to NR2B (Bayer et al., 2006), CaMKII self-association (Hudmon et al., 2005a), and perhaps binding to other synaptic partners including NR1, NR2A, densin-180, and α-actinin (Colbran, 2004). Depending on the stimulus and extracellular Ca++ level, synaptic CaMKII accumulation can either rapidly reverse or stably persist (Bayer et al., 2006; Shen et al., 2000). Following selective activation of synaptic NMDA receptors in a chemical LTP paradigm, the mobility of CaMKII is persistently reduced maintaining high levels at the synapse (Sharma et al., 2006).
Here, we tested whether local dendritic stimulation might induce locally restricted accumulation of CaMKII at synapses. We report instead an unexpected propagating accumulation of CaMKII at synapses, starting locally and spreading to the entire dendrite and usually entire neuron. This global synaptic accumulation of CaMKII by activation of a small contiguous set of synapses represents a novel form of heterosynaptic plasticity.
RESULTS
Rat hippocampal neurons in culture were transfected to express GFP-CaMKII α or β (Shen and Meyer, 1999) and imaged at 12-24 DIV. As in previous experiments (Sharma et al., 2006), expression level of the transfected CaMKII was well below that of the endogenous CaMKII (Figure S1). During time-lapse imaging, pyramidal neurons were stimulated locally with a single 15 msec puff of 100 μM glutamate plus 10 μM glycine delivered from a pipette placed near the dendrite (Figure 1A Phase); immediately after the stimulus, the pipette was withdrawn from the continuously perfusing bath. We initially observed a local clustering of CaMKII along the stimulated portion of the dendrite. Unexpectedly, CaMKII clustering did not remain confined to the stimulation site but propagated in a wave-like phenomenon along the dendrite (Figure 1, Figures S2-3, Movies 1-4). At each dendritic site, CaMKII in the shaft appeared to translocate to nearby spines. CaMKII translocation to spines typically proceeded first distally from the puff site, reaching the end of the stimulated dendrite by 2-3 min after local stimulation. Finally, by 5-10 min, the propagating wave crossed the soma and CaMKII translocation spread to the entire dendritic arbor. The increase in punctate spine-associated CaMKII persisted for the entire imaging period, typically 15 min and up to 40 min. GFP-CaMKII α and β behaved indistinguishably; at the low expression level used here, tagged subunits are expected to multimerize with endogenous subunits in mixed holoenzymes. Experiments in which the distal dendritic arbor was oriented in all possible directions from the puff site relative to the bath flow and relative to the pipette orientation show CaMKII translocation always propagated first distally in the dendrite (Figure S4).
Figure 1. Locally-Induced Propagating Synaptic (L-IPS) Translocation of CaMKII to the Entire Neuron Dendritic Arbor.
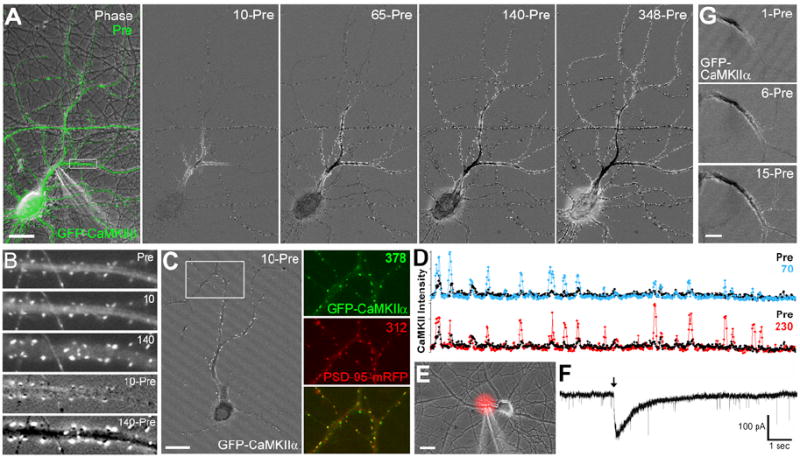
(A) A pyramidal neuron expressing CaMKIIβ was stimulated locally with glutamate (100 μM) plus glycine (10 μM) followed by live-cell imaging (numbers indicate sec following stimulation). In the subtraction images, white indicates an increase in CaMKII concentration and black a decrease (10-Pre; the Pre image was subtracted from the 10 sec image and a constant grey value added). (B) Enlargement of boxed region in panel A. (C) A pyramidal neuron expressing GFP-CaMKIIα and PSD-95-mRFP was locally stimulated at the base of the apical dendrite. Colocalization with PSD-95 in the boxed region demonstrates that CaMKIIα translocated to synaptic sites. (D) Linescans of peak GFP-CaMKIIα intensity from a distal portion of the stimulated dendrite (~110-160 μm from the soma) in panel C comparing pre-stimulation with 70 or 230 sec after stimulation. (E) Another neuron in which GFP-CaMKII translocated to synapses throughout the dendrite arbor is shown with Alexa 594 (red) co-released with the local glutamate plus glycine to indicate the puff zone. (F) Representative trace from whole-cell voltage clamp recording during local glutamate plus glycine stimulation; arrow indicates time of puff. The recording was performed in ECS + TTX + SR95531 (without glycine). Thus the locally-induced current is superimposed on a baseline of AMPA mEPSCs. (G) More rapid timecourse imaging of GFP-CaMKIIα following local stimulation with glutamate plus glycine shows the initial gradual spread of CaMKII redistribution. Scale bars A,C,E 20 μm; G 10 μm.
The size of the stimulated region was determined by co-imaging Alexa 594 included in the stimulating pipette (Figure 1E). For neurons in which L-IPS CaMKII accumulation spread to the entire neuron (n = 8), the region accessed by half-maximal Alexa 594 was an ellipse of 28.6 ± 2.7 μm by 24.0 ± 3.1 μm diameter containing 21.3 ± 2.7 μm primary dendrite and 33.0 ± 5.8 μm total dendrite length. The number of synapses within this region, determined by counting CaMKII clusters following stimulation, was 23.3 ± 3.6 (range 12-32), consistent with the previously determined mean excitatory synapse density of 0.6/μm (Harms and Craig, 2005). Alexa 594 released from the pipette remained locally elevated for 546 ± 100 msec (range 120-900). Difference images of 1 sec post-stimulation minus pre-stimulation also show increased CaMKII at 16.5 ± 3.4 (n = 4; range 10-26) clusters with punctate and/or diffuse levels altered along 33.7 ± 4.1 μm of dendrite length, presumably corresponding to the initial unit of direct stimulation.
The local stimulus was further characterized by whole cell patch voltage clamp recordings (Figure 1F). A typical local puff of glutamate plus glycine induced an inward current with a timecourse characteristic of NMDA responses (mean peak amplitude 96.0 ± 20.2 pA, 10-90% rise time 33 ± 7 msec, width at 50% peak 293 ± 78 msec, n = 11). Based on a comparison with NMDA mEPSCs recorded in similar cultures (Gomperts et al., 1998), activation of approximately 20-30 synapses is expected to yield a similar NMDA response. NMDA-mediated EPSCs similar to these locally induced currents have also been recorded in slice CA1 neurons in response to short bursts of high frequency stimulation that imitate natural stimulus patterns (Lozovaya et al., 2004). Thus our local stimulation of glutamate receptors may mimic naturally occurring simultaneous bursts by inputs localized nearby on the dendritic arbor.
To determine whether the propagating clustering of CaMKII occurred at synapses, neurons were cotransfected to express GFP-CaMKII and PSD95-mRFP, a marker for glutamatergic postsynaptic sites (Okabe et al., 1999). As indicated by colocalization with PSD-95, CaMKII translocated primarily to synaptic sites following local dendritic stimulation (Figure 1C). The distribution of PSD-95 did not change following stimulation; only small scale movement as previously reported under baseline conditions was observed. Occasional clusters of CaMKII not colocalizing with PSD-95 were also observed following local stimulation, particularly in the soma, perhaps indicating self-association (Hudmon et al., 2005a).
Interneurons expressing GFP-CaMKIIα or GFP-CaMKIIβ also exhibited synaptic accumulation of CaMKII following local dendritic stimulation. Since interneurons lack CaMKIIα ((Sik et al., 1998), and confirmed in our cultures), the propagating synaptic translocation of GFP-CaMKIIβ in interneurons indicates a mechanism that does not require any features specific to the α isoform. In many interneurons, CaMKII synaptic translocation spread to the entire cell. However, in some interneurons, GFP-CaMKII synaptic translocation spread only to the dendrite arbor distal to the puff site but not proximally to the soma or to other primary dendrites (Figure 2A). Clustering of CaMKII occurred in the dendrite shafts of the aspiny interneurons, and the induced clusters colocalized with PSD95-mRFP (Figure S2D), indicating synaptic translocation. Distally restricted propagating translocation also occurred more rarely in pyramidal neurons. In one such pyramidal neuron (Figure 2B), the region accessed by half-maximal co-released Alexa 594 was 11.3 μm diameter and contained 7 synapses, a smaller region than that resulting in whole cell translocation. Distally restricted propagation in interneurons was also sensitive to the pipette size, occurring less frequently with a larger pipette. These observations suggest there are different thresholds for stimulation, with a lower threshold for distally restricted propagation and higher for whole cell propagation.
Figure 2. NMDAR-Mediated CaMKII Translocation Restricted to the Stimulated Distal Dendrite.

(A) In an interneuron expressing GFP-CaMKIIα, CaMKII clustering spread from the puff site (arrow) distally along the stimulated dendrite, but not proximal to the puff site or to other dendrites of the same cell, even those crossing the path of the stimulated dendrite. (B) In a pyramidal neuron, GFP-CaMKIIα also accumulated (white) at spines only distally along the branched stimulated dendrite (arrow) and not on other dendrites (the Pre image was subtracted from the 910 sec image and a constant grey value added). (C) The rate of spread of L-IPS translocation of CaMKII distally is plotted for 6 pyramidal cells (black diamonds) and 5 interneurons (grey squares). Distance indicates the total distance from the point of stimulation to the most distal dendritic site at which an increase in clustered CaMKII was observed. Time zero is the first image after stimulation. A linear fit of all points yields an estimated rate of spread of 0.76 μm/sec. (D) L-IPS CaMKII translocation was inhibited by MK801 (5 µM) but not by TTX (0.5 µM) or NBQX (10 µM). Given long access to glutamate plus glycine it is likely that flicker of the bound NMDA channel could lead to sufficient cumulative depolarization to remove the Mg++ block. Local stimulation with NMDA (200 µM) plus glycine (10 μM) was sufficient to induce L-IPS translocation. Stimulation of local synaptic NMDA receptors with glycine alone (10 μM) in 0 Mg2+ ECS with strychnine (5 μM) was sufficient to induce L-IPS CaMKII translocation, but not in the presence of TTX (0.5 μM). Post-stimulation GFP-CaMKII peak intensities were divided by pre-stimulation intensities for the same regions (with background subtraction) and multiplied by 100. Grey line marks no change in CaMKII distribution. One-way ANOVA revealed a significant difference between conditions (F(6, 51) = 6.31, p<0.0001). Dunnett’s post-hoc analyses show differences from glutamate plus glycine in ECS for MK801 and for glycine-alone in 0 Mg2+ ECS with TTX (** = p<0.01; otherwise p>0.05). Scale bars A, 20 μm; B, 10 μm.
The rate of distal spread of CaMKII synaptic translocation following local stimulation was determined by measuring the linear distance from the site of stimulation to the farthest point at which increased synaptic CaMKII was observed (Figure 2C). Lines of best fit from rates of individual cells yield a mean rate of spread of 0.93 ± 0.17 μm/sec. Pyramidal neurons showed a more consistent rate of spread (range 0.67 - 1.37 μm/sec) compared with interneurons (range 0.22 -2.18 μm/sec), perhaps reflecting the greater diversity of interneurons.
We tested whether NMDA receptor activation is necessary and sufficient for L-IPS translocation of CaMKII (Figure 2D, Figure S5), as it is for globally induced translocation (Shen and Meyer, 1999). The NMDA receptor open channel blocker MK-801 blocked the effects of local glutamate plus glycine on CaMKII distribution. Conversely, locally applied NMDA plus glycine was sufficient to induce L-IPS CaMKII accumulation. In contrast, the AMPA receptor antagonist NBQX did not block L-IPS CaMKII redistribution. Pre- and co-incubation with tetrodotoxin (TTX) voltage-gated sodium channel antagonist did not inhibit L-IPS accumulation of CaMKII, indicating independence from action potential dependent network activity and back-propagating action potentials.
Bath application of glycine in 0 Mg++ ECS is a means to selectively activate synaptic NMDA receptors and not extrasynaptic receptors, depending on the normal synaptic release of glutamate (Lu et al., 2001). We perfused neurons in 0 Mg++ ECS (plus strychnine to inhibit glycine receptors) and locally applied glycine alone; this glycine-alone stimulus was sufficient to induce L-IPS accumulation of CaMKII (Figure 2D, Figure S5). Thus activation of only synaptic NMDA receptors at a neighboring set of synapses is a sufficient trigger. This same stimulus, locally applied glycine in ECS with 0 Mg++, in the additional presence of TTX was not sufficient. TTX would block the synchronous release of glutamate from neighboring terminals that is dependent on network activity, leaving only random single mEPSC events (Figure S5G,H). Thus activation of single synapses is not sufficient to mediate L-IPS accumulation of CaMKII.
Fortuitous crossing of different primary dendrites of a single interneuron revealed CaMKII clustering spread distally in the stimulated dendrite but not to the crossing dendrite (Figure 2A). This observation indicates propagation via an intracellular mechanism. In cells where CaMKII translocated to all postsynaptic sites, the pattern of spread with a temporal lag of a few minutes to cross the soma and no effect on CaMKII in axons, is also consistent with an intracellular somatodendritic mechanism.
An obvious intracellular signal arising from NMDA receptor activation is Ca++ influx. Pre-loading with the membrane permeable Ca++ chelator BAPTA-AM effectively blocked L-IPS CaMKII translocation (Figure 3A, Figure S6). Calcium-induced Ca++ release from intracellular stores was not essential for L-IPS CaMKII translocation, but activation of CaMKII itself and the resultant kinase activity was necessary. To more directly assess the spatial and temporal parameters of the Ca++ elevation, we performed Ca++ imaging with fluo-4FF (Figure 3B). Local stimulation resulted in an increase in Ca++ spreading to the entire dendritic arbor of the stimulated cell over a mean 168 ± 22 msec (n = 10). The total time Ca++ was elevated anywhere in the cell was mean 807 ± 104 msec before returning to baseline, after which no further elevations were observed. The Ca++ rise was restricted to the somatodendritic domain of stimulated cells, axons did not show Ca++ elevation. Thus, local stimulation results in a Ca++ spike that spreads through the somatodendritic domain.
Figure 3. L-IPS Translocation Follows a Ca++ Spike and Requires Ca++ Influx, L-type Ca2+ Channel Activity, and CaMKII Activation.

(A) L-IPS translocation of CaMKII by glutamate plus glycine was inhibited by BAPTA-AM (10 μM) or the CaMKII inhibitor KN93 (0.5 μM) but not by inhibitors of Ca++ release from intracellular stores CICR (combinations of 2-amino-ethoxydiphenylborate (2-APB; 100 μM), ryanodine (10 μM), thapsigargin (1 μM), and cyclopiazonic acid (CPA; 20 μM)). One-way ANOVA revealed a significant difference between groups (F(3, 28) = 4.34, p<0.05). Dunnett’s post-hoc analyses show differences from ECS control for BAPTA-AM and KN93 (* = p<0.05). (B) Calcium imaging with fluo-4FF AM in ECS + TTX indicated that a typical local stimulation results in a rise in Ca++ spreading to the entire somatodendritic domain. In the subtraction image of 165 msec after stimulation minus pre-stimulation, white indicates Ca++ elevation. Note that the non-stimulated cell at right shows no Ca++ elevation. (C) Nifedipine (10 μM) did not block the effect of a 1-min bath application of glutamate (100 μM) and glycine (10 μM) to increase CaMKII clustering (t(13) = 1.12, p>0.1). (D) Nifedipine blocked L-IPS CaMKII translocation (t(19) = 3.11, **p<0.01). Scale bar B 20 μm; C,D 10 μm.
It has previously been reported that blockade of VGCCs does not affect synaptic CaMKII translocation following bath application of glutamate and glycine (Thalhammer et al., 2006). We confirmed that the L-type Ca++ channel blocker nifedipine does not block globally induced CaMKII translocation upon bath stimulation with glutamate and glycine, both by live cell imaging of GFP-CaMKII expressing neurons (Figure 3C) and by population analysis of endogenous CaMKIIα by immunocytochemistry (Figure S7). If locally-induced propagating CaMKII translocation occurs by the same cellular mechanism as CaMKII translocation following bath stimulation, we would expect nifedipine to have no effect. However, nifedipine blocked propagating L-IPS CaMKII translocation (Figure 3D). Thus L-IPS CaMKII translocation requires VGCC activation and is a distinct cellular process compared to bath-induced CaMKII translocation.
To determine if locally-induced synaptically translocated CaMKII is biochemically active, we used a recombinant synaptic reporter of active CaMKII (Tsui and Malenka, 2006). PSD95-Vim-CFP consists of PSD-95 to mediate postsynaptic localization fused to the head region of vimentin containing a dedicated CaMKII phosphorylation site (Inagaki et al., 2000) fused to CFP (Tsui and Malenka, 2006). We co-expressed PSD95-Vim-CFP and YFP-CaMKII in neurons, stimulated one dendrite with local glutamate plus glycine, and fixed within 2 min. At this time, CaMKII translocated to synaptic sites distally in the stimulated dendrite but not across the soma in non-stimulated dendrites. The intensity ratio of pVim phosphorylated substrate to total PSD95-Vim-CFP substrate fluorescence, reflecting CaMKII activity, was significantly higher in stimulated dendrites compared to corresponding control dendrites (Figure 4A,B, Figure S8A). Extrapolating to the situation where L-IPS CaMKII translocation proceeds to the entire dendrite arbor is expected to result in increased CaMKII activity at all synapses.
Figure 4. CaMKII Activity and AMPA Receptor GluR1 Increase at Synaptic Sites Following L-IPS Translocation.
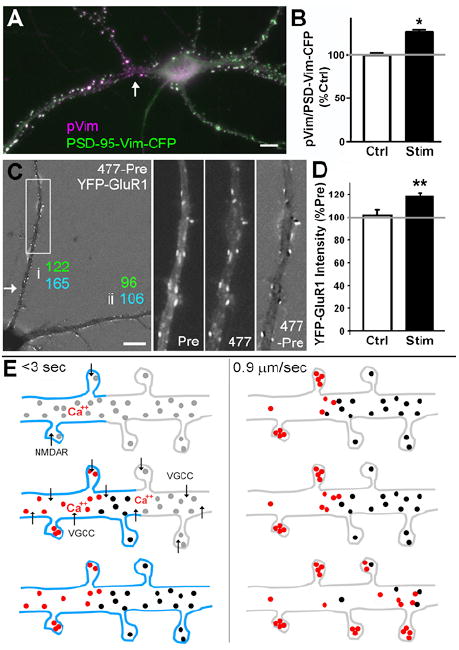
(A) Neurons expressing YFP-CaMKII and the synaptically localized specific CaMKII substrate PSD-Vim-CFP were locally stimulated with glutamate plus glycine (arrow) and fixed 90 sec later. CaMKII-mediated phosphorylation indicated by phospho-vimentin (pVim) immunoreactivity preferentially appears in the stimulated dendrite. (B) Quantification of CaMKII-mediated phosphorylation as intensity of pVim divided by intensity of PSD-Vim-CFP substrate. Stimulated dendrites were compared with non-stimulated dendrites of the same cells (t(4) = 3.41, *p<0.05). (C) A neuron co-expressing CFP-CaMKII and AMPA receptor subunit YFP-GluR1 was locally stimulated with glutamate plus glycine (arrow). CaMKII accumulated at clusters (165% increase) along the stimulated dendrite (i) but not the non-stimulated dendrite (ii, 106%). GluR1 co-accumulated along the stimulated dendrite (122%) but not the non-stimulated dendrite (96%). (D) In another series of locally stimulated cells, L-IPS accumulation of CFP-CaMKII spread to all synapses. YFP-GluR1 co-accumulated compared to control cells that were similarly co-transfected and imaged without local stimulation to account for any bleaching during imaging (t(9) = 3.25, **p<0.01). (E) Model for mechanism of L-IPS accumulation of CaMKII involving a rapid priming step (<3 sec) and a slower translocation phase spreading at 0.9 μm/sec. Blue indicates membrane depolarization, grey circles basal CaMKII, black circles primed CaMKII, and red circles activated CaMKII. Scale bars 10 μm.
Finally, we assessed whether L-IPS accumulation of CaMKII is associated with other synaptic alterations. We focused on AMPA receptors, in particular GluR1, given its key role in synaptic transmission and prior evidence that active CaMKII stimulates synaptic insertion of GluR1 (Hayashi et al., 2000), a widely accepted mechanism underlying LTP. CFP-CaMKII and YFP-GluR1 were co-expressed and imaged both before and after local stimulation with glutamate plus glycine (Figure 4C,D, Figure S8B). GluR1 appeared to become slightly more clustered following local stimulation although the effect was not as obvious as for CaMKII. A series of puncta located distally on the stimulated dendrite were chosen for quantitation based on the post-stimulation CaMKII image for each cell. The intensity of CFP-CaMKII at these distal puncta increased by mean 164 + 11% and YFP-GluR1 by 118.3 + 2.9%. This 118.3% increase in synaptic accumulation of GluR1 is significant compared with the mean change of 101.9 + 4.4% for GluR1 measured from a series of co-transfected cells imaged over the same timecourse without local stimulation (p<0.01). Furthermore, in one neuron in which local stimulation resulted in distally restricted propagation of CFP-CaMKII clustering without spread to a non-stimulated dendrite, the intensity of YFP-GluR1 clusters also selectively increased distally in the stimulated dendrite and not in the non-stimulated dendrite. Thus, local stimulation results in propagating synaptic accumulation of CaMKII and an associated synaptic accumulation of GluR1.
DISCUSSION
We report here propagating translocation of CaMKII upon local stimulation of NMDA receptors at 7-32 neighboring synapses. CaMKII moves from shaft to spine first locally and then in a spreading wave distally along the dendrite and often to the entire somatodendritic domain. Propagating CaMKII translocation follows a somatodendritic Ca++ spike and requires L-type Ca++ channel activation. L-IPS CaMKII accumulation is associated with a corresponding increase in synaptic CaMKII activity and accumulation of GluR1. These results identify a novel form of heterosynaptic molecular dynamics.
The first step in L-IPS translocation is activation of total or just synaptic NMDA receptors over a stretch of dendrite bearing 7-32 synapses. Previous experiments applying glutamate or NMDA to short regions of dendrites, or by current injection or synaptic stimulation, report induction of dendritic Na+, Ca++, or NMDA spikes (Schiller and Schiller, 2001; Schwindt and Crill, 1997; Spruston, 2008). In our experiments, the lack of effect of TTX rules out a requirement for Na+ channels, and absence of glycine from the bath restricts a role for NMDA receptors to the stimulated region. However, the selective requirement for L-type Ca++ channels for L-IPS CaMKII translocation supports a requirement for Ca++ spikes. Calcium spikes can propagate to the entire somatodendritic domain or can propagate distally but not proximally (Cai et al., 2004; Spruston, 2008), matching our two patterns of L-IPS accumulation of CaMKII. There is a final slow step in L-IPS CaMKII accumulation, spreading distally along the stimulated dendrite at a mean rate of 0.93 ± 0.17 μm/sec. This rate is not consistent with spread via diffusion of Ca++ but consistent with diffusion of a larger molecule such as CaMKII itself, or with spread via a molecular motor. However, it seems necessary that the slowly spreading intracellular signal be regenerative in nature rather than simply diffusing; it is difficult to envision how a single locally generated signal could spread at sufficient concentration to activate translocation in an entire dendrite. Finally, the evidence indicates that CaMKII activity is essential.
The precise mechanism of this slowly propagating accumulation remains to be determined but is likely to involve the complex regulatory properties of CaMKII, including a regenerative step such as autophosphorylation. CaMKII autophosphorylation at T286 occurs when adjacent subunits in the holoenzyme are bound to Ca++/CaM and can prolong synaptic association. T253 autophosphorylation also promotes postsynaptic association and is stimulated by activity. Whereas T286 autophosphorylation can occur within 10 sec, T253 autophosphorylation occurs slowly over 1-4 min (Migues et al., 2006), consistent with the timecourse of L-IPS translocation. Direct binding of CaMKII to L-type VGCC Cav1.2 α and β subunits (Grueter et al., 2006; Hudmon et al., 2005b) may participate by enhancing CaMKII activation. The greatest level of CaMKII activation would occur within the directly stimulated region of dendrite, promoting synaptic accumulation. Elsewhere in the dendrite arbor, CaMKII is not sufficiently activated to mediate synaptic accumulation, but may be partially autophosphorylated by the Ca++ spike and left in a primed state. Shen and Meyer (2000) showed the existence of a similar primed state of CaMKIIα lasting for >3 min; after strong stimulation, primed CaMKII more readily accumulates at synapses upon further weak stimulation. Following our local stimulus, some of the activated CaMKII would diffuse down the dendrite, encountering primed CaMKII. The abundance of CaMKII in hippocampal neurons, ~2% of total protein (Hudmon and Schulman, 2002), and the interaction of CaMKII holoenzymes promoted by Ca++/CaM (Hudmon et al., 2005a) will serve to bring together activated and primed forms. We suggest that CaMKII activity is regenerated via association of these holoenzymes, leading to activation of primed CaMKII by intersubunit interactions. This could involve autophosphorylation at T286 and/or T253 between subunits of different holoenzymes, similar to that observed between concentrated truncated monomers (Hudmon and Schulman, 2002), or perhaps exchange of monomers among holoenzymes, as speculated based on structural data (Rosenberg et al., 2006). This slow step in CaMKII accumulation might be considered a sequential “bumper car” type activation, generating a form of CaMKII that associates with postsynaptic sites.
The higher incidence of distally-restricted propagation in interneurons may be explained in part by their high resting Ca++ level and high Ca++-binding capacity (Lee et al., 2000; Rozsa et al., 2004) limiting whole cell propagation. Furthermore, many interneurons exhibit a distal distance-dependent increment in dendritic Ca++ transients (Rozsa et al., 2004); this would promote distal CaMKII translocation. The absence of proximal spread in some interneurons may result in selective sparing under pathological conditions of glutamate release from neighboring degenerating neurons (Oliva et al., 2002).
Evidence suggests that L-IPS accumulation of CaMKII would also occur under non-pathological conditions. Dendritic Ca++ spikes have been recorded in the apical dendrites of CA1 pyramidal neurons in the intact anaesthetized rat brain, most consistently during sharp wave events that are associated with population activity during consummatory behaviors and slow wave sleep (Kamondi et al., 1998). Ca++ spikes have also been observed spontaneously in neocortical dendrites in vivo (Helmchen et al., 1999). Dendritic Ca++ spikes that reliably propagate to induce somatic action potentials have been generated in pyramidal neurons in slice by synaptic stimulation, similar to effects of focal glutamate (Cai et al., 2004; Spruston, 2008).
L-IPS accumulation of CaMKII results in elevated synaptic CaMKII activity and AMPA receptor GluR1 at synapses both within the activated region of dendrite and at non-activated synapses. Thus L-IPS accumulation of CaMKII is a form of heterosynaptic plasticity. Effects may be multiple, perhaps related to heterosynaptic LTP, reduction in threshold for LTP (Harvey and Svoboda, 2007), or reversal of LTD (Muller et al., 1995) . Heterosynaptic spread of LTP to non-stimulated synapses has been reported previously, developing more slowly than homosynaptic LTP of the stimulated pathway (Bauer and LeDoux, 2004), but is not common. However, LTP is typically induced by stimulation of distributed inputs, not clustered inputs as with our local puff. In the strongest demonstration of heterosynaptic spread of LTP in CA1 pyramidal neurons, (Engert and Bonhoeffer, 1997) used local perfusion to activate synapses within a ~30 μm diameter region, and observed a loss of input specificity 70 μm away. Our findings suggest that potentiation of clustered inputs may be most likely to generate heterosynaptic plasticity via propagating molecular redistribution of CaMKII.
EXPERIMENTAL PROCEDURES
Hippocampal sandwich cultures with glial feeders were prepared from E18 rats (Harms et al., 2005). GFP-CaMKII (Shen and Meyer, 1999) was transfected at least one day prior to imaging at 14-24 days in vitro (DIV) or transduced by lentiviral vector at 3-5 DIV. For live cell imaging, coverslips were placed in a custom-built chamber and continuously perfused with extracellular solution (ECS in mM: 168 NaCl, 2.4 KCl, 10 HEPES, 10 D-glucose, 1.3 CaCl2, 1.3 MgCl2, pH 7.4, osmolality adjusted with sucrose) at room temperature (RT). Imaging was performed on a Nikon TE300 microscope with a 40X 1.0 NA oil objective and Retiga EXi cooled CCD camera using Metamorph software and Chroma excitation and emission filters in Sutter filter wheels. For local stimulation, a 3-10 MΩ pipette filled with 100 μM glutamate and 10 μM glycine was positioned very close to the dendrite and a single pressure ejection of 10-20 psi and 10-25 msec generated via a General Valve Picospritzer. Immunocytochemistry was performed essentially as described (Sharma et al., 2006). For Ca++ imaging, neurons were incubated 30 min in 5 μM Fluo-4FF in ECS at 37°C, washed 30 min in 0 Ca++ ECS, then imaged in ECS with 0.5 μM TTX at RT. Whole cell patch clamp was performed with 2-4 MΩ electrodes filled with (in mM): 140 Kgluconate, 10 HEPES, 8 NaCl, 2 MgCl2, 6.23 CaCl2, 2 Na2ATP, and 0.1 Na2GTP, pH 7.4. Neurons were held at -70 mV in ECS with 0.5 μM TTX and 10 μM SR95531. See Supplemental Experimental Procedures for further detail.
Supplementary Material
Acknowledgments
We thank Xiling Zhou, Amanda Rooyakkers, and Huaiyang Wu for excellent technical assistance. We thank Drs. Rachel Wong, Tim Murphy, Paul De Koninck, Ulli Bayer, and Frederick Dobie for helpful discussions. This work was supported by CIHR MOP69096, NIH NS33184, Canada Research Chair (AMC) and Michael Smith Foundation for Health Research (AMC & JKR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–9512. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Colbran RJ. Targeting of calcium/calmodulin-dependent protein kinase II. Biochem J. 2004;378:1–16. doi: 10.1042/BJ20031547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Synapse specificity of long-term potentiation breaks down at short distances. Nature. 1997;388:279–284. doi: 10.1038/40870. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Harms KJ, Craig AM. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J Comp Neurol. 2005;490:72–84. doi: 10.1002/cne.20635. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk W, Tank DW. In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci. 1999;2:989–996. doi: 10.1038/14788. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005a;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005b;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Nishizawa M, Arimura N, Yamamoto H, Takeuchi Y, Miyamoto E, Kaibuchi K, Inagaki M. Activation of Ca2+/calmodulin-dependent protein kinase II within post-synaptic dendritic spines of cultured hippocampal neurons. J Biol Chem. 2000;275:27165–27171. doi: 10.1074/jbc.M003751200. [DOI] [PubMed] [Google Scholar]
- Kamondi A, Acsady L, Buzsaki G. Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J Neurosci. 1998;18:3919–3928. doi: 10.1523/JNEUROSCI.18-10-03919.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa K, Irvine EE, Giese KP, Kullmann DM. NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J Physiol. 2007;584:885–894. doi: 10.1113/jphysiol.2007.137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Rosenmund C, Schwaller B, Neher E. Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol. 2000;525(Pt 2):405–418. doi: 10.1111/j.1469-7793.2000.t01-3-00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze T, Feng B, Monaghan DT, Krishtal OA. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’ afterburst EPSC in rat hippocampus. J Physiol. 2004;558:451–463. doi: 10.1113/jphysiol.2004.063792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Migues PV, Lehmann IT, Fluechter L, Cammarota M, Gurd JW, Sim AT, Dickson PW, Rostas JA. Phosphorylation of CaMKII at Thr253 occurs in vivo and enhances binding to isolated postsynaptic densities. J Neurochem. 2006;98:289–299. doi: 10.1111/j.1471-4159.2006.03876.x. [DOI] [PubMed] [Google Scholar]
- Muller D, Hefft S, Figurov A. Heterosynaptic interactions between LTP and LTD in CA1 hippocampal slices. Neuron. 1995;14:599–605. doi: 10.1016/0896-6273(95)90316-x. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H. Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci. 1999;2:804–811. doi: 10.1038/12175. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Lam TT, Swann JW. Distally directed dendrotoxicity induced by kainic Acid in hippocampal interneurons of green fluorescent protein-expressing transgenic mice. J Neurosci. 2002;22:8052–8062. doi: 10.1523/JNEUROSCI.22-18-08052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J. Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. Febs J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- Rozsa B, Zelles T, Vizi ES, Lendvai B. Distance-dependent scaling of calcium transients evoked by backpropagating spikes and synaptic activity in dendrites of hippocampal interneurons. J Neurosci. 2004;24:661–670. doi: 10.1523/JNEUROSCI.3906-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343–348. doi: 10.1016/s0959-4388(00)00217-8. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Local and propagated dendritic action potentials evoked by glutamate iontophoresis on rat neocortical pyramidal neurons. J Neurophysiol. 1997;77:2466–2483. doi: 10.1152/jn.1997.77.5.2466. [DOI] [PubMed] [Google Scholar]
- Sharma K, Fong DK, Craig AM. Postsynaptic protein mobility in dendritic spines: long term regulation by synaptic NMDA receptor activation. Mol Cell Neurosci. 2006;31:702–712. doi: 10.1016/j.mcn.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Sik A, Hajos N, Gulacsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci U S A. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR, Chang BH, Brickey DA. Cellular Signaling Through Multifunctional Ca2+/calmodulin-dependent Protein Kinase II. J Biol Chem. 2000;28:28. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008;9:206–221. doi: 10.1038/nrn2286. [DOI] [PubMed] [Google Scholar]
- Thalhammer A, Rudhard Y, Tigaret CM, Volynski KE, Rusakov DA, Schoepfer R. CaMKII translocation requires local NMDA receptor-mediated Ca2+ signaling. Embo J. 2006;25:5873–5883. doi: 10.1038/sj.emboj.7601420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui J, Malenka RC. Substrate localization creates specificity in calcium/calmodulin-dependent protein kinase II signaling at synapses. J Biol Chem. 2006;281:13794–13804. doi: 10.1074/jbc.M600966200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.