

Abstract
The liver is the main organ that clears lipopolysaccharide (LPS), and hepatocytes are a major cell-type involved in LPS uptake. LPS-tolerance, or desensitization, is important in negative regulation of responses to LPS, but little is known about its mechanisms in hepatocytes. Primary isolated C57BL/6 hepatocytes, and liver in vivo, internalized fluorescent LPS, and this was dependent on TLR4 at the cell surface but not on TLR4-TIR signaling through MyD88. LPS clearance from plasma was also TLR4-dependent. Pretreatment of C57BL/6 hepatocytes with LPS prevented uptake of LPS 24h later and this LPS-mediated suppression was dependent on TLR4 signaling through MyD88. Many regulators of TLR4 signaling have been identified and implicated in LPS desensitization, including SOCS1. SOCS1 mRNA and protein expression increased after LPS stimulation in hepatocytes and in whole liver. LPS uptake in hepatocytes and liver was significantly reduced following infection with adenoviral vectors overexpressing SOCS1. Similarly, inhibition of SOCS1 using siRNA mediated knockdown prevented LPS desensitization in hepatocytes. SOCS1 is known to interact with TIRAP and cause TIRAP ubiquitination and degradation, which regulates TLR signaling. We have also shown previously that TIRAP regulates LPS uptake in hepatocytes. SOCS1 co-immunoprecipitated with TIRAP in WT hepatocyte cell lysates up to 8h after LPS stimulation, but not at later time points. In the same samples ubiquitinated TIRAP was detected after 4h and up to 8h after LPS-stimulation, but not at later time points.
Conclusions
These data indicate hepatocytes are desensitized by LPS in a TLR4 signaling dependent manner. LPS-induced SOCS1 upregulation increases degradation of TIRAP and prevents subsequent LPS uptake. The exploitation of these mechanisms of LPS desensitization in the liver may be important in future sepsis therapies.
Keywords: LPS desensitization, LPS-tolerance, uptake, TIRAP
Lipopolysaccharide (LPS), a cell wall component of Gram negative bacteria, binds to a complex of receptor proteins comprised of Toll-like receptor 4 (TLR4), CD14 and MD2 at the cell surface (1–3). Binding of LPS activates intracellular signaling pathways via TLR4, which leads to activation of mitogen-activated protein kinases (MAPK), transcription factors (e.g. NFκB), and initiation of a proinflammatory response. LPS-binding at the cell surface also results in LPS internalization through mechanisms that have yet to be fully elucidated. The function of LPS uptake into cells has also not been completely determined. For example, it is still unclear whether LPS-internalization is a mechanism for removal of LPS and therefore downregulation of LPS mediated signaling, or whether internalized LPS has separate and/or adjuvant signaling effects (4–7).
LPS signaling is tightly controlled and there are multiple known proteins that regulate the signaling pathways. These regulatory proteins can be induced following stimulation with LPS, and subsequently regulate continued LPS-signaling. Alternatively, regulatory proteins can competitively associate with main TLR4 signaling partners to alter responses to LPS without requiring de novo induction. TLR4 regulators act at multiple points in the signaling pathway. Known negative regulators of TLR4 signaling include: single immunoglobulin interleukin-1 receptor related molecule (SIGIRR) and ST2 (interleukin-1 receptor-like protein) which associate with TLR4 at the cell membrane (8); Toll-interacting protein (TOLLIP) and interleukin-1 receptor-associated kinase (IRAK)-M which regulate the activation of IRAK4 and IRAK1 as part of the downstream signaling pathway of TLR4 (9;10); suppressor of cytokine signaling (SOCS)1 and SOCS3 which are induced by LPS and block downstream TLR4 signaling (11–14).
The sustained negative regulation of LPS-signaling that occurs after LPS stimulation can suppress cell activation in response to a second LPS stimulus. This phenomenon has been termed LPS desensitization or LPS tolerance (15–19). Many of the proteins regulating LPS signaling have also been implicated in the molecular mechanism of LPS desensitization (17;20;21). The exact physiological role of LPS desensitization is also under investigation and may differ depending on cell type and experimental model. Some studies that show an initial stimulus, or priming, with LPS can result in diminished proinflammatory responses to experimental paradigms of sepsis or trauma, with outcomes that can be either beneficial or detrimental (22;23). Other studies have determined that pretreatment with LPS leads to increased bacterial clearance which can be beneficial in models of sepsis (24;25). Defining the regulatory pathways of LPS-signaling and LPS desensitization has the potential to lead to strategies to modulate the host response during sepsis.
The primary cell type whose responses to LPS have been investigated is the monocyte/macrophage although many other cell types respond to LPS (26;27). We have focused on the responses of hepatocytes and liver (28;29). In the present study we delineate the mechanism of LPS desensitization in liver. The liver is the main site for LPS-clearance and hepatocytes play an important role in this process (30;31). Hepatocytes express the TLR4/CD14/MD2 LPS recognition complex and respond to LPS with the activation of MAPK and NFκB, as well as upregulation of acute phase proteins (28;29;32). We have recently shown that hepatocytes take up LPS through a process that involves the TLR4/CD14/MD2 complex in association with β2-integrins and TLR-interleukin-1 receptor associated protein (TIRAP) at the cell surface (33). We show here that LPS uptake and signaling in hepatocytes and liver is suppressed by LPS-pretreatment. This desensitization to LPS requires TLR4 signaling through MyD88 and SOCS1 upregulation. We further show that SOCS1 negatively regulates LPS-signaling through interactions with TIRAP.
MATERIALS AND METHODS
Reagents
Ultrapure LPS (Escherichia coli 0111:B4) was from List Biological Laboratories, Inc. (Vandell Way, CA). This LPS does not contain a significant amount of contaminating proteins that could stimulate Toll-like receptor 2 (TLR2) non-specifically. Alexa-488 Fluor™ E.coli LPS was from Molecular Probes (Carlsbad, CA). All LPS was tested for purity by separation on Silver stained SDS-PAGE gels and no detectable TNF was produced in TLR4-null (TLR4−/−) macrophages in response to any of the LPS used. Williams Medium E was from Gibco-BRL (Grand Island, NY); fetal calf serum from Hyclone Laboratories (Logan, UT). Rabbit anti-mouse TIRAP was from eBioscience (San Diego, CA). NF-κB consensus oligonucleotides were from Promega (Madison, WI). Anti-SOCS1, anti-SOCS3 antibodies were purchased from (Zymed, Invitrogen Corp.). AdSOCS1, AdSOCS3 (both myc-tagged) were a kind gift from Dr. Akihiko Yoshimura, Kyushu University, Fukuoka, Japan. Anti-MD2 antibody was from Abcam (Cambridge, MA)
Animals
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. TLR4-mutant mice (C3H/HeJ) and their control mice (C3H/HeOuJ), C57BL/10, and C57BL/10ScNJ (TLR4 knockout), and C57BL/6 mice were all purchased from Jackson Laboratories (Bar Harbor, ME). MyD88−/− mice were a kind gift from Dr. R Medzhitov, HHMI, Yale. These mice and matched control C57BL/6 mice were treated with sulfamethoxazole (40mg/mL) and trimethoprim (4mg/mL) orally in drinking water for the first 8 weeks of life. Mice were used two weeks after cessation of antibiotic treatment. LPS2−/−(TRIF−/−) mice, a kind gift from Dr. Bruce Beutler (Scripps Research Institution), were backcrossed at least 6 times and bred in our facility. All mice used were specific pathogen-free, between 6 and 8 weeks old and allowed rodent chow and water ad libitum.
Hepatocyte isolation and cell culture
Hepatocytes were isolated from mice by an in situ collagenase (type VI; Sigma) perfusion technique, modified as described previously (34). Hepatocyte purity exceeded 99% by flow cytometric assay, and viability was typically over 95% by trypan blue exclusion. Hepatocytes (150,000 cells/mL) were plated on gelatin-coated culture plates or coverslips pre-coated with Collagen I (BD Pharmingen) in Williams medium E with 10% calf serum, 15mM HEPES, 10−6M insulin, 2mM L-glutamine, 100U/ml penicillin, 100U/mL streptomycin. Hepatocytes were allowed to attach to plates overnight and prior to treatments cell culture media was changed to serum-free media.
LPS uptake by hepatocytes
LPS uptake was assessed as previously (33). Briefly100 ng/mL Alexa-488 Fluor™ E.coli LPS was added to hepatocytes plated on coverslips for times up to 90min. Cells were then washed and fixed with 2% paraformaldehyde and nuclei stained with Hoechst stain. LPS uptake was visualized by fluorescent microscopy, and quantified using Metamorph™ imaging software. Fluorescence intensity was linear over the dose range of LPS of 100ng/mL to 5μg/mL (Supplemental Figure). Trypan blue quenching was used to quench extracellular fluorescence and reduced fluorescence by up to 15%, suggesting the majority of the fluorescence recorded is intracellular LPS. No LPS uptake was seen after pretreatment of LPS with an excess of Polymyxin B. For LPS pretreatment experiments, cells were washed twice in PBS and media containing 5% FBS was replaced. Cells were then pretreated for the stated number of hours with 100ng/mL ultrapure E.coli LPS. Cells were then washed twice in PBS and serum free media was replaced before assessement of LPS-uptake as detailed above.
LPS uptake into liver in vivo
Mice were anesthetized and subjected to laparotomy. The hepatic portal vein was exposed and 5mg/kg Alexa-488 Fluor™ E.coli LPS was injected directly into the hepatic portal vein. Mice remained anesthetized until harvesting of the liver and blood for plasma at time points up to 90min after LPS injection. Liver tissue was fixed immediately in 2% paraformaldehyde for 4h, followed by overnight rehydration in 30% sucrose. Tissues were then frozen down in 2-methyl butane and stored at −80°C until tissue sectioning of 4 μm using a cryostat. LPS uptake was visualized using Olympus Provis fluorescent microscopy.
Assessment of LPS clearance from plasma
C57BL/6, C57BL/10, TLR4−/−, HeJ and HeOuJ mice were given 5mg/kg LPS in a total volume of 200μL saline, or control saline injection via tail vein. Blood was collected at time points up to 48h via cardiac puncture in heparinized tubes. Plasma samples were assayed using LAL Chromogenic assay (Hycult Biotechnologies, Netherlands) to semi-quantitatively determine clearance of LPS from plasma.
Preparation of cell lysates and Western blotting analysis
Treated hepatocytes were washed twice in PBS, and lysed with 1x cell lysis buffer (Cell Signaling Technologies) containing 20mM Tris-HCl (pH 7.5), 150mM NaCl, 1mM Na2EDTA, 1mM EGTA, 1% Triton, 2.5mM sodium pyrophosphate, 1mM β-glycerolphosphate, 1 mM Na3VO4, 1μg/mL leupeptin and 1 μg/mL phenylmethylsulfonyl fluoride (PMSF) on ice for 10min. Cell lysates were scraped and transferred into microcentrifuge tubes, sonicated for 10s prior to centrifugation at 10,000xg for 10min at 4°C. Protein content in supernatant was determined by BCA protein assay (Pierce, Rockford, IL.). For Western equal protein amounts were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked for 1 hour in PBS-Tween (0.1%) with 5% milk, followed by immunostaining with optimized dilutions of primary antibody in 1% milk in PBS-Tween overnight at 4°C. Horseradish peroxidase-conjugated secondary antibodies were then used in a standard enhanced chemiluminescence reaction according to manufacturer’s instructions (Pierce).
Total RNA isolation and Northern blot analysis
Total RNA was extracted using RNEasy extraction kits from Qiagen (Valencia, CA) according to the manufacturer’s protocol. For Northern blot analysis, total RNA (20μg per well) was resolved by electrophoresis in a 1% agarose gel containing 2.2M formaldehyde prior to being transferred to a GeneScreen™ membrane (Dupont, NEN research products, Boston, MA). The RNA was cross-linked to the membrane with UV Stratalinker™ (Stratagene, San Diego, CA), and then hybridized with either 32P-labeled mouse SOCS1 or SOCS3 probe. Full length cDNA probes from plasmid vectors obtained as a kind gift from Dr. T. Willson (The Walter and Eliza Hall Institute of Medical Research, Victoria, Australia). The probe was labeled using a random primed labeling kit (Roche Molecular Biochemicals, Indianapolis, IN). Hybridization was carried out at 43°C for 16–18. in a buffer containing 50% deionized formamide, 0.25M sodium phosphate (pH 7.2), 0.25M NaCl, 1mM EDTA, 7% SDS, and 100μg/mL denatured salmon sperm DNA. Blots were washed 4 times for 5 minutes in 2X SSC (1X SSC: 0.015M NaCl, 0.015M sodium citrate)-0.5% SDS at room temperature, and 3 times for 10 min in 0.1X SSC-0.5% SDS at 53°C prior to exposure to X-ray film for autoradiography.
Quantitative PCR
Total RNA was isolated from primary isolated hepatocytes +/− LPS for up to 24h using RNEasy Mini kits (Qiagen) according to manufacturer’s instructions. An on-column DNase digestion using RNase-free DNase (Qiagen) was performed to rid the samples of genomic DNA. cDNA was synthesized using 1μg RNA and oligo dT primers (Qiagen) and Omniscript™ reverse transcriptase (Qiagen). PCR reaction mixtures were prepared using SYBR Green PCR master mix (PE Applied Biosystems, Foster City, CA). SYBR Green two-step real-time RT-PCR was performed using forward and reverse primer pairs prevalidated and specific for SIGIRR, TOLLIP, SOCS1, SOCS3, ST2, and SARM (Qiagen). All samples were run in duplicate and repreated three times using cell isolated from separate experimental groups of mice. The level of gene expression for each sample was normalized to β-actin mRNA expression using the comparative Ct method and all groups were calibrated to levels of gene expression in untreated RAW 264.7 macrophage-like cells as a comparison.
Preparation of nuclear extracts and electrophoretic mobility shift assay (EMSA)
Cells were washed twice with ice-cold PBS and then scraped into 1mL PBS using a cell scraper. Cells were centrifuged at 5000 × g for 5 minutes at 4°C. The cell pellet was resuspended in 0.3mL of buffer A [10mM HEPES (pH 7.9), 10mM KCl, 1.5mM MgCl2, 0.5mM dithiothreitol (DTT), 0.2mM PMSF, 0.5% NP-40], and incubated on ice for 15 min before being vigorously vortexed for 10 s and centrifuged at 13,000 × g for 5 min. Nuclei were washed once in buffer A. Nuclear proteins were extracted by gentle resuspension of the nuclei in 75μL buffer C [20mM HEPES (pH7.9), 10mM KCl, 1.5mM MgCl2, 10% glycerol, 0.2mM EDTA, 0.5mM DTT, 0.2mM PMSF] together with 25μL buffer D (as for buffer C except 400mM KCl) added in a dropwise fashion. Samples were kept on ice for a further hour, followed by centrifugation at 13,000 × g for 15 min and collection of the supernatants. Protein concentration was determined by BCA protein assay according to manufacturer’s instructions (Pierce). Double-stranded NFκB-specific oligonucleotide was end-labeled by incubation with [γ-32P]ATP using T4 polynucleotide kinase (U.S. Biochemicals, Cleveland, Ohio) at 37°C for 30 min, and purified on a G-50 Sephadex column at 3000 × g for 10 min. Nuclear proteins (5μg per well) were incubated with 50,000 cpm of 32P-labeled oligonucleotide for 45 minutes at room temperature in a reaction mixture containing 1μg poly(dI-dC), 12.5mM Tris-HCl (pH 7.5), 50% glycerol, 0.25mM EDTA, 1.25% NP40, and 0.25mM DTT (final volume 20μL). The DNA-protein complexes were resolved on a 4% nondenaturing polyarylamide gel in 0.5X Tris-borate-EDTA (TBE) buffer. The gels were dried using a vacuum-assisted gel-dryer and then subjected to autoradiography.
Immunofluoresecence
Hepatocytes plated on coverslips were treated with 100ng/mL E.coli LPS for up to 24h then fixed as described above. Cells were then permeabilized with 0.1% Triton X, washed in PBS and PBB (0.5% BSA in PBS), and blocked with 2% BSA in PBS for 1h. Anti-MD2 antibody was added at 1:200 dilution for 1h at RT. Secondary antibody was goat anti-mouse Fab1 fragments (Cy3, 1:1000 dilution). Coverslips were visualized by confocal microscopy.
Transfection with recombinant adenoviral vectors
AdSOCS1, AdSOCS3 and AdDNSOCS1 were infected into hepatocytes at MOI of 10. Control adenoviral vectors (AdΨ5) were also infected into similar groups of hepatocytes. Infection was allowed to occur overnight before cell treatment. In vivo adenovirus was given to mice at a dose of 3×1011VP/mouse via tail vein injection 24h before intraportal LPS administration.
Transient siRNA transfection
Cells were plated at 4×105 cells per well on 6-well plates the day before the transfection. Prevalidated siRNA targeting SOCS1, SOCS3 or a control, non-targeting siRNA were purchased from Ambion (Austin, TX). Transfection into hepatocytes was performed using Lipofectamine 2000™-reagent according to manufacturer’s instructions (Gibco-BRL) at a final concentration of 30nM siRNA/well. After 24h cells were washed with PBS and media replaced with serum-free media prior to treatments.
Immunoprecipitation analysis
Whole cell lysate was obtained from isolated hepatocytes using RIPA buffer (Sigma) with added proteinase inhibitors. To preclear the lysate, 10μL of Protein A/G Sepharose Plus beads (Santa Cruz) was added to 200μg of lysate and incubated at 4°C for 1h on a rotating wheel before centrifugation at 3000rpm for 5min. After preclearance, 2.5 μg of primary antibody (SOCS1 or SOCS3) was added per sample and incubated for a 1–2h at 4°C. 20μL of Protein A/G Sepharose Plus beads per sample were added and samples incubated overnight, rotating at 4°C. Samples were centrifuged for 5min at 3000rpm and the pellet washed twice with lysis buffer on ice. 30μL sample buffer was added per sample and samples boiled for 5 min, followed by centrifugation (5min/3000rpm). Samples were then loaded and separated by size on SDS-PAGE gels, and immunoblotted for TIRAP, SOCS1 and SOCS3. Whole cell lysates from hepatocytes treated with 100ng/mL LPS for 8h was used as a positive control in immunoblots.
Polyubiquitination assay
Hepatocytes treated with 100ng/mL LPS for up to 24h were lysed in RIPA buffer containing protease inhibitors. Ubiquitination was determined after using Pierce Ubiquitin Enrichment Kit on cell lysates. Briefly, samples are diluted 1:1 in sample buffer and after addition of ubiquitin affinity resin samples are incubated at 4°C overnight. Samples were then centrifuged through an affinity column and washed three times before elution of the ubiquitin enriched fraction. Fractions were separated by SDS-PAGE and immunoblotted with anti-ubiquitin antibody (positive control) and anti-TIRAP antibody.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM). Experimental results are analyzed for their significance by Student’s t-test. Significance was established at the 95% confidence level (P <0.05).
RESULTS
Hepatocytes take up LPS and are desensitized to LPS in a TLR4-dependent manner
Hepatocytes have been shown in previous studies to play a role in uptake of LPS together with Kupffer cells (30;31). More recently we have shown that hepatocyte LPS uptake requires the presence of TLR4, CD14, MD2 and β2-integrin at the cell surface (33). Using primary hepatocytes from C57BL/6 mice we confirmed that these cells take up LPS in the absence of serum. LPS uptake, measured by fluorescence, could be detected by 15 min and became maximal between 60 and 90min (data not shown; (33)).
To determine whether hepatocyte LPS uptake could be desensitized by LPS pretreatment, we pretreated C57BL/6 hepatocytes with 100ng/mL LPS (or PBS control) 24h prior to assessment of LPS uptake. Control (PBS) pretreated hepatocytes were able to take up LPS measured at 90min. However, LPS-pretreated hepatocytes did not take up LPS (Fig. 1A, left images). No significant LPS uptake was determined in LPS-pretreated cells even after 4h, or after exposure to higher doses of fluorescent LPS (1–10μg/mL LPS; data not shown). This finding suggests that mechanisms of desensitization of LPS uptake cannot be overcome with higher LPS concentrations.
Figure 1.

Desensitization of LPS uptake in C57BL/6 hepatocytes +/− pretreatment with 100ng/mL LPS followed by stimulation with 100ng/mL Alexa Fluor™ 488 LPS for up to 90min (A, left images). LPS uptake at 90min in C57BL/6 hepatocytes at time points (4,8,18,24,36, and 48h) after pretreatment with 100ng/mL LPS or control (no pretreatment). Results show fluorescence in cells relative to background determined by Metamorph™ (A, top right). EMSA showing activation of NFκB in C57BL/6 hepatocytes 60min after stimulation with up to 10mg/mL LPS and pretreatment with cell culture media alone (Medium) or 100ng/mL LPS for 24h. (A, bottom right). Uptake at 90min of 100ng/mL Alexa Fluor™ 488 LPS in hepatocytes from C57BL/10, TLR4−/−, C3H/HeOuJ or C3H/HeJ (TLR4-mutant) mice. Results show fluorescence relative to background (B, upper panels). NFκB activation in WT, TLR4−/−, C3H/HeOuJ and C3H/HeJ hepatocytes at time points up to 60min after stimulation with 100ng/mL LPS (B, lower images). LPS uptake in hepatocytes from C3H/HeOuJ and C3H/HeJ mice at time points (4,8,18,24,36, and 48h) after pretreatment with 100ng/mL LPS or control (no pretreatment)(C). All results shown are represnentative of results obtained in at least three separate experiments.
We then determined a time course for LPS desensitization of hepatocytes. We pretreated hepatocytes with 100ng/mL LPS for 4, 8, 18, 24, 36 and 48h before assessment of LPS uptake of 100ng/mL Alexa Fluor™ 488 LPS for 90min. Results were compared with controls (no LPS-pretreatment). Decreased LPS uptake was observed after only 4h of LPS-pretreatment and uptake was significantly suppressed with pretreatments given up to 36h prior to experimentation. However, hepatocytes regained the ability to take up LPS by 48h after LPS pretreatment (Fig. 1A, right graph).
We have previously shown that hepatocytes express CD14/TLR4/MD2 (LPS-receptor complex) and that intracellular signaling molecules such as MAPK and NFκB are activated after LPS stimulation in these cells (28;29;32). We therefore wanted to determine if these pathways were also desensitized by pretreatment with LPS. Isolated hepatocytes from C57BL/6 mice were pretreated for 24h with media alone or with 100ng/mL LPS followed by stimulation with 0, 0.01, 0.1, 1.0, 10μg/mL LPS for 60min. Cells were collected and nuclei were extracted for assessment of NFκB DNA binding activity. Significant NFκB activation was observed in media-treated hepatocytes stimulated with 0.1μg/mL or higher concentrations of LPS (Fig 1A, lower right). However, little or no activation of NFκB was detected in hepatocytes pretreated with LPS, even when stimulated with 10μg/mL of LPS (Fig 1A, lower right). These data suggest that LPS-signaling in hepatocytes is desensitized similarly to LPS uptake and that the mechanisms of desensitization cannot be overcome with excess LPS stimulation.
LPS is known to signal through pathways involving TLR4 and we hypothesized that LPS desensitization in hepatocytes would also require TLR4. First we confirmed our previous work showing that LPS-uptake is dependent on the presence of TLR4 but not on signaling through TIR domain/MyD88 signaling. To do this we isolated hepatocytes from TLR4−/−(C57BL/10ScN) mice and their C57BL/10 controls, as well as from TLR4 signaling mutant mice (C3H/HeJ) and their controls (C3H/HeOuJ). C3H/HeJ mice have a mutation in the TIR domain of TLR4 (P712H) and are unable to bind and activate MyD88, although this mutated domain is able to bind to TIRAP (35). LPS uptake at 90min was then assessed using 100ng/mL Alexa Fluor™ 488 LPS. Consistent with our previous findings we found decreased uptake of LPS in TLR4−/− hepatocytes compared with controls, but similar uptake of LPS in hepatocytes from C3H/HeJ and C3H/HeOuJ mice (Fig. 1B). Also as expected there was significantly decreased activation of NFκB in response to LPS in cells from TLR4−/− or C3H/HeJ mice.
We then determined whether the LPS uptake observed in hepatocytes from C3H/HeJ mice, where TLR4 signaling is attenuated, was likewise subject to LPS desensitization. We therefore pretreated isolated hepatocytes from C3H/HeJ and C3H/HeOuJ mice with 100ng/mL LPS for 4, 8, 18, 24, 36 and 48h prior to assessment of uptake of 100ng/mL Alexa Fluor™ 488 LPS at 90min. Results were compared to uptake in control hepatocytes (no LPS-pretreatment). Similarly to results obtained in hepatocytes from C57BL/6 mice, LPS pretreatment of C3H/HeOuJ hepatocytes for 4 to 24h significantly decreased their ability to take up LPS. By 48h, there was no effect of LPS pretreatment on LPS uptake, and the ability to take up LPS was fully restored (Fig. 1C). However, no decrease in LPS uptake was observed in LPS pretreated C3H/HeJ cells (Fig. 1C). These data suggest that TLR4 signaling is essential for desensitization of hepatocyte LPS uptake in contrast to the lack of such a requirement for initial LPS uptake.
Uptake of LPS in vivo and clearance of LPS from plasma is similarly TLR4-dependent
To determine whether similar uptake occurred in hepatocytes in vivo, we injected WT (C57BL/10), TLR4−/−, C3H/HeJ and C3H/HeOuJ mice with 5mg/kg fluorescent LPS via the hepatic portal vein following laparotomy under anesthesia. Liver tissue was fixed at time points after LPS injection and 4μm sections were visualized by fluorescent microscopy. The dose of LPS used (5mg/kg) is non-lethal, but was sufficient to produce measurable increases in plasma TNF-α levels (data not shown). As seen previously in isolated hepatocytes, LPS was taken up by parenchymal liver cells of WT, C3H/HeOuJ and TLR4-mutant C3H/HeJ mice, but not in the livers of TLR4−/− mice (Fig. 2A). These in vivo findings confirm LPS-uptake in the liver as a physiological finding.
Figure 2.

LPS uptake in liver of WT (C57BL/10), TLR4−/−, C3H/HeOuJ and C3H/HeJ mice after portal vein injection of 5mg/kg LPS (A). Liver was collected 90min after LPS injection, fixed and frozen. Four micron sections were made and uptake was visualized using Olympus fluoresenct microscope (x40 magnification). Images are representative of at least four separate experiments (A). LPS level in plasma of WT (BL10), TLR4−/−, C3H/HeOuJ and C3H/HeJ mice at time points after tail vein injection of 5mg/kg E.coli LPS (B). Whole blood was collected by cardiac puncture at time points after injection, and plasma LPS was measured using chromogenic limulus amebocyte assay. Results shown are representative of three experiments performed using separate groups of mice.
We also examined clearance of LPS from plasma of WT (C57BL/10), TLR4−/−, C3H/HeOuJ and C3H/HeJ mice injected with 5mg/kg E.coli LPS via the tail vein. At time points after LPS injection, blood was collected via cardiac puncture and LPS levels in plasma were determined using a commercially available limulus amebocyte chromogenic assay. TLR4−/− mice had significantly higher levels of LPS in their plasma compared with their WT controls at 4, 24 and 36h after LPS injection (Fig. 2B). However, C3H/HeJ mice showed similar ability to clear LPS as C3H/HeOuJ mice (Fig. 3C). These data suggest in vivo clearance of LPS from the plasma is TLR4-dependent but, similarly to hepatic uptake, does not require TLR4-signaling through MyD88.
Figure 3.
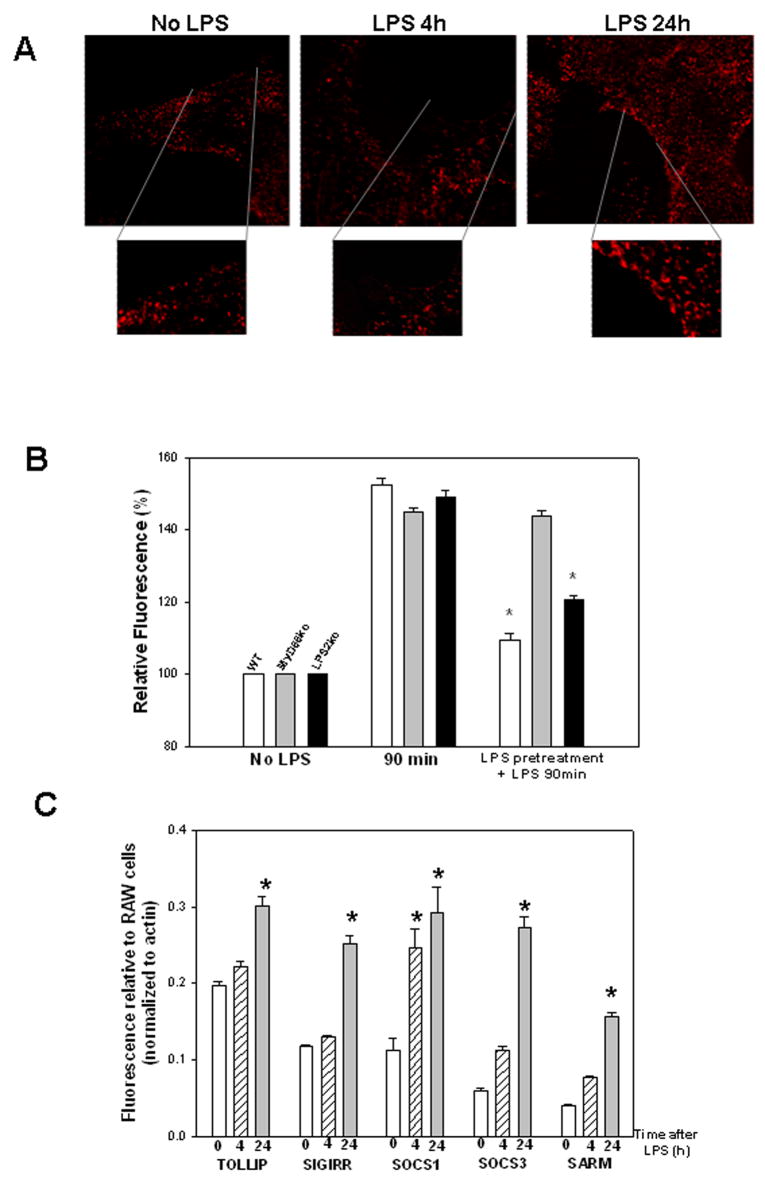
Cell surface expression of MD2 on isolated mouse hepatocytes at baseline (No LPS), and at 4h and 24h hours after 100ng/mL LPS (A). Isolated hepatocytes were treated with LPS and at time points after treatment were washed thoroughly and fixed in paraformaldehyde. Immunofluorescence was then done, using anti-MD2 antibody primary and Cy3 labeled secondary antibody, and imaged by confocal microscopy (x60 magnification). Images represent multiple high power fields and experiments were repeated at least twice in similar hepatocytes from separate mice (A). Uptake of LPS in isolated hepatocytes from WT, MyD88−/− and LPS2−/− (TRIF−/−) mice after 100ng/mL Alexa Fluor 488 LPS for 90min, or 100ng/mL E.coli LPS pretreatment for 24h followed by 100ng/mL Alexa Fluor 488 LPS for 90min (B). Uptake was visualized by fluorescent microscopy, and quantitation performed using Metamorph™. Experiments were repeated three times and graph represents results from a single representative experiment. (* p<0.05 vs no pretreatment; Students t-test)(B). Upregulation of expression of TOLLIP, SIGIRR, SOCS1, SOCS3, SARM mRNA in hepatocytes after stimulation with 100ng/mL LPS for 4 or 24h or control (time 0) (C). Quantitative RT-PCR was done using specific primers. Results were normalized to actin and expression is shown relative to levels in unstimulated RAW 264.7 cells for comparison. Results are from three separate experiments. (* p<0.05 vs baseline expression; Students t-test) (C).
Cell surface TLR4/MD2 expression transiently decreases after LPS stimulation
Previous studies have suggested that decreased expression of TLR4 at the cell surface after LPS stimulation may be a contributing factor in the mechanism of LPS desensitization. We therefore investigated cell surface levels of TLR4-associated MD2 in WT hepatocytes at baseline, and at 4, 8, and 24h after LPS stimulation. Good cell surface expression of MD2 was seen at baseline in hepatocytes (Fig. 3A, left). Cell surface MD2 expression decreased after 4h of LPS treatment (Fig. 3A, middle) but was restored by 8 and 24h after LPS stimulation (Fig. 3A, right). Total TLR4 protein expression was also measured by Western blot of whole cell lysates, and no overall change in TLR4 expression was observed after LPS stimulation of hepatocytes. These data therefore suggest that saturation of TLR4 or decreased expression of TLR4 after LPS stimulation is unlikely to be a main cause of LPS desensitization in hepatocytes.
Desensitization of hepatocytes to LPS is MyD88, but not TRIF, dependent
Results showing that hepatocytes from C3H/HeJ mice did not undergo LPS desensitization of LPS uptake suggested that LPS desensitization was dependent on MyD88 signaling via the TIR domain of TLR4. To investigate this further, and to determine whether the other main TLR4-signaling pathway via TRIF was involved, we used hepatocytes isolated from MyD88−/− and LPS2−/− (TRIF−/−) mice. Hepatocytes from all experimental groups were able to take up LPS when given as a single first challenge, but only WT and LPS2−/− hepatocytes were desensitized to a subsequent LPS challenge 24h after initial LPS pretreatment (Fig. 3B). MyD88−/− hepatocytes were not desensitized and still took up LPS on the second challenge (Fig. 3B). These data confirmed the importance of MyD88, rather than TRIF, in the signaling pathway leading to LPS desensitization in hepatocytes.
Upregulation of regulatory proteins after LPS-stimulation in hepatocytes
Many signaling proteins have been associated with regulation of TLR4 signaling and LPS desensitization. These include TOLLIP, SIGIRR, SOCS1, SOCS3, ST2, and SARM (8;9;11;13;36;37). These proteins have been shown to act on different parts of various LPS-signaling pathways and regulate signaling in diverse ways. To determine whether these regulatory intermediates affect desensitization of LPS uptake in hepatocytes we used quantitative RT-PCR to determine their respective mRNA expression levels in hepatocytes after LPS-stimulation. Hepatocytes were isolated from C57BL/6 mice and stimulated with 100ng/mL LPS for 0, 4, or 24h. Total RNA was collected and, after cDNA synthesis, quantitative RT-PCR was performed using commercially available prevalidated specific primers for each regulatory molecule studied (Fig. 3C). There was good baseline expression of all the potential signaling regulators except ST2, which was expressed only at a very low level (data not shown). The expression level of all of the signaling intermediates significantly increased by 24h after LPS-stimulation, but only SOCS1 expression was significantly upregulated at the early 4h time point. Taken together these data suggest that SOCS1 might play a role in LPS desensitization in hepatocytes, at least at early time points.
SOCS1 upregulation of expression in hepatocytes after LPS is TLR4-dependent
To further investigate expression of SOCS1 and SOCS3 mRNA and protein in hepatocytes and liver in response to LPS, we injected 2.5mg/kg LPS intraperitoneally into C57BL/6 mice and then harvested liver after 0 (no LPS), 3, 6, and 12h. Total RNA was isolated from liver and Northern analysis confirmed qRT-PCR results in hepatocytes showing upregulation of SOCS1 and SOCS3 expression in liver after only 3h of LPS-stimulation (Fig. 4A, left). Upregulation of SOCS1 and SOCS3 protein expression was also confirmed by Western blot in isolated hepatocytes at time points up to 48h after 100ng/mL LPS stimulation. SOCS1 protein level increased as early as 1h after stimulation and remained elevated to 24h. SOCS1 expression then decreased by 48h. SOCS3 protein expression increased after 8h and remained elevated to 48h after LPS stimulation (Fig. 4A, right).
Figure 4.

SOCS1 and SOCS3 RNA expression by Northern blot in C57BL/6 mouse liver at time points up to 12h after intraperitoneal injection of LPS (2.5mg/kg) (A, left image). SOCS1 and SOCS3 protein expression by Western blot in C57BL/6 isolated hepatocytes at time points up to 48h after stimulation with 100ng/mL LPS (A, right image). SOCS1 and SOCS3 RNA expression by Northern blot in primary isolated hepatocytes from C3H/HeOuJ, C3H/HeJ, C57BL/10 and TLR4−/− mice at time points after stimulation with 100ng/mL LPS (B). SOCS1 and SOCS3 protein expression by Western blot in primary isolated hepatocytes from C3H/HeOuJ, C3H/HeJ, C57BL/10 and TLR4−/− mice at time points after stimulation with 100ng/mL LPS (C). All results are representative of results obtained in at least three separate experiments.
As we had already determined that LPS desensitization in hepatocytes required TLR4 signaling, we assessed SOCS1 and SOCS3 expression in hepatocytes from C57BL/10, TLR4−/−, C3H/HeOuJ, and C3H/HeJ mice. Isolated hepatocytes were stimulated for up to 24h with 100ng/mL LPS and SOCS1 and SOCS3 mRNA expression was determined by Northern analysis. Both SOCS1 and SOCS3 expression were upregulated in C3H/HeOuJ hepatocytes, but only SOCS3 and not SOCS1 expression was increased in TLR4-mutant (C3H/HeJ) hepatocytes (Fig. 4B). Protein expression of SOCS1 was also not upregulated in either TLR4−/− or C3H/HeJ hepatocytes compared with controls (Fig. 4C). These data suggest that increased SOCS1 expression, but not SOCS3 expression, is dependent on TLR4 signaling through MyD88.
SOCS1 expression regulates desensitization of LPS uptake in hepatocytes and liver
To determine if either SOCS1 or SOCS3 were important in the mechanism of desensitization of LPS uptake in hepatocytes, we overexpressed SOCS1 and SOCS3 in hepatocytes using adenoviral vectors. Isolated C57BL/6 hepatocytes were pretreated for 24h with AdSOCS1, AdSOCS3 or control adenovirus (AdΨ5) at MOI of 10, and LPS uptake at 90min was assessed by stimulation with 100ng/mL Alexa Fluor™ 488 LPS. LPS uptake was not affected by overexpression of SOCS3 (Fig. 5A). However, overexpression of SOCS1 prevented LPS uptake in hepatocytes (Fig. 5A). Overexpression of SOCS1 and SOCS3 was confirmed by Western blot of whole cell lysates (Fig. 5A right). Additionally, overexpression of SOCS1 but not SOCS3 decreased activation of NFκB in hepatocytes stimulated with 0, 100, or 1000ng/mL LPS. Taken together these data suggest that SOCS1 regulates LPS uptake through a mechanism involving inhibition of TLR4 signaling. Similar experiments were carried out in vivo to confirm the physiological importance of SOCS1 in liver LPS desensitization. WT mice were injected via the tail vein with adenovirus 24h prior to assessment of LPS uptake in the liver. Similarly to above, mice overexpressing SOCS1 had decreased LPS-uptake in the liver compared to control mice of mice overexpressing SOCS3 (Fig. 5B).
Figure 5.
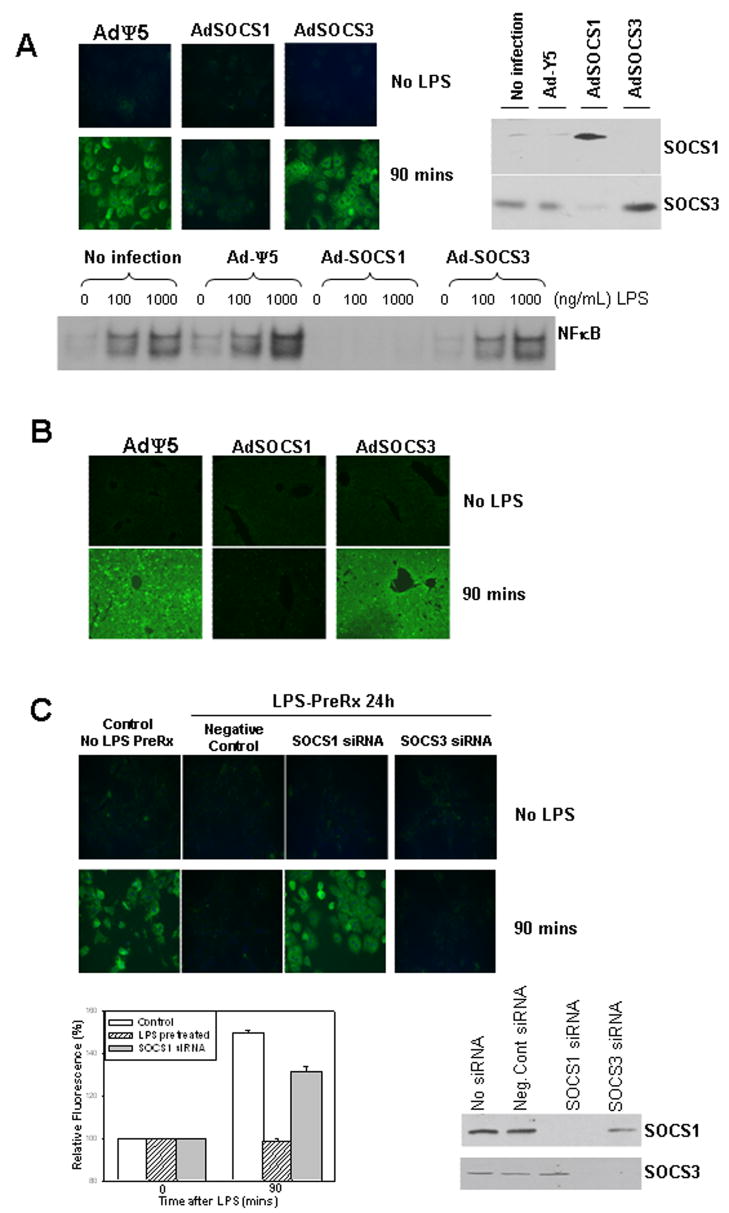
Uptake after 90min of fluorescent LPS in primary isolated hepatocytes from C57BL/6 mice pretreated for 24h with adenoviral control vector (AdΨ5), or vectors expressing SOCS1 (AdSOCS1) or SOCS3 (AdSOCS3). Protein expression of SOCS1 and SOCS3 in hepatocytes after pretreatment with AdSOCS1 or AdSOCS3 respectively was confirmed by Western blot of whole cell lysates (A, upper images). Activation of NFκB by EMSA in C57BL/6 hepatocytes treated for 60min with 0 (control, PBS), 100ng/mL, or 1000ng/mL LPS (A, lower image). LPS uptake in liver of WT mice injected with AdΨ5, AdSOCS1, or AdSOCS3 via tail vein 24h prior to injection of 5mg/kg Alexa Fluor 488 LPS via portal vein (B). Liver was harvested and fixed after 90min, and uptake visualized by fluorescent microscopy (x40 magnification). Uptake after 90min of fluorescent LPS in primary isolated C57BL/6 hepatocytes. Uptake in control cells (no siRNA, no LPS pretreatment) was compared with uptake in cells pretreated for 24h with either non-targeting control siRNA, siRNA targeting SOCS1, or siRNA targeting SOCS3 followed by pretreatment with 100ng/mL LPS. Fluorescence levels were quantified using Metamorph™ and results shown are fluorescence relative to baseline. (C, upper panels and lower left). Knockdown of SOCS1 or SOCS3 was confirmed by Western blot of whole cell lysates (C, lower right). All images representative of uptake in at least three separate experiments.
We also used a knockdown strategy with siRNA specific for SOCS1 or SOCS3 to further confirm the role of SOCS1 in LPS uptake and LPS desensitization in hepatocytes. C57BL/6 hepatocytes were treated for 24h with either non-targeting (negative control) siRNA, or siRNA targeting SOCS1 or SOCS3. Knockdown of SOCS1 and SOCS3 was confirmed in separately treated hepatocytes by Western analysis of whole cell lysates. Cells were then pretreated with 100ng/mL LPS for 24h before LPS uptake was assessed using 100ng/mL Alexa Fluor™ 488 LPS. Results were compared in parallel with uptake in control hepatocytes that had not been pretreated with either siRNA or LPS. As shown previously, LPS uptake at 90min was seen in control cells with no LPS pretreatment and there was no significant LPS uptake in cells given negative control siRNA and pretreated with LPS (Fig. 5C). Knockdown of SOCS3 also did not affect LPS uptake in hepatocytes after LPS-pretreatment, but knockdown of SOCS1 was able to overcome the effects of LPS-pretreatment and enabled LPS uptake in these cells (Fig. 5C images and graph). These data confirm the significance of SOCS1 in the mechanism of desensitization of LPS uptake in hepatocytes.
SOCS1 association with TIRAP causes TIRAP degradation and decreased LPS uptake
Previous work by others (14) has shown that one mechanism of SOCS1-mediated LPS desensitization in macrophages is through binding to the TLR4 signaling adaptor TIRAP (38). TIRAP is essential for LPS-signaling mainly through association with another TLR adaptor molecule, MyD88 (38). SOCS1 binding to TIRAP has been shown to lead to TIRAP ubiquitination and degradation by the proteasome to negatively impact LPS-signaling through TLR4 (14). We have also shown that TIRAP signaling, independently of MyD88, is essential for LPS uptake in hepatocytes (33). To determine whether SOCS1 or SOCS3 bind to TIRAP in hepatocytes, cell lysates were collected from C57BL/6 hepatocytes stimulated with 100ng/mL LPS for up to 24h. SOCS1 or SOCS3 were immunoprecipitated and samples were separated by SDS-PAGE and immunoblotted for TIRAP, SOCS1 and SOCS3. SOCS1, but not SOCS3, associated with TIRAP in hepatocytes as early as 1h after LPS-stimulation (Fig. 6A). This association occurs up to the 8h time point but was no longer seen by 24h (Fig. 6A). Similarly, TIRAP protein expression in hepatocyte whole cell lysates decreased after 1h and was not detected at 24h. Expression was partially restored by 48h (Fig. 6B). Additionally we assessed TIRAP protein levels in hepatocytes pretreated for 24h with AdSOCS1 or control AdΨ5 and then stimulated for up to 24h with 100ng/mL LPS. TIRAP was detected by Western blot in control treated cells and levels decreased significantly by 4h after LPS-treatment (Fig. 6B). No TIRAP expression was detected in cells overexpressing SOCS1 (Fig. 6B). Similar cell lysates from LPS-treated hepatocytes were analyzed for ubiquitination of TIRAP using an immunoprecipitation method. TIRAP ubiquitination was seen by 1h and was maximal by 8h after LPS-stimulation (Fig. 6C). This corresponded with a decrease in TIRAP protein expression. Taken together these data suggest that SOCS1 expression leads to degradation of TIRAP to undetectable levels, and this in turn downregulates the uptake of LPS in hepatocytes.
Figure 6.

Immunoprecipiation (IP) of SOCS1 but not SOCS3 with TIRAP in primary isolated C57BL/6 hepatocytes at time points up to 24h after treatment with LPS (A). Immunoprecipitates were immunoblotted with anti-TIRAP, SOCS1 or SOCS3 antibodies, using whole cell lysates from hepatocytes treated with 100ng/mL LPS for 8h as a positive control (A). Protein expression by Western blot of TIRAP in C57BL/6 hepatocytes treated with LPS for up to 48h (B, upper images), and actin loading controls. Similar groups of C57BL/6 hepatocytes were pretreated for 24h with adenovirus expressing control vector (AdΨ5) or SOCS1 (AdSOCS1) and then stimulated for up to 24h with LPS. Whole cell lysates were then immunoblotted with anti-TIRAP antibody and actin as a loading control (B, lower images). TIRAP ubiqitination in C57BL/6 hepatocytes at time points up to 24h after stimulation with 100ng/mL LPS, using total ubiquitin as a positive control (C). All results are representative of at least three separate experiments using hepatocytes from separate mice.
DISCUSSION
The phenomenon of LPS desensitization, or tolerance, has been well described in macrophages and monocytes where it is characterized by decreased cytokine production in response to LPS (26;27). Desensitization is part of the larger phenomenon of preconditioning, where stimulation with a second inflammatory stimulus of the same, or different, proinflammatory agent or stress can lead to three possible outcomes: similar response to the primary challenge upon secondary challenge; priming - where the secondary challenge produces a greater response than the primary challenge; desensitization - where the secondary response is diminished compared with the primary response (39;40). In this study we describe two distinct aspects of LPS desensitization in hepatocytes and liver, the first characterized by decreased LPS signaling through TLR4, and the second characterized by decreased uptake of LPS.
The liver plays a major role in responses to sepsis including the production of acute phase proteins, complement factors, clotting factors and primarily Kupffer cell-derived cytokines (41). Additionally multiple liver cell types contribute to the clearance of bacteria and microbial products such as LPS (30;31). The exact role of hepatocytes during sepsis continues to be defined, but we have shown previously that hepatocytes respond to LPS in multiple ways both in vivo and in vitro (28;29;32). With this in mind it does not seem surprising that parenchymal cells of the liver, which make up the main part of liver mass, also regulate responses to LPS and can be desensitized. In this study we have also determined that hepatocytes in vivo are desensitized by LPS and this may subsequently affect LPS clearance from plasma. Other liver cells such as liver sinusoidal epithelial cells and intrahepatic biliary cells are also desensitized by LPS (42;43). Regulation and desensitization of LPS responses are especially important given the constant exposure of the liver to endotoxin from the gut (44).
The physiological role of endotoxin desensitization has yet to be fully defined. Similar ‘tolerance’ effects are seen clinically in patients after shock, trauma, burns and sepsis. These patients become immunosuppressed and monocytes isolated from them show a similar type of decreased responsiveness to LPS (39;45;46). The relative refractory state of the immune system is thought to contribute to the immunosuppression and decreased ability to defend against a subsequent bacterial challenge. The molecular basis for the observed tolerance continues to be the subject of many studies. No single mechanism for LPS desensitization has yet been described, although it is known that disruptions to LPS stimulated signaling through TLR4 are important in this process (47;48). Multiple parts of the signaling pathway can be regulated and desensitization affects multiple downstream transcriptional targets (49). However, not all of the negative regulators of LPS have been shown to also be involved in LPS-tolerance (50).
Regulation of LPS-signaling and desensitization is not only dependent on the dose and timing of LPS (18;51;52) but may also be specific to the cell type and experimental model being examined. These specificities may explain the seemingly contradictory findings in much of the LPS tolerance literature. This is especially true when considering the question of whether or not LPS tolerance results in increased bacterial clearance (24;25;53) or a decreased responsiveness to bacteria (17;22). Hepatocytes are not known to take up bacteria directly, however they do contribute to LPS-clearance and the innate immune response to bacteria and bacterial products. We speculate that one reason hepatocytes regulate LPS uptake is because of continued activation of the cell in some way after internalization. Continued LPS-signaling has been shown previously in other cell types after internalization (5;7). Contradictory findings have also been published suggesting LPS-internalization is a mechanism for restricting continued signaling, as well as allowing degradation of LPS through deacylation in macrophages and monocytes (4;6;54). However, Kupffer cells and liver sinusoidal endothelial cells show decreased LPS-responsiveness but paradoxical increases in scavenger and phagocytic properties (25;42). Further work is required to define differences in cell type specificity of response to LPS and how these responses are integrated into the entire innate immune response.
Multiple proteins have been determined to negatively regulate TLR signaling and responses to TLR ligands. SOCS1 has been implicated as both a negative regulator of TLR-signaling and an important mediator of LPS tolerance (11;14;55). SOCS1 interacts with TIRAP and leads to its degradation (14). Degradation of signaling proteins such as TIRAP severely limits LPS signaling through TLR4. Additionally, regulation of signaling occurs through changes in TIRAP and TLR4 phosphorylation that can regulate this pathway at other levels (22;23). Regulation of IRAK proteins through increased expression of the negative regulator IRAK-M (56), or degradation of IRAK1 or IRAK4 may also be important in regulation and desensitization (37;57). It is likely that more than one regulatory mechanism is responsible for LPS tolerance in any cell type. These multiple layers of regulation could represent redundancy or could allow for fine specificity of regulation of cell function during LPS desensitization. In hepatocytes, we have shown that regulation of TIRAP is key to the regulation of LPS uptake (33), and so induction of SOCS1 is key to LPS desensitization in these cells. We have yet to determine whether other regulatory pathways are important in other hepatocyte functions during sepsis. Importantly, however, we have identified TIRAP as pivotal to the regulation of both signaling through TLR4 to activate NFκB, and signaling to initiate uptake and clearance of LPS through the CD14/TLR4/MD2 complex at the cell surface (33). This separation of the signaling pathways of NFκB upregulation and LPS uptake may allow for dissociation of the two processes under certain circumstances as well as integration through regulatory agents such as SOCS1.
LPS desensitization is an important regulatory mechanism in the innate immune response to sepsis. In this report we have expanded the characterization of LPS desensitization to include a role for LPS uptake and clearance in hepatocytes. Further work is required in order to determine other hepatocyte functions affected by LPS desensitization, and other pathways that might be involved. Taken together this delineation of the mechanisms and physiological relevance of LPS desensitization in the liver may be important in providing future treatment options for immunosuppressed septic patients.
Acknowledgments
Financial Support: This work was supported in part by NIH grant R01-GM-05441 and through Infectious Diseases Society of America Young Investigator Award (to MS).
Grateful thanks to Hong Liao, Carol Meiers, and Danielle Reiser for technical assistance.
List of Abbreviations
- LPS
lipopolysaccharide
- TLR4
Toll-like receptor 4
- MAPK
mitogen associated protein kinase
- NFκB
nuclear factor kappa B
- SIGGIRR
single immunoglobulin interleukin-1 receptor related molecule
- SARM
sterile alpha and HEAT-Armadillo motifs
- ST2
interleukin-1 receptor-like protein
- TOLLIP
toll interacting protein
- IRAK
interleukin 1 receptor associated kinase
- SOCS
suppressor of cytokine signaling
- TIRAP
Toll/IL-1 receptor associated protein
References
- 1.Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Gangloff SC, Zahringer U, Blondin C, Guenounou M, Silver J, Goyert SM. Influence of CD14 on ligand interactions between lipopolysaccharide and its receptor complex. J Immunol. 2005;175:3940–3945. doi: 10.4049/jimmunol.175.6.3940. [DOI] [PubMed] [Google Scholar]
- 4.Fukuda I, Tanamoto K, Kanegasaki S, Yajima Y, Goto Y. Deacylation of bacterial lipopolysaccharide in rat hepatocytes in vitro. Br J Exp Pathol. 1989;70:267–274. [PMC free article] [PubMed] [Google Scholar]
- 5.Cowan DB, Noria S, Stamm C, Garcia LM, Poutias DN, del Nido PJ, et al. Lipopolysaccharide internalization activates endotoxin-dependent signal transduction in cardiomyocytes. Circ Res. 2001;88:491–498. doi: 10.1161/01.res.88.5.491. [DOI] [PubMed] [Google Scholar]
- 6.Latz E, Visintin A, Lien E, Fitzgerald KA, Espevik T, Golenbock DT. The LPS receptor generates inflammatory signals from the cell surface. J Endotoxin Res. 2003;9:375–380. doi: 10.1179/096805103225003303. [DOI] [PubMed] [Google Scholar]
- 7.Husebye H, Halaas O, Stenmark H, Tunheim G, Sandanger O, Bogen B, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neill LA. Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases. Curr Opin Pharmacol. 2003;3:396–403. doi: 10.1016/s1471-4892(03)00080-8. [DOI] [PubMed] [Google Scholar]
- 9.Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, et al. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 12.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 13.Fang M, Dai H, Yu G, Gong F. Gene delivery of SOCS3 protects mice from lethal endotoxic shock. Cell Mol Immunol. 2005;2:373–377. [PubMed] [Google Scholar]
- 14.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi KS, Flavell RA. Shielding the double-edged sword: negative regulation of the innate immune system. J Leukoc Biol. 2004;75:428–433. doi: 10.1189/jlb.0703321. [DOI] [PubMed] [Google Scholar]
- 16.Chan C, Li L, McCall CE, Yoza BK. Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J Immunol. 2005;175:461–468. doi: 10.4049/jimmunol.175.1.461. [DOI] [PubMed] [Google Scholar]
- 17.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133–150. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 18.Day J, Rubin J, Vodovotz Y, Chow CC, Reynolds A, Clermont G. A reduced mathematical model of the acute inflammatory response II. Capturing scenarios of repeated endotoxin administration. J Theor Biol. 2006;242:237–256. doi: 10.1016/j.jtbi.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 20.Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 21.Butchar JP, Parsa KV, Marsh CB, Tridandapani S. Negative regulators of toll-like receptor 4-mediated macrophage inflammatory response. Curr Pharm Des. 2006;12:4143–4153. doi: 10.2174/138161206778743574. [DOI] [PubMed] [Google Scholar]
- 22.Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, et al. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piao W, Song C, Chen H, Wahl LM, Fitzgerald KA, O’Neill LA, et al. Tyrosine phosphorylation of MyD88 adapter-like (Mal) is critical for signal transduction and blocked in endotoxin tolerance. J Biol Chem. 2008;283:3109–3119. doi: 10.1074/jbc.M707400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphey ED, Fang G, Varma TK, Sherwood ER. Improved bacterial clearance and decreased mortality can be induced by LPS tolerance and is not dependent upon IFN-gamma. Shock. 2007;27:289–295. doi: 10.1097/01.shk.0000245024.93740.28. [DOI] [PubMed] [Google Scholar]
- 25.Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, et al. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. 2008 doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Virca GD, Kim SY, Glaser KB, Ulevitch RJ. Lipopolysaccharide induces hyporesponsiveness to its own action in RAW 264.7 cells. J Biol Chem. 1989;264:21951–21956. [PubMed] [Google Scholar]
- 27.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 28.Vodovotz Y, Liu S, McCloskey C, Shapiro R, Green A, Billiar TR. The hepatocyte as a microbial product-responsive cell. J Endotoxin Res. 2001;7:365–373. [PubMed] [Google Scholar]
- 29.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, et al. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopf U, Ramadori G, Moller B, Galanos C. Hepatocellular clearance function of bacterial lipopolysaccharides and free lipid A in mice with endotoxic shock. Am J Emerg Med. 1984;2:13–19. doi: 10.1016/0735-6757(84)90105-0. [DOI] [PubMed] [Google Scholar]
- 31.Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology. 1995;109:1969–1976. doi: 10.1016/0016-5085(95)90765-3. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Khemlani LS, Shapiro RA, Johnson ML, Liu K, Geller DA, et al. Expression of CD14 by hepatocytes: upregulation by cytokines during endotoxemia. Infect Immun. 1998;66:5089–5098. doi: 10.1128/iai.66.11.5089-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott MJ, Billiar TR. beta 2-integrin induced p38MAPK activation is a key mediator in the CD14/TLR4/MD2-dependent uptake of LPS by hepatocytes. J Biol Chem. 2008 doi: 10.1074/jbc.M803905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- 35.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 36.Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7:1074–1081. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 37.Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N, Hazeki O. IRAK-4-dependent degradation of IRAK-1 is a negative feedback signal for TLR-mediated NF-kappaB activation. J Biochem. 2008;143:295–302. doi: 10.1093/jb/mvm234. [DOI] [PubMed] [Google Scholar]
- 38.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 39.West MA, Heagy W. Endotoxin tolerance: A review. Crit Care Med. 2002;30(1 Supp):S64–S73. [PubMed] [Google Scholar]
- 40.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 41.Kmiec Z. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol. 2001;161:III–151. doi: 10.1007/978-3-642-56553-3. [DOI] [PubMed] [Google Scholar]
- 42.Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, et al. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol. 2005;77:626–633. doi: 10.1189/jlb.0604332. [DOI] [PubMed] [Google Scholar]
- 43.Harada K, Isse K, Sato Y, Ozaki S, Nakanuma Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006;26:935–942. doi: 10.1111/j.1478-3231.2006.01325.x. [DOI] [PubMed] [Google Scholar]
- 44.Jacob AI, Goldberg PK, Bloom N, Degenshein GA, Kozinn PJ. Endotoxin and bacteria in portal blood. Gastroenterology. 1977;72:1268–1270. [PubMed] [Google Scholar]
- 45.Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, et al. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood. 1995;85:1341–1347. [PubMed] [Google Scholar]
- 46.Lehner MD, Hartung T. Endotoxin tolerance-mechanisms and beneficial effects in bacterial infection. Rev Physiol Biochem Pharmacol. 2002;144:95–141. doi: 10.1007/BFb0116586. [DOI] [PubMed] [Google Scholar]
- 47.Sato S, Takeuchi O, Fujita T, Tomizawa H, Takeda K, Akira S. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int Immunol. 2002;14:783–791. doi: 10.1093/intimm/dxf046. [DOI] [PubMed] [Google Scholar]
- 48.Cuschieri J, Billigren J, Maier RV. Endotoxin tolerance attenuates LPS-induced TLR4 mobilization to lipid rafts: a condition reversed by PKC activation. J Leukoc Biol. 2006;80:1289–1297. doi: 10.1189/jlb.0106053. [DOI] [PubMed] [Google Scholar]
- 49.Mages J, Dietrich H, Lang R. A genome-wide analysis of LPS tolerance in macrophages. Immunobiology. 2007;212:723–737. doi: 10.1016/j.imbio.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 50.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 51.Vodovotz Y, Csete M, Bartels J, Chang S, An G. Translational systems biology of inflammation. PLoS Comput Biol. 2008;4:e1000014. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An G. Introduction of an agent-based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theor Biol Med Model. 2008;5:11. doi: 10.1186/1742-4682-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphey ED, Fang G, Sherwood ER. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock. 2008;29:512–518. doi: 10.1097/shk.0b013e318150776f. [DOI] [PubMed] [Google Scholar]
- 54.Shao B, Lu M, Katz SC, Varley AW, Hardwick J, Rogers TE, et al. A host lipase detoxifies bacterial lipopolysaccharides in the liver and spleen. J Biol Chem. 2007;282:13726–13735. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 55.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 56.van’ V, van den Pangaart PS, van Zoelen MA, de Kruif M, Birjmohun RS, Stroes ES, et al. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;179:7110–7120. doi: 10.4049/jimmunol.179.10.7110. [DOI] [PubMed] [Google Scholar]
- 57.Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis. 2003;35:555–562. doi: 10.1080/00365540310015683. [DOI] [PubMed] [Google Scholar]