

Abstract
Apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1) is the redox regulator of multiple stress-inducible transcription factors, such as NF-κB, and the major 5’-endonuclease in base excision repair (BER). We utilized mice containing heterozygous gene-targeted deletion of APE1/Ref-1 (Apex+/-) to determine the impact of APE1/Ref-1 haploinsufficiency on the processing of oxidative DNA damage induced by 2-nitropropane (2-NP) in the liver tissue of mice. APE1/Ref-1 haploinsufficiency results in a significant decline in NF-κB DNA binding activity in response to oxidative stress in liver. In addition, loss of APE1/Ref-1 increases the apoptotic response to oxidative stress where a significant increase in GADD45g expression, p53 protein stability and caspase activity are observed. Oxidative stress displays a differential impact on monofunctional (UDG) and bifunctional (OGG1) DNA glycosylase initiated BER in liver of Apex+/- mice. APE1/Ref-1 haploinsufficiency results in a significant decline in the repair of oxidized bases (e.g., 8-OHdG), while removal of uracil is increased in liver nuclear extracts of mice using an in vitro BER assay. Apex+/- mice exposed to 2-NP displayed a significant decline in 3’-OH-containing single-strand breaks and an increase in aldehydic lesions in their liver DNA suggesting an accumulation of repair intermediates of failed bifunctional DNA glycosylase initiated BER.
Keywords: Apurinic/apyrimidinic endonuclease 1/redox factor-1, Redox activity, Base Excision Repair, oxidative DNA damage, NF-κB, apoptosis, Liver
Introduction
The imbalance between pro-oxidants and anti-oxidants within the cellular milieu promotes a chronic state of oxidative stress which can damage DNA and other macromolecules within the cell [1]. The steady-state accumulation of oxidative damage is thought to be an important mechanism underlying aging and age-related diseases such as cancer [2,3]. In order to maintain DNA integrity, cells employ elaborate DNA repair mechanisms of which base excision repair (BER) is the most versatile and the pathway of choice for repairing oxidative damage, single-strand breaks, and other small, non helix-distorting DNA damage [4-6].
Apurinic/apyrimidinic (AP) endonuclease 1 (APE1) was originally characterized as an endonuclease that cleaves the backbone of double-stranded DNA containing AP sites [7,8]. Subsequently, APE1 was shown to possess 3’-phosphodiesterase, 3’-phosphatase, and 3’→5’ exonuclease activities [9]. APE1 was also independently characterized as redox factor-1 (Ref-1), a redox activator of cellular transcription factors [10,11]. APE1/Ref-1 participates in cellular signaling via activation of multiple transcription factors involved in the cellular stress response, such as NF-κB [12]. Studies of NF-κB DNA binding indicate a mechanism that is redox regulated by and dependent upon APE1/Ref-1. For example, APE1/Ref-1 was shown to enhance the DNA binding activity of NF-κB in vitro as well as NF-κB-dependent transcriptional activation in vivo [13]. Furthermore, deletion of the redox-sensitive domain of APE1/Ref-1 significantly inhibited TNF-induced NF-κB activation [14]. Loss of APE1/Ref-1 also resulted in decreased NF-κB DNA binding and transcriptional activation, in addition to increased susceptibility to TNF-induced apoptosis [15,16]. These findings establish APE1/Ref-1 as an essential upstream signaling molecule regulating NF-κB.
Research focused on understanding the role of APE1/Ref-1 in the BER response to oxidative stress provides insight into its multifunctional activity. Initially, BER was believed to be a simplistic linear pathway involving damage recognition and removal, followed by base insertion and nick-sealing activity, requiring only four enzymatic reactions [17]. However, recent studies have indicated that BER is a dynamic and environmentally responsive DNA repair pathway [18, 19], with individual BER enzymes being induced by oxidizing agents [20, 21]. Our laboratory has demonstrated that both BER activity and DNA polymerase β (β-pol) levels increase in response to the oxidative stress [22]. Research has also shown that APE1/Ref-1 expression is inducible by oxidative stress [23,24], while its down regulation increased sensitivity to DNA damaging agents [25,26] and its overexpression protected against oxidative stress-induced genotoxicity [27]. Recently, Fung and Demple [28] showed that APE1/Ref-1 repair activity is essential for cellular viability and indicate that APE1/Ref-1 redox activity may be dispensable. However, Izumi et al. [29] and Vasko et al. [30] present data supporting the notion that both functions of APE1/Ref-1, repair and redox, are essential for cell survival.
The objective of this study is to determine the functional importance of APE1/Ref-1 in the repair of oxidative damage in vivo. We provide evidence that in response to in vivo exposure to oxidative stress, an increase in BER activity, β-pol, and APE1/Ref-1 protein levels are observed. We also present data demonstrating increased activation of NF-kB in response to oxidative stress in vivo. To determine whether BER activity in response to oxidative stress is affected by reduced APE1/Ref-1, we utilized a mouse model of APE1/Ref-1 haploinsufficiency [Apex+/-] previously characterized by our lab [31]. We find that oxidative stress displays a differential impact on monofunctional (UDG) and bifunctional (OGG1) DNA glycosylase initiated BER in Apex+/- mice. Oxidative stress results in a significant increase in UDG initiated BER activity, but a significant decline in the repair of oxidized bases (8-OHdG). We also observed reduced DNA binding activity of NF-kB in Apex+/- mice exposed to oxidative stress establishing a significant role for APE1/Ref-1 redox function in the activation of NF-κB in response to oxidative stress in vivo. Our data has relevant translational implications since APE1/Ref-1 variants have been identified in the human population [32], and variants in BER have been associated with increased cancer risk [33,34].
MATERIALS AND METHODS
Animals
The experiments were performed in young (3-6 month), wild-type and APE1/Ref-1 heterozygous heterozygous (Apex+/-) male C57BL/6 specific pathogen-free mice in accordance with NIH guidelines for the use and care of laboratory animals. Mice were backcrossed to C57BL/6 background. The Wayne State University Animal Investigation Committee approved the animal protocol. Mice were maintained on a 12-hr light/dark cycle and fed standard mouse chow and water ad libitum. Mice were anesthetized in a CO2 chamber and sacrificed by cervical dislocation. Harvested liver was flash frozen in liquid nitrogen and stored at -70°C for further analysis.
DNA damage induction
Experimental mice were i.p. injected with 100 mg/kg body weight 2-nitropropane (2-NP; Aldrich Chemical Company, Chem. Abstr. Serv. Reg. No. [79-46-9]) dissolved in olive oil. Control mice were injected with olive oil vehicle. Mice were sacrificed after 24-hr. The dose and exposure time were based on previous studies characterizing the effect of 2-NP on DNA damage and repair induction [22].
Gene expression profiling
The mRNA expression level of APE1/Ref-1 was quantified using a real-time PCR-based pathway focused on gene expression profiling of mouse DNA damage. Total RNA was extracted from liver tissue of control and 2-NP-treated mice using TRIzol® Reagent [GibcoBRL, Rockville, MD]. First strand cDNA was synthesized from 1 μg RNA using random primers and purified using QIAquick PCR purification kit (Qiagen, Valencia, California). Gene-profiling was analyzed using Realtime PCR array (SuperArray, Frederick, MD) according to manufacturer’s instructions. Briefly, a cocktail of cDNA samples was prepared using a supplied master mix and aliquoted into each well of a 96-well plate containing primer pairs specific for 84 genes involved in the DNA damage pathway, including 5 housekeeping genes. Among the 84 genes analyzed, changes in the expression of genes related to BER pathway were confirmed using real time PCR and reported herein. Expression of β-pol, UNG, OGG1 and GADD45g were also quantified using real time PCR with RNA extracted from liver tissue of control and 2-NP treated mice. UNG primers were designed to detect both UNG1 and UNG2 message. Primer sequences used for β-pol, GADD45g, APE-1/Ref-1, UNG, OGGI, GAPDH and β-actin transcripts are detailed in Table 1. External standards for all the genes were prepared by subcloning the amplicons, synthesized using the primers listed in Table 1, into PGEM-T easy vector. The vectors were linearized using Ecor1 to make the standard curves. All gene transcripts were normalized to both GAPDH and β-actin.
Table 1.
Primer Sequences for Real-Time PCR
| Gene | Sense Primer Sequence | Antisense Primer Sequence |
|---|---|---|
| Ape1/Ref-1 | 5’-ATGAAGAAATTGACCTCCGTAACC-3’ | 5’-GTGTAAGCGTAAGCAGTGTTG-3’ |
| β-actin | 5’-ACCAACTGGGACGACATGGAGAAG-3’ | 5’-TACGACCAGAGGCATACAGGGACT-3’ |
| β-pol | 5’-CTGGAAAAGGGCTTCACAATCAATG-3’ | 5’-GCGCCACTGGATGTAATCAAAAATG-3’ |
| GADD45g | 5’-AGTTCCGGAAAGCACAGCCAGGATG-3’ | 5’-GCCAGCACGCAAAAGGTCACATTGT-3’ |
| GAPDH | 5’-GCACAGTCAAGGCCGAGA-3’ | 5’-TACGACCAGAGGCATACAGGGACT-3’ |
| OGG1 | 5’-CGGCTGGCATCCTAAGACATC-3’ | 5’-AACAGGCTTGGTTGGCGAAGG-3’ |
| UNG | 5’-AAGAGCTGTCTACAGACATCGA-3’ | 5’-ATAAGAGCCCCAGAGGAGGAA-3’ |
Nuclear protein isolation
Nuclear proteins were isolated as previously described [31]. Briefly, nuclear extracts were isolated using transfactor extraction kit (Clontech, Mountain View, CA). The kit uses a hypotonic buffer to lyse the cell allowing the removal of cytosolic fractions and is followed by the extraction of nuclear proteins by a high salt buffer. All samples and tubes were handled and chilled on ice, and all solutions were made fresh according to manufacturer’s protocol. Low molecular weight contaminants were removed from extracts by dialysis in 1L dialysis buffer (20 mM Tris-HCl, pH 8.0; 100 mM KCl; 10 mM NaS2O5; 0.1 mM DTT; 0.1 mM PMSF; 1 μg/ml Pepstatin A) for 4-hr at 4°C using Slide-A-Lyzer® mini-dialysis units (Pierce Biotechnology, Rockford, IL) with a molecular weight cut off of 3.5kDa. Dialyzed extracts were aliquoted and flash frozen in liquid nitrogen and stored at -70°C for subsequent analyses. Protein concentrations were determined according to Bradford using Protein Assay Kit I (Bio-Rad, Hercules, CA).
Protein expression analysis
Western blot analysis was performed using 200 μg nuclear protein as previously described [31]. Upon completion of SDS-PAGE, the region containing the protein(s) of interest was excised and prepared for western blot analysis while the remaining portion of the gel was stained with GelCode®Blue Stain Reagent (Pierce Biotechnology, Rockford, IL) to ensure equal protein loading. Manufacturer recommended dilutions of anti-sera developed against APE1/Ref-1 (Clone 13B8E5C2, Novus Biologicals, Littleton, CO), β-pol (Ab-1 Clone 18S, NeoMarkers, Fremont, CA) and p53(Pab 240, Santa Cruz Biotechnology, Delaware, CA) were used to detect proteins of interest followed by incubation with HRP-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). As an internal control to ensure equal protein transfer, membranes were reprobed with anti-Lamin B antibody (Santa Cruz Biotechnology, Santa Cruz, CA) The bands were visualized and quantified using a ChemiImager™ System (AlphaInnotech, San Leandro, CA) after incubation in SuperSignal® West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL). Data are expressed as the integrated density value (I.D.V.) of the band per μg of protein loaded.
Electrophoretic mobility shift assay (EMSA)
A non-radioisotopic EMSA was used to determine the NF-κB and CREB DNA binding activity of nuclear extracts isolated from liver tissue of control and 2-NP-treated Apex+/+ and Apex+/- mice according to the manufacturer’s protocol (LightShift® Chemiluminescent EMSA kit, Pierce Biotechnology, Rockford, IL). Briefly, 40 fmol biotin-end-labeled DNA containing an NF-κB consensus sequence (5’ AGTTGAGGGGACTTTCCCAGG 3’BTN from Panomics, Redwood City, CA) and CRE sequence with β-pol flanking region (-36..AGCCTGGCGCGTGACGTCAC CGCGCTGCGC..-7) was incubated with 10 μg nuclear extract in a 20 μl reaction mixture containing 1X binding buffer (100 mM Tris, 500 mM KCl, 10 mM DTT; pH 7.5), 2.5% glycerol, 5 mM MgCl2, 50 ng/μl poly (dI-dC), and 0.05% NP-40. Negative controls (all components except nuclear extract) were included in all experiments. In competitive assays, 100X molar excess of unlabeled oligonucleotide was added to the reaction mixture. Samples were incubated for 20 min at room temperature then resolved on a 6% non-denaturing polyacrylamide gel in 0.5X TBE buffer. After electrophoresis, samples were transferred from the gel to a positively charged nylon membrane and cross-linked. Biotin-labeled protein/DNA complexes were detected by chemiluminescence and quantified using a ChemiImager™ System (AlphaInnotech, San Leandro, CA). Data are expressed as the integrated density value (I.D.V.) of the band per μg of protein loaded.
DNA base excision repair activity assay
The G:U mismatch repair assay is developed to measure monofunctional glycosylase-initiated base excision repair (BER) activity. Purified radio-end-labeled 30-bp oligonucleotides (upper strand: 5’-ATATACCGCGGUCGGCCGATCAAGCTTATTdd-3’; lower strand: 3’ ddTATATGGCGCCG GCCGGCTAGTTCGAATAA-5’) containing a G:U mismatch and a Hpa II restriction site (CCGG) were incubated in a BER reaction mixture containing 50 μg nuclear protein as previously described [31]. This repair assay uses a 30bp long oligonucleotide with G:U mismatch as no significant difference was seen in the catalytic efficiency of the in vitro assay when a plasmid or oligonucleotide was used as a substrate [35]. Repair of the G:U mismatch to a correct G:C base pair was determined via treatment of the duplex oligonucleotide with 20U of HpaII (Promega, Madison, WI) for 1-hr at 37°C and analysis by electrophoresis on a 20% denaturing 19:1 acrylamide/bis-acrylamide gel (SequaGel® Sequencing System, National Diagnostics, Atlanta, GA). Repair activity (presence of a 16-mer band) was visualized and quantified using a Molecular Imager® System (Bio-Rad, Hercules, CA) by calculating the ratio of the 16-mer product with the 30-mer substrate (product/substrate). Data are expressed as machine counts per μg of protein.
The 8-OH G:C repair assay is utilized to measure bifunctional glycosylase-initiated BER activity. Fluorescein-end-labeled 30-bp oligonucleotides (upper strand: 5’-ATATACCGC GGGCG*GCCGATCAAGCTTATTdd-3’; lower strand:3’ ddTATATGGCGCCGGCCGGCTAGTT CGAATAA-5’,* G=8-hydroxydeoxyguanine) containing a Hpa II restriction site (CCGG) were incubated in a BER reaction mixture containing 50 μg nuclear protein as described previously [31]. The repair activity was determined as described above.
DNA damage analysis: Random oligonucleotide primed synthesis [ROPS] Assay
The relative number of single-strand breaks containing a 3’-OH group was quantified using a Klenow (exo-) incorporation ROPS assay as previously described [31,36]. This assay is based on the ability of Klenow to initiate DNA synthesis from 3’-OH ends of single-strand DNA. Incorporation of α (32P) dCTP was quantified using a Packard scintillation counter. DNA for the ROPS assay was isolated using Qiagen (Valencia, CA) gravity tip columns as described in the manufacturer’s protocol. This method generates large fragments of DNA (up to 150-kb) while minimizing shearing.
ASB Assay
Detection of aldehydic DNA lesions (ADLs) was carried out by ASB as described previously [37] with slight modifications. DNA [8ug] from liver tissue was incubated in 30ul of phosphate-buffered saline with 2 mM aldehyde reactive probe (Dojindo Laboratories, Kumamoto, Japan) at 37 °C for 10 min. DNA was precipitated by the cold ethanol method and resuspended in 1X TE buffer overnight at 4 °C. DNA was heat-denatured at 100 °C for 10 min, quickly chilled on ice, and mixed with an equal volume of 2 M ammonium acetate. The nitrocellulose membrane (Schleicher & Schuell) was prewet in deionized water and washed for 10 min in 1 mM ammonium acetate. DNA was immobilized on the pretreated nitrocellulose membrane using an Invitrogen filtration manifold system. The membrane was washed in 5X SSC for 15 min at 37 °C and then baked under vacuum at 80 °C for 30 min. The dried membrane was incubated in a hybridization buffer (20 mM Tris, pH 7.5, 0.1 M NaCl, 1 mM EDTA, 0.5% (w/v) casein, 0.25% (w/v) bovine serum albumin, 0.1% (v/v) Tween 20) for 30 min at room temperature. The membrane was then incubated in fresh hybridization buffer containing 100 ul of streptavidin-conjugated horseradish peroxidase (BioGenex, San Ramon, CA) at room temperature for 45 min. Following incubation in horseradish peroxidase, the membrane was washed three times for 5 min each at 37 °C in TBS, pH 7.5 (0.26 M NaCl, 1 mM EDTA, 20 mM Tris, pH 7.5, 0.1% Tween 20). Membrane was incubated in ECL (Pierce) for 5 min at room temperature and visualized using a ChemiImager™ system (AlphaInnotech, San Leandro, CA).
Caspase Activity
Caspase-3 activity was measured using Enzchek Caspase-3 Assay kit No.1 (Molecular probes Eugene, OR). Briefly, Liver tissues were homogenized, and cytosolic extracts were isolated using transfactor extraction kit (Clonetech, Mountain View, CA). The extracts (250 μg protein) were incubated for 2hr at room temperature in the working solution (25mM PIPES, pH 7.4, 5mM EDTA and 0.25% CHAPS) containing synthetic Caspase-3 substrate, Z-DEVD-AMC. Caspase mediated proteolytic cleavage of the substrate yields a bright blue-fluorescent product. An additional control assay was performed using reversible aldehyde inhibitor Ac-DEVD-CHO to confirm that the fluorescence observed in the sample assay was due to caspase activity. The fluorescence was measured using a fluorescence microplate reader (Genios plus, Tecan) at excitation:342nm, emission:441nm. The caspase activity was determined using an AMC (7-amino-4-methylcoumarin) standard curve (0-100uM), and reported as fluorescence per microgram of protein.
Statistical analysis
Statistical significance between means was determined using ANOVA followed by the Fisher’s least significant difference test where appropriate [38]. P-values less than 0.05 were considered statistically significant.
RESULTS
Analysis of the liver tissue for APE1/Ref-1 expression and redox activity in response to 2-Nitropropane
Using the hepatocarcinogen 2-nitropropane (2-NP), we analyzed the impact of oxidative stress on the expression and redox activity of APE1/Ref-1 in vivo. Metabolism of 2-NP in liver generates reactive oxygen species (ROS) and promotes oxidative DNA damage, e.g., 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-OHdG), both of which are believed to be one of the causative factors behind 2-NP-induced carcinogenesis [39]. 2-NP has also been shown to be genotoxic in vitro, inducing mutations in bacteria and unscheduled DNA synthesis in hepatocytes [40]. Our laboratory has demonstrated that 2-NP (100mg/kg body weight) induces levels of 8-OHdG [by 4-5 fold, p< 0.01], followed by a concomitant increase in BER activity and β-pol protein levels [50%, p< 0.01] in liver tissues of mice and rats [22]. In addition, 2-NP is also shown to increase mutation frequency in liver tissues of these animals [22]. Using RT-PCR and Western blot analyses, we analyzed the expression of APE1/Ref-1 in response to 2-NP treatment. Our data show that 2-NP induces APE1/Ref-1 mRNA (Fig. 1A) and protein levels significantly (Fig. 1B) in the liver. Thus, we confirmed previous reports from cultured cells e.g. HeLa S3 tumor cells and WI 38 primary fibroblast [24] where expression of APE1/Ref-1 was shown to be inducible by oxidative stress and extended this observation to in vivo study establishing APE1 as a stress response gene,. Consequently, we examined the impact of 2-NP on DNA binding activity of NF-κB (Fig. 1C). As expected, NF-κB DNA binding activity was significantly increased in response to oxidative stress in liver nuclear extracts. Furthermore, increase in NF-κB DNA binding activity was correlated with an increase in expression/activity of APE1/Ref-1, the redox activator of NF-κB. To determine the role of APE1/Ref-1 and its redox function in this process, we evaluated the activation of NF-κB DNA binding activity in response to 2-NP in mice heterozygous for the APE1/Ref-1 gene, i.e., Apex+/- mice.
Fig. 1.
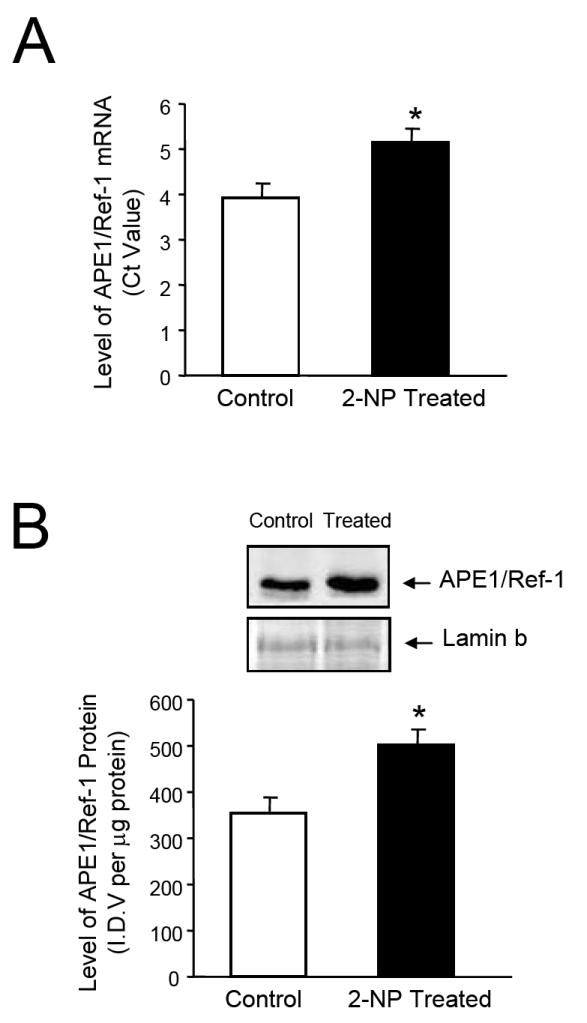
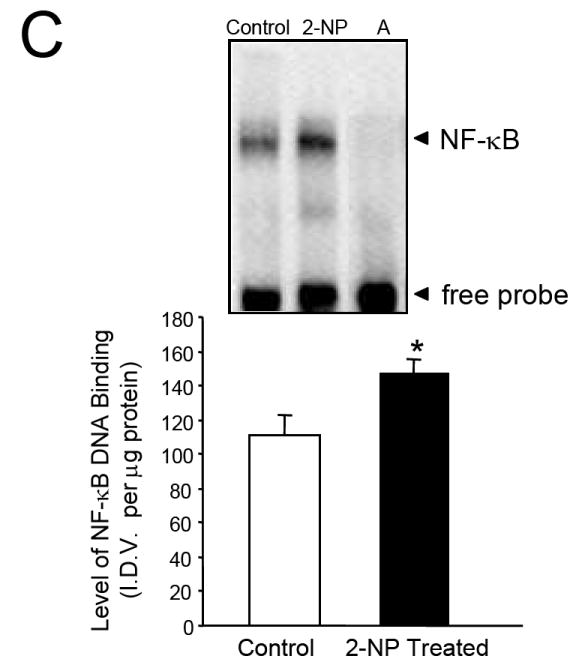
Expression and activity of APE1/Ref-1 in response to 2-NP in vivo. (A) APE1/Ref-1 mRNA expression was quantified using a real-time PCR and the data were normalized using GAPDH. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group (B) The level of the 37-kDa APE1/Ref-1 protein in 200 μg of liver nuclear extract as determined by western blot analysis. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group (C) The level of APE1/Ref-1 redox-activation of NF-κB in 10 μg of live nuclear extract as determined using EMSA. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate independent experiments. (I.D.V.): integrated density value corresponding to the level of APE1/Ref-1 protein as quantified by an AlphaInnotech ChemiImager™; Lane A: nuclear extracts incubated in the presence of 100X molar excess of unlabeled NF-kB consensus DNA to confirm binding specificity; (*): value significantly different from control, wild-type mice at P < 0.05; (**): value significantly different from control, Apex+/- mice at P < 0.05. Lamin B protein level served as nuclear protein loading control. The picture depicts the representative sample from each group.
Analysis of APE1/Ref-1 expression and redox activity in response to oxidative DNA damage in liver nuclear extracts of Apex+/- mice
We determined whether reduced level of the APE1/Ref-1 gene in Apex+/- mice would impact the activation of NF-κB in response to oxidative damage generated by 2-NP treatment. APE1/Ref-1 protein level was significantly reduced in Apex+/- mice (Fig. 2), in addition, NF-κB DNA binding activity was reduced in Apex+/- mice suggesting that the redox activation of NF-κB and its consequent DNA binding activity is significantly reduced when the expression of APE1/Ref-1 is compromised (Fig. 3). The specificity of the NF-κB DNA binding activity in our assay is established as the shifted band is completely abolished using an oligonucleotide containing the NF-κB unlabeled consensus sequence (Fig. 3, Lane A). In agreement with our previous data [31], we confirmed that APE1/Ref-1 heterozygosity promoted haploinsufficiency with respect to APE1/Ref-1 gene expression. In this study we established that wild-type mice exposed to 2-NP showed a significant increase in APE1/Ref-1 protein and redox activation of NF-kB in vivo, while mice haploinsufficient for APE1/Ref-1 showed similar response to oxidative stress via their intact allele, the ultimate level of induction was significantly lower in heterozygous mice reducing its redox capacity (Figs. 2 and 3). This suggests that the induction in APE1/Ref-1 expression and increased NF-κB activation in response to oxidative stress are dependent on APE1/Ref-1 genotype. Based on these data, it appears that the level of APE1/Ref-1 protein is instrumental in redox activation of NF-κB and its DNA binding activity in vivo.
Fig. 2.
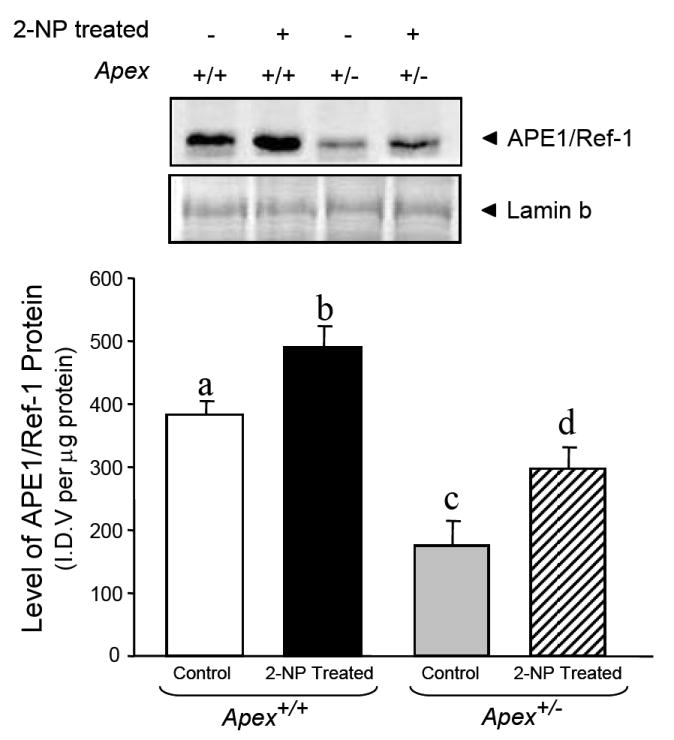
Effect of 2-NP on APE1/Ref-1 expression levels in Apex+/- mice. The level of the 37-kDa APE1/Ref-1 protein in 200 μg of liver nuclear extract was determined by western blot analysis. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate independent experiments. (SDS-PAGE]: the level of APE1/Ref-1 protein was normalized based on the amount of protein loaded on each gel. (I.D.V.): integrated density value corresponding to the level of APE1/Ref-1 protein as quantified by an AlphaInnotech ChemiImager™; Values with different letter superscripts indicate significant differences at P < 0.05. Lamin B protein level served as nuclear protein loading control. The picture depicts the representative sample from each group.
Fig. 3.
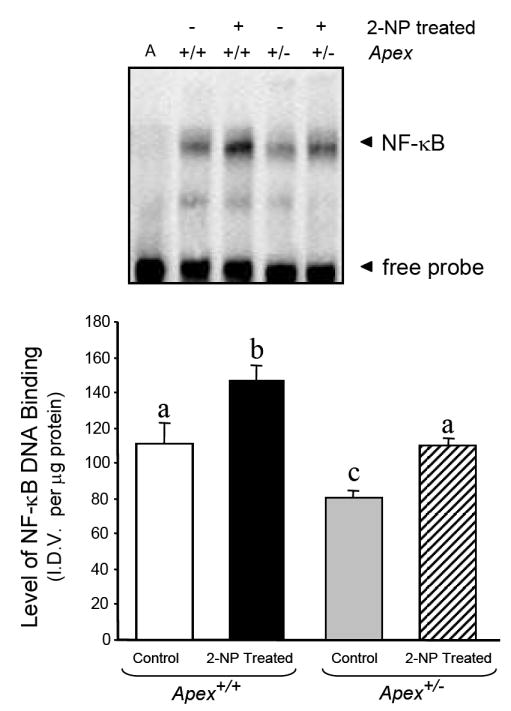
Effect of 2-NP on APE1/Ref-1 redox activation of NF-κB in Apex+/- mice. The level of NF-κB DNA binding in 10 μg of liver nuclear extract was determined using EMSA. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate independent experiments. (I.D.V.): integrated density value corresponding to the level of NF-κB DNA binding as quantified by an AlphaInnotech ChemiImager™; Lane A: nuclear extracts incubated in the presence of 100X molar excess of unlabeled NF-κB consensus DNA to confirm binding specificity; Values with different letter superscripts indicate significant differences at P < 0.05. The picture depicts the representative sample from each group.
Analysis of DNA damage intermediates in the liver tissue of Apex+/- mice
Data from our laboratory and other labs indicate that down regulation of APE1/Ref-1 promotes a damage hypersensitive phenotype [26, 31]. Thus, it was essential to determine the impact of APE1/Ref-1 haploinsufficiency on the level of DNA damage. We have previously reported the levels of AP sites, single-strand breaks and Aldehydic lesions in DNA isolated from liver of young Apex+/- mice under normal condition. Interestingly, no significant difference in DNA damage in Apex+/- mice as compared to wildtype counterparts was observed [36]. In this study, wild-type mice exposed to oxidative stress displayed a 2-fold induction in the level of 3’-OH-containing single-strand breaks (Fig. 4a) and no significant increase in the level of Aldehydic lesions (Fig. 4b). Interestingly, the level of 3’-OH-containing single-strand breaks was significantly lower in Apex+/- mice exposed to similar treatment as compared to their wildtype counterparts (Fig. 4a). However, the level of aldehydic lesions was significantly higher in Apex+/- mice exposed to oxidative stress as compared to wildtype animals (Fig 4b). We suggest that the processing of oxidized bases by a bi-functional DNA glycosylase such as OGG1 (8-oxoguanine DNA glycosylase) could result in generation of aldehydic blocking lesions at 3’ end. Inability to process these 3’ blocking groups in the absence of the 3’-phophoesterase activity of Apex in Apex+/- mice [41], could result in lower detection of endonuclease-mediated single-strand breaks in the heterozygous animal.
Fig. 4.

DNA Damage analysis in liver DNA of Apex+/- mice injected with 2-NP. (A) The level of 3’-OH single-strand breaks as determined by the ROPS assay in liver of wild-type (Apex+/+) and Apex+/- mice treated with 100 mg/kg body weight 2-NP. (B) The level of Aldehydic lesions as determined by the ASB assay in liver of wild-type (Apex+/+) and Apex+/- mice treated with 100 mg/kg body weight 2-NP.Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate identical experiments. (C.P.M.): machine counts per minute corresponding to the level of α-[32P]dCTP incorporation as quantified by a Packard scintillation counter; (I.D.V.): integrated density value corresponding to the level of DNA as quantified by an AlphaInnotech ChemiImager™; Values with different letter superscripts indicate significant differences at P < 0.05.
Nevertheless, the decrease in the detection of 3’-OH-containing single-strand breaks in Apex+/- mice could also arise from an increase in β-pol-dependent BER capacity and rapid removal of oxidized bases and their repair-intermediates. Based on the findings that APE1 is not the rate limiting enzyme in Uracil initiated BER pathway [31, 41] and emergence of AP endonuclease-independent BER pathway for repair of oxidized bases [42], upregulation in BER pathway could be the plausible mechanism for decline in 3’-OH-containing single strand breaks in Apex+/- mice exposed to oxidative stress. Accordingly, it has become important to evaluate BER capacity using a BER assay and determine the expression of its rate limiting enzyme, β-pol, in Apex+/- mice in response to oxidative stress. Understanding this mechanism is important as these data potentially sheds light on the means by which APE1/Ref-1 haploinsufficiency alters the DNA damage signal in Apex+/- mice.
Analysis of the BER response to oxidative DNA damage in the liver nuclear extracts of Apex+/- mice
We examined whether loss of APE1/Ref-1 affects the BER activity in liver in response to 2-NP-induced oxidative DNA damage in vivo. Using a G:U mismatch repair assay where APE1 endonuclease activity is essential, we analyzed the in vitro BER activity in Apex+/- mice and their wild-type counterparts in response to 2-NP treatment. For this assay we utilized a 30bp long oligonucleotide as no significant difference has been observed in the catalytic efficiency of the G:U mismatch assay when a plasmid or small oligonucleotide were used as substrate [35]. As expected, BER activity was significantly increased in response to oxidative stress in wildtype mice (Fig. 5A) with a concomitant increase in β-pol protein (Fig. 5B). We confirmed previous reports that mice haploinsufficient for APE1/Ref-1 display reduced in vitro BER activity (Fig. 5A) and β-pol expression (Fig. 5B). However, while the BER activity significantly declined in Apex+/- mice, much to our surprise this activity was significantly higher in Apex+/- mice treated with 2-NP as compared to its wildtype littermates. In other words, APE1/Ref-1 haploinsufficiency resulted in a significant increase in BER activity in response to oxidative stress. In addition, we provide evidence that 2-NP treatment resulted in a significantly higher induction in β-pol expression/protein stability as compared to wildtype animals (Fig. 5B).
Fig. 5.

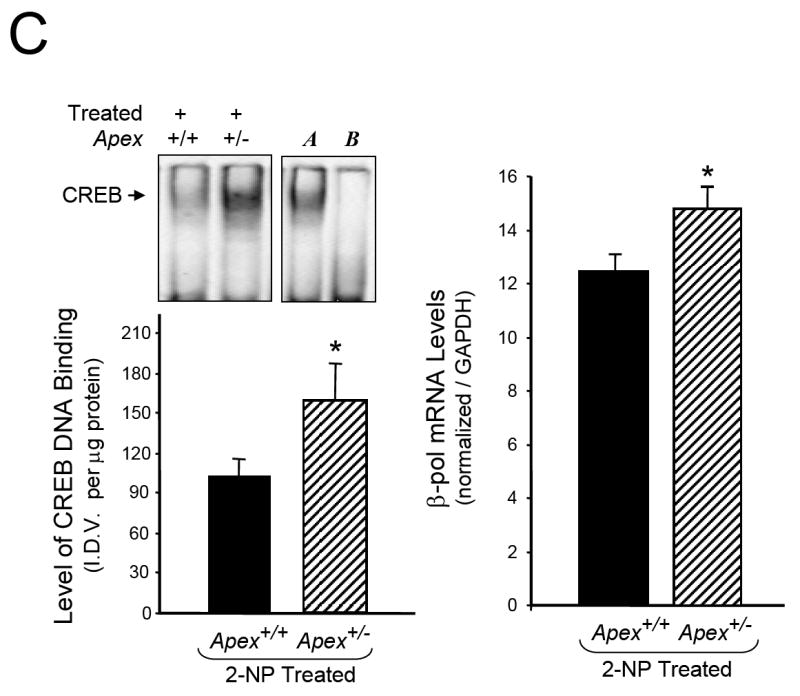
Effect of APE1/Ref-1 haploinsufficiency and 2-NP on G:U mismatch BER, DNA polymerase β expression and CREB DNA binding activity. (A) The in vitro G:U mismatch BER was conducted using nuclear extracts from liver of control and 2-NP treated Apex+/+ and Apex+/- mice. Base excision repair (BER) reaction products were resolved on a sequencing gel and visualized by autoradiography. Repair activity is visualized by the appearance of a 16b fragment. The relative level of BER was quantified using a Bio-Rad Molecular Imager® System. The data were normalized based on the amount of protein used in each reaction and expressed as machine counts per μg of protein. (B) The level of the 39-kDa β-pol protein in 200 μg of liver nuclear extract was determined by western blot analysis. (SDS-PAGE): the level of β-pol protein was normalized based on the amount of protein loaded on each gel. (C) The level of CREB DNA binding in 10 μg of liver nuclear extract was determined using EMSA and β-pol mRNA expression level as quantified using a real-time PCR and normalized with GAPDH Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate identical experiments. (M): molecular weight standard; (-): negative control, G:U mismatch oligonucleotide incubated in the absence of nuclear extract and treated with HpaII restriction endonuclease; (+): positive control, G:U mismatch oligonucleotide incubated with recombinant β-pol and treated with HpaII restriction endonuclease; (G:C): positive control, G:C oligonucleotide incubated with nuclear extract and treated with HpaII restriction endonuclease; (I.D.V.): integrated density value corresponding to the level of β-pol protein and the level of CREB DNA binding as quantified by an AlphaInnotech ChemiImager™; Lane A & B: nuclear extracts incubated in the presence of 100X molar excess of mutated and unlabeled CRE sequence with β-pol flanking region respectively to confirm binding specificity; Lamin B protein level served as nuclear protein loading control. Values with different letter superscripts indicate significant differences at P < 0.05. (*): value significantly different from wildtype (Apex+/+) mice treated with 2-NP at P < 0.05. The picture depicts the representative sample from each group.
Regulation of eukaryotic gene expression is controlled, in part, though interaction of cis-elements within promoters of the genes with their associated DNA-binding factors. One of the candidates responsible for triggering induction of a specific gene by oxidative stress is the cAMP responsive element (CRE). The CRE sequence is present within the promoter of both the APE1/Ref-1 as well as the β-pol genes. Interestingly, mutational inactivation of CRE sequence in the APE1/Ref-1 promoter completely eliminates APE promoter activity in response to oxidative stress in cells [43]. In addition, the CRE sequence in human β-pol promoter has been shown to play a key role in the basal expression as well as induction of β-pol in response to DNA-alkylating agents [44, 45]. Based on these findings, we propose that attenuation in the redox function of APE1/Ref-1 in Apex+/- mice impacts the handling of oxidative stress perhaps via alterations in activation of factors such as NF-κB and consequently results in increased CREB binding activity that impacts the expression of the β-pol gene. In support of this notion, we determined whether APE1/Ref-1 haploinsufficiency alters ATF/CREB binding activity to CRE sequence within the β-pol promoter. In gel retardation experiments, the protein binding capacity of the CRE element was significantly increased in Apex+/- mice in response to oxidative stress as compared to their wildtype littermates (Fig. 5C). The shifted band is completely abolished using an oligonucleotide containing the consensus CRE sequence as competitor (Fig 5C, Lane B) while using an oligonucleotide with mutational inactivation of the CRE site does not compete with the shifted band (Fig 5C, Lane A). Interestingly, the increase in CREB/CRE binding activity within the β-pol promoter in Apex+/- mice corresponds to an increase in β-pol mRNA levels in heterozygous animals (Fig 5C). Based on these results, it appears that alteration in the redox function of APE1/Ref-1 enzyme impacts the handling of oxidative stress and consequently results in an oxidative DNA damage repair response, e.g., increased expression of β-pol gene.
Further, to directly determine the role of APE1/Ref-1 in the repair of oxidized bases, we used a 8-OHdG:C repair assay where APE1 3’-phosphoesterase activity are reported to be rate limiting [41]. As expected, the wildtype animals displayed an increase in 8-OHdG:C repair activity when exposed to 2-NP (Fig. 6A). However, while the Apex+/- mice showed similar response to 2-NP, the overall repair activity remained considerably lower for Apex+/- mice when compared to their wildtype counterparts (Fig. 6A). Additionally, to confirm the role of APE1/Ref-1 in repairing the oxidized base, the 8-OHdG:C BER assay was performed in the presence of APE1/Ref-1 purified protein. When purified APE1/Ref-1 (Novus biologicals, Littleton, CO) was added to the reaction mixture, the reduced BER activity was restored in control and 2-NP treated Apex+/- mice, while the wildtype mice displayed no noticeable differences (Fig. 6B). Taken together, these results confirm that APE1/Ref-1 plays an important role in the repair of oxidized bases (e.g., 8-OHdG) and that APE1/Ref-1 haploinsufficiency results in a significant decline in this repair activity, while monofunctional DNA glycosylase dependent BER activity is increased in these mice.
Fig. 6.

Effect of APE1/Ref-1 haploinsufficiency and 2-NP on 8 OH G:C BER and the consequence of hApe1 enrichment on the repair capacity. (A) The in vitro 8-OH G:C BER was conducted using nuclear extracts from liver of control and 2-NP treated Apex+/+ and Apex+/- mice in the presence of 1.6 U of ogg1 enzyme (New England Biolab, MA). Base excision repair (BER) reaction products were resolved on a sequencing gel. Repair activity is visualized by the appearance of a 16b fragment. The relative level of BER was quantified using an Bio-rad ChemiImager™. The data were normalized based on the amount of protein used in each reaction and expressed as machine counts per μg of protein. (B) The in vitro 8-OH G:C BER was conducted with human Ape1/Ref-1 (hApe1) enrichment. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate identical experiments. (M): molecular weight standard; (-): negative control, G:U mismatch oligonucleotide incubated in the absence of nuclear extract and treated with HpaII restriction endonuclease; Values with different letter superscripts indicate significant differences at P < 0.05. The picture depicts the representative sample from each group.
Analysis of Glycosylases in the liver of Apex+/- mice in response to oxidative DNA damage
To further confirm the impact of APE1/Ref-1 heterozygosity on the BER pathway, we determined the expression of two glycosylases responsible for initiating the repair of oxidative damage. Using Real time PCR we determined the expression of OGGI, a bi-functional glycosylase and UNG (Uracil DNA glycosylase), a monofunctional glycosylase. Both these glycosylases are involved in the removal of oxidized bases from DNA. There are two types of 8-Oxoguanine DNA glycosylase; OGG1 and OGG2 [46]. Hazra et al, [46] have demonstrated that both these enzymes are involved in processing oxidized bases, with OGG1 being involved in the removal of 8-OHdG paired with cytosine, thymine and guanine and OGG2 in responsible for the removal of 8-OHdG paired with Adenine. UNG has two isoforms arising from alternative splicing; UNG1 and UNG2 [47, 48]. UNG 1 is a mitochondrial enzyme whereas UNG2 is found in the nucleus [47, 48]. In addition to initiating uracil removal from DNA, UNG is also involved in the repair of Uracil derivatives like isodialuric acid, 5-hydroxyuracil and alloxan, derived from oxidative damage to cytosine residues in the DNA [49], thus playing an important role in repair of oxidative damage. Interestingly, UNG expression follows the profile of β-pol and the G:U mismatch BER activity. In other words, mice treated with 2-NP showed significant increase in UNG expression with maximum induction seen in Apex+/- mice (Fig. 7A). Using a UDG activity assay as described by our laboratory [36], we determined the impact of oxidative stress on UNG activity in liver tissue of mice treated with 2-NP. Interestingly, oxidative stress resulted in a significant increase in UNG activity following the expression of UNG gene and BER activity (data not shown). However, OGG1 mRNA levels did not change significantly among the control and experimental groups (Fig. 7B). Interestingly, addition of purified human OGG1 (Novus biologicals, Littleton, CO) protein to BER reaction mixture did not increase OGG1 initiated BER activity in 2-NP treated Apex+/- mice (data not shown) suggesting that APE1/Ref-1 activity is essential for bifunctional DNA glycosylase initiated BER. Thus, upregulation in UNG expression and simultaneous increase in β-pol protein explain the increase in G:U mismatch BER activity in 2-NP treated Apex+/- mice (Fig. 5A). Conversely, lack of induction of OGG1 initiated BER in the liver tissue of Apex+/- mice exposed to 2-NP (Fig. 6A) are expected to increase oxidative DNA damage that drives apoptosis.
Fig. 7.

Effect of 2-NP on UNG and OGG1 expression in Apex+/- mice. (A) UNG mRNA expression level in control and 2-NP treated Apex+/+ and Apex+/- mice as quantified using real-time PCR and normalized against GAPDH. (B) OGG1 mRNA expression level in control and 2-NP treated Apex+/+ and Apex+/- mice as quantified using real-time PCR and normalized against GAPDH. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate identical experiments; Values with different letter superscripts indicate significant differences at P < 0.05.
Analysis of Apoptosis in the liver of Apex+/- mice in response to oxidative stress
To characterize the effect of Ape1/Ref-1 heterozygosity on cell cycle arrest and apoptosis during oxidative stress, we analyzed three factors associated with apoptosis; GADD45g (Growth Arrest and DNA-damage-inducible, gamma), p53 and Caspase-3. GADD45g, a genotoxic stress inducible gene associated with cell cycle arrest and apoptosis was analyzed by real time PCR. The mRNA level of GADD45g was significantly increased in Apex+/+ mice in response to 2-NP (Fig. 8A). Under control conditions, there was no effect of genotype on GADD45g expression (Fig 8A), while in response to oxidative stress, expression of GADD45g was significantly greater in Apex+/- mice as compared to their wildtype counterparts. As a p53 responsive gene, we wanted to evaluate the role of p53 in GADD45g gene response. Using western blot assay, we determined the effect of Ape1/Ref-1 heterozygosity on p53 stability in response to oxidative stress in the liver nuclear extracts. p53, a tumor suppressor protein is a well established determinant of cell cycle arrest and apoptosis. p53 protein stability followed the same trend as GADD45g expression, with 2-NP treated Apex+/- mice displaying the highest level of p53 protein stability (Fig. 8B). We further analyzed the activity of caspase-3, one of the final effectors in the apoptotic pathway under the same conditions using liver cytosolic extracts. Caspase-3 activity was significantly induced in the 2-NP treated Apex+/- mice compared to its untreated counterpart (Fig. 8C). Thus, loss of APE1/Ref-1 increases the apoptotic response to oxidative stress.
Fig. 8.

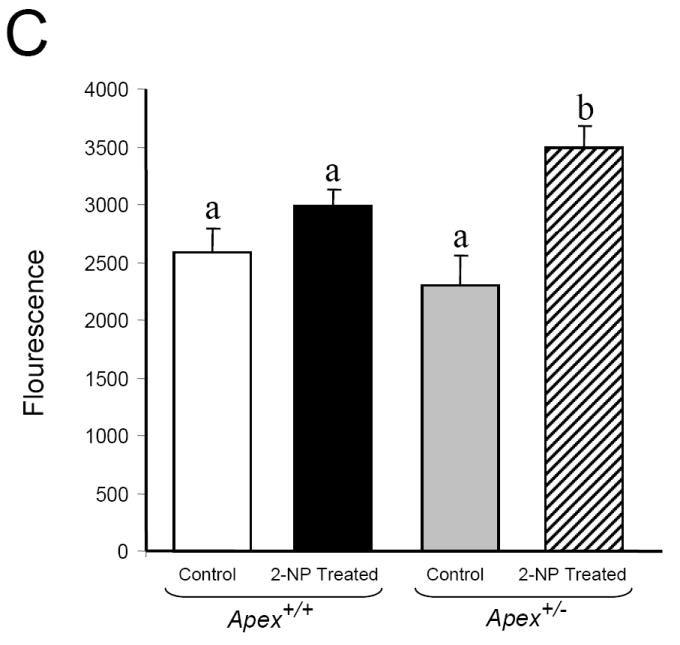
Effect of 2-NP on apoptosis in Apex+/- mice. (A) GADD45g mRNA expression level was quantified using real-time PCR and normalized with GAPDH. (B) The level of the p53 protein in 200 μg of liver nuclear extract was determined by western blot analysis. (SDS-PAGE): the level of p53 protein was normalized based on the amount of protein loaded on each gel. (C) The level of Caspase-3 activity in liver cytosolic extract as determined by Caspase-3 enzyme assay. Values represent an average (± S.E.M.) for data obtained from 5 mice in each group and are representative of separate identical experiments; Values with different letter superscripts indicate significant differences at P < 0.05. Lamin B protein level served as nuclear protein loading control. The picture depicts the representative sample from each group.
DISCUSSION
APE1/Ref-1 is a multifunctional protein involved in the maintenance of genomic integrity and in the regulation of gene expression. In addition to its role in BER as the major 5’-endonuclease and 3’phosphoesterase, APE1/Ref-1 was independently characterized as a redox activator of cellular transcription factors. Although research has demonstrated that APE1/Ref-1 is involved in the cellular response to oxidative DNA damage, it is currently unclear whether this response is strictly due to APE1/Ref-1 repair activity, APE1/Ref-1 redox regulatory activity, or both. A study by Fritz et al. [50] suggests that APE1/Ref-1 repair activity is constitutively expressed, while APE1/Ref-1 redox activity is subject to regulation under various stimuli. Furthermore, Hsieh et al. [51] present data indicating that APE1/Ref-1 redox activity is mediated by phosphorylation in response to oxidative stress.
Reports to date have shown that APE1/Ref-1 expression is inducible by various forms of oxidative stress in vitro [23-30]. For example, HeLa cells exposed to damaging agents like hypochlorite or methyl methane sulfonate show an increase in both the APE1/Ref-1 expression and activity [51]. Additionally, Ramana et al. [24] have shown induction of APE1/Ref-1 mRNA and protein levels with increased translocation of the protein into the nucleus when HeLa S3 tumor cells and WI 38 primary fibroblasts were exposed to ROS generators. Ape1/Ref-1 protein has also been shown to be translocated from the cytoplasm to the nucleus during H2O2 and ATP stimulation [52]. In order to determine the effect of oxidative stress on APE1/Ref-1 in vivo, we used mice containing a heterozygous gene-targeted deletion of APE (Apex+/-). Homozygous deletion of the APE1/Ref-1 gene (Apex-/-) is embryonic lethal, but heterozygous mice survive and are fertile [26, 53, 54]. These APE1/Ref-1 haploinsufficient (Apex+/-) mice show tissue specific differences in BER capacity characterized using an in vitro G:U mismatch repair assay. In addition Huamani et al. [55] have demonstrated that Apex+/- mice show a significant increase in spontaneous mutagenesis in lacI transgenes in liver and spleen. Furthermore, MEF’s and brain cells obtained from Apex+/- mice have been shown to be more susceptible to oxidizing agents [26, 56]. In this study we used 2-nitropropane (2-NP), a well known hepatocarcinogen [39, 57] to induce oxidative stress in vivo. 2-NP, an industrial solvent, is found in paints, varnishes and cigarette smoke [39, 57]. 2-NP promotes formation of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-OHdG) and 8-aminoguanine through its metabolites N-isopropylhydroxylamine and hydroxylamine-O-sulfonic acid, which are believed to be the causative factors behind 2-NP-induced carcinogenesis [39]. In addition, 2-NP occurs as Propane 2-nitronate (P2N) at physiological pH and promotes genotoxicity through production of NO species [57]. NO is shown to induce genotoxicity by deaminating DNA and increasing uracil to cytosine ratio [48, 58]. 2-NP has also been shown to be genotoxic in vitro, inducing mutations in bacteria and increasing unscheduled DNA synthesis in cultured hepatocytes [40]. Additionally, our laboratory has previously demonstrated that 2-NP-treatment results in an increase in the levels of 8-OHdG in vivo in the liver of mice and rats, followed by a concomitant increase in β-pol expression and BER activity [22]. Here we verify that APE1/Ref-1 is indeed an inducible protein (Figure 1B). While fold increase is the same in response to oxidative stress across the genotypes, the total accumulative level of APE1/Ref-1 protein is lower in the liver of Apex+/- mice, i.e., even though the intact allele is induced in response to 2-NP, it does not compensate for the lost allele.
APE1/Ref-1 has been shown to be an essential component of transcription complexes by directly interacting with other transcription factors such as HIF-1 and p300 [59, 60]. Research has shown that APE1/Ref-1 enhances the DNA binding activity of NF-κB in vitro as well as NF-κB-dependent transcriptional activation in vivo [13, 14]. Deletion of the redox-sensitive domain of APE1/Ref-1 has been shown to inhibit TNF-induced NF-κB activation, while loss of APE1/Ref-1 is shown to result in a significant decline in NF-κB activity and increased susceptibility to TNF-induced apoptosis [15, 16]. Furthermore, down-regulation of APE1/Ref-1 by soy isoflavones in vitro and in vivo resulted in concomitant reduction of NF-κB activity, while a 2-fold increase in APE1/Ref-1 expression, obtained by cDNA transfection, resulted in a concomitant 2-fold increase in NF-κB activity, confirming the cross-talk between these molecules [61]. In line with these findings the Apex+/+ mice showed a significant increase in APE1/Ref-1 redox activation of NF-κB when exposed to 2-NP, while increase in NF-κb activation is the same in response to oxidative stress across the genotypes, the total NF-κb DNA binding activity is lower in the liver of Apex+/- mice, i.e., the ultimate level of NF-κB activation was significantly attenuated in the heterozygous animals. It is well established that NF-κB is a mediator of inflammatory responses promoting cell proliferation and survival by inhibiting cell cycle arrest & apoptosis [62, 63], thus reduced activation of NF-κB and other redox-regulated transcription factors in response to oxidative stress in Apex+/- mice may prove detrimental due to alterations in the signaling mechanisms necessary to differentiate between repair and apoptosis.
We have previously reported tissue-specific differences in BER capacity in APE1/Ref-1 haploinsufficient mice [31]. Moreover, previous studies have indicated that down-regulation of APE1/Ref-1 may promote a damage hypersensitive phenotype [26-28]. Therefore, it was essential to analyze the effect of reduced APE1/Ref-1 on the level of DNA damage production and BER. In this study, we demonstrate that the liver tissue of Apex+/- mice expresses a BER phenotype that is more susceptible to accumulation of DNA damage in response to oxidative stress as a result of reduced APE1/Ref-1 3’-phoshoesterase activity in vivo. Here we show the differential impact of APE1/ref-1 heterozygosity on monofunctional and bifunctional glycosylase initiated BER. The traditional BER response to certain DNA base damages like uracil and alkylated bases, involves a monofunctional glycosylase-mediated removal of the damaged base resulting in the generation of an abasic site (AP site) that serves as a substrate for APE1/Ref-1 endonuclease activity. APE1/Ref-1 subsequently promotes formation of a transient single-strand break that serves as a substrate for β-pol-mediated nucleotide insertion and is followed by DNA ligase-mediated BER completion. β-pol, dRP lyase activity serves as the rate limiting step in this pathway [64]. On the other hand, other damages like oxidized bases (e.g. 8-hydroxyguanosine and thymine glycol) requires a bi-functional glycosylase mediated removal of the damage. These glycosylases have associated AP lyase activity resulting in 3’blocking ends. DNA polymerase requires a 3’OH group as its substrate for repair synthesis. Therefore 3’-blocking end trimming requires the 3’-phosphoesterase activity of APE1/Ref-1. β-pol and DNA ligase subsequently complete the repair process. The 3’-phoshoesterase activity of APE1/Ref-1 is a crucial step in this bi-functional glycosylase mediated BER [41]. In this study, we show the difference in the requirement of Ape1/Ref-1 in BER pathway in vivo. Our Apex+/- mice exposed to 2-NP show significant upregulation in the UNG message with concomittant increase in the β-pol protein and G:U mismatch BER capacity in the liver tissue. We suggest that 2-NP, via its NO generation leads to increased uracil levels in the DNA resulting in the upregulation of UNG, a monofunctional glycosylase which is also associated in the processing of oxidized cytosine derivatives [49]. Since β-pol is the rate limiting enzyme in this pathway, the liver extracts of Apex+/- mice treated with 2-NP show significant induction in the G:U mismatch repair. Apex+/- mice treated with 2-NP showed increased CREB binding activity, a stress response transcriptional activator of β-pol promoter, potentially explaining the induction of β-pol expression [44]. It is widely accepted that β-pol protein expression and activity strongly correlate with BER activity in response to a variety of stressors [22, 65-67]. Moreover, the endonuclease activity of APE1/Ref-1 is 100 fold more efficient than the 3’ phosphoesterase activity [68], thus we suggest that the available APE1/Ref-1 protein in apex+/- mice is adequate to sustain the monofunctional glycosylase initiated BER. In addition, increased p53 level can stabilize the APE1/Ref-1 and β-pol protein complex on the DNA abasic site which would enhance BER activity [69, 70].
Alternatively, the liver tissues of the 2-NP treated Apex+/- mice displayed significantly lower 8-OH G:C BER activity compared to the wildtype animals. OGG1 is a bifunctional glycosylase known to be involved in the removal of 8-OHdG. While, OGG1 mRNA and protein levels are shown to be unaffected during oxidative stress, the activity of this enzyme is enhanced by the increased APE1/Ref-1 protein [71]. In line with these findings, our 2-NP treated Apex+/- mice did not show any significant difference in the OGG1 mRNA levels. Furthermore, addition of purified human OGG1 protein to BER reaction mixture did not increase OGG1 initiated BER activity in 2-NP treated Apex+/- mice. However, addition of purified APE1/Ref-1 protein to BER reaction mix resulted in a significant increase in the 8-OH G: C BER activity in these mice confirming the significant role played by APE1/Ref-1 in the removal of 3’blocking groups. These data indicate that APE1/Ref-1 3’ phosphoesterase activity is critical in removing the 3’blocking lesion created by a bifunctional glycosylase, e.g., OGG1. Therefore, APE1/Ref-1 heterozygosity compromises this function resulting in reduced detectable single strand break, increased aldehydic lesions and reduced bifunctional DNA glycosylase initiated BER activity.
Previously, we have measured the presence of AP sites, single-strand breaks and aldehydic lesions in isolated liver DNA from APE1/Ref-1 haploinsufficient mice and observed no significant difference in DNA damage accumulation as a result of reduced APE1/Ref-1 [31]. The lack of damage accumulation in untreated Apex+/- mice suggested that APE1/Ref-1 haploinsufficiency in liver does not cause an accumulation of genotoxic DNA repair intermediate products under baseline conditions. In line with previous studies from our laboratory [22] and others [72], we have demonstrated a significant increase in 3’-OH-containing single-strand breaks in response to oxidative stress. However the level of detectable single strand breaks (SSB’s) in the liver tissue of 2-NP treated Apex+/- mice was found to be significantly lower than its wildtype counterpart while the level of aldehydic lesion was significantly higher. We suggest that the processing of oxidized bases by a bi-functional DNA glycosylase such as OGG1 (8-oxoguanine DNA glycosylase) could result in generation of aldehydic blocking lesions at 3’ end. Inability to process these 3’ blocking groups in the absence of the 3’-phophoesterase activity of Apex in Apex+/- mice [41], could result in lower detection of endonuclease-mediated single-strand breaks in the heterozygous animal.
Herein, we demonstrate that Ape1/Ref-1 haploinsufficiency does not promote a concomitant decline in G:U mismatch BER activity in the liver in response to oxidative stress, it rather displays a significant increase in this activity. While, the high efficiency of APE1/Ref-1 endonuclease activity, upregulation in UNG and β-pol expression appear to be the reason behind the significant increase in G:U mismatch repair activity, alternatively, recent studies report an APE1/Ref-1-independent BER pathway in human cells [42]. While mono-functional DNA glycosylases such as SMUG1 and TDG and bi-functional DNA glycosylases/lyases such as OGG1 and NTH require APE1/Ref-1 to process AP sites and the 3’-OH aldehydic groups, respectively, APE1/Ref-1 backup enzymes have been identified for the repair of oxidative damage in vivo [73]. The recently discovered mammalian DNA glycosylase/AP lyases NEIL1 and NEIL2 recognize oxidative damage and generate DNA strand breaks with 3’-phosphate termini. Removal of the 3’-phosphate termini was shown to be dependent upon polynucleotide kinase (PNK) activity, not APE1/Ref-1, with NEIL1 serving as a backup enzyme. Additionally, a study by Das et al. [74] has demonstrated that NEIL1 is induced by oxidative stress. Therefore, it is likely that the observed increase in BER may be due to increased processing of DNA damage by an APE-independent β-pol regulated BER pathway, however, this alternative pathway does not rescue the observed decline in 8OHdG:C BER capacity in APE1/REF-1 haploinsufficient mice.
In conclusion, we suggest that the optimal balance between the stress response transcription factors, notably NF-κB, and the DNA repair pathway required for cell survival is attenuated in APE1/Ref-1 haploinsufficient mice exposed to oxidative stress. Reduced redox activity of APE1/Ref-1 leads to reduced activation of NF-κB potentially leading to cell cycle arrest and apoptosis. This notion is supported by the augmentation of GADD45g expression, a cell cycle arrest gene, observed in the liver of 2-NP treated Apex+/-mice. GADD45g has also been implicated in the promotion of apoptosis under environmental stress via the p38K-Jun NH2-terminal Kinase pathway [75, 76]. Furthermore, Apex+/- mice exposed to 2-NP show significant induction in the p53 protein levels and Caspase-3 activity in the liver, both being markers of apoptosis. Therefore, when APE1/Ref-1 is compromised the cells are more susceptible to oxidative stress mainly due to reduced redox and 3’phosphodiestrase activity impacting cell survival, pushing it towards apoptosis. This finding has therapeutic importance as increased APE1/Ref-1 levels in tumor cells have been shown to confer resistance to chemotherapeutic drugs perhaps via decrease in apoptosis [77]. In line with previous findings, inhibition of DNA repair or redox or both activities of APE1/Ref-1 using inhibitors could potentially sensitize the tumor cells to the therapeutic agents [77] making APE1/Ref-1 an attractive molecular target in the treatment of cancer.
Acknowledgments
This work was supported by grants from the National Institutes of Health [R01 CA121298, to A.R.H.] and the American Institute for Cancer Research [03A061, to A.R.H.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Mullaart E, Lohman PHM, Berends F, Vijg J. DNA damage metabolism and aging. Mutat Res. 1990;237:189–210. doi: 10.1016/0921-8734(90)90001-8. [DOI] [PubMed] [Google Scholar]
- 3.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 4.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 5.Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–251. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and Mutagenesis. Washington D.C: ASM Press; 2006. pp. 169–226. [Google Scholar]
- 7.Ljungquist S, Lindahl T. A mammalian endonuclease specific for apurinic sites in double-stranded deoxyribonucleic acid. I. Purification and general properties. J Biol Chem. 1974;249:1530–1535. [PubMed] [Google Scholar]
- 8.Ljungquist S, Andersson A, Lindahl T. A mammalian endonuclease specific for apurinic sites in double-stranded deoxyribonucleic acid. II. Further studies on the substrate specificity. J Biol Chem. 1974;249:1536–1540. [PubMed] [Google Scholar]
- 9.Lindahl T. Inroads into base excision repair I: The discovery of apurinic/apyrimidinic [AP] endonuclease. DNA Repair. 2004;3:1521–1530. doi: 10.1016/j.dnarep.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans AR, Limp-Foster M, Kelley MR. Going APE over Ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu N, Sugimoto K, Tang J, Nishi T, Sato I, Hiramoto M, Aizawa S, Hatakeyama M, Ohba R, Hatori H, Yoshikawa T, Suzuki F, Oomori A, Tanaka H, Kawaguchi H, Watanabe H, Handa H. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000;18:877–881. doi: 10.1038/78496. [DOI] [PubMed] [Google Scholar]
- 14.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 15.Hall JL, Wang X, Adamson V, Zhao Y, Gibbons GH. Overexpression of Ref-1 inhibits hypoxia and tumor necrosis factor-induced endothelial cell apoptosis through nuclear factor-κB-independent and-dependent pathways. Circ Res. 2001;88:1247–1253. doi: 10.1161/hh1201.091796. [DOI] [PubMed] [Google Scholar]
- 16.Guan Z, Basi D, Li Q, Mariash A, Xia YF, Geng JG, Kao E, Hall JL. Loss of redox factor 1 decreases NF-kappaB activity and increases susceptibility of endothelial cells to apoptosis. Arterioscler Thromb Vasc Biol. 2005;25:96–101. doi: 10.1161/01.ATV.0000150418.14698.75. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T. Keynote: past, present, and future aspects of base excision repair. Nucleic Acid Res Mol Biol. 2001;68:xvii–xxx. doi: 10.1016/s0079-6603(01)68084-x. [DOI] [PubMed] [Google Scholar]
- 18.Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 19.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nature Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 20.Wilson DM, III, Sofinowski TM, McNeill DR. Repair mechanisms for oxidative DNA damage. Front Biosci. 2003;8:d963–981. doi: 10.2741/1109. [DOI] [PubMed] [Google Scholar]
- 21.Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S, Hazra TK. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 22.Cabelof DC, Raffoul JJ, Yanamadala S, Guo Z, Heydari AR. Induction of DNA polymerase β-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis. 2002;23:1419–1425. doi: 10.1093/carcin/23.9.1419. [DOI] [PubMed] [Google Scholar]
- 23.Grosch S, Fritz G, Kaina B. Apurinic endonuclease [Ref-1] is induced in mammalian cells by oxidative stress and is involved in clastogenic adaptation. Cancer Res. 1998;58:4410–4416. [PubMed] [Google Scholar]
- 24.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci U S A. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ono Y, Furuta T, Ohmoto T, Akiyama K, Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 26.Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, Grundy S, Jialal I, Friedberg EC. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- 27.Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 29.Izumi T, Brown DB, Naidu CV, Bhakat KK, MacInnes MA, Saito H, Chen DJ, Mitra S. Two essential but distinct functions of mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Raffoul JJ, Cabelof DC, Nakamura J, Meira LB, Friedberg EC, Heydari AR. Apurinic/apyrimidinic endonuclease [APE/REF-1] haploinsufficient mice display tissue-specific differences in DNA polymerase beta-dependent base excision repair. J Biol Chem. 2004;279:18425–18433. doi: 10.1074/jbc.M313983200. [DOI] [PubMed] [Google Scholar]
- 32.Hadi MZ, Coleman MA, Fidelis K, Mohrenweiser HW, Wilson DM., III Functional characterization of Ape1 variants identified in the human population. Nucleic Acids Res. 2000;28:3871–3879. doi: 10.1093/nar/28.20.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Boer JG. Polymorphisms in DNA repair and environmental interactions. Mutat Res. 2002;509:201–210. doi: 10.1016/s0027-5107(02)00217-8. [DOI] [PubMed] [Google Scholar]
- 34.Mohrenweiser HW, Wilson DM, III, Jones IM. Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res. 2003;526:93–125. doi: 10.1016/s0027-5107(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 35.Hou EW, Prasad R, Asagoshi K, Masaoka A, Wilson SH. Comparative assessment of plasmid and oligonucleotide DNA substrates in measurement of in vitro base excision repair activity. Nucleic Acids Res. 2007;35:1–10. doi: 10.1093/nar/gkm639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabelof DC, Raffoul JJ, Nakamura J, Kapoor D, Abdalla H, Heydari AR. Imbalanced base excision repair in response to folate deficiency is accelerated by polymerase beta haploinsufficiency. J Biol Chem. 2004;279:36504–36513. doi: 10.1074/jbc.M405185200. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 38.Sokal RR, Rohlf FJ. Biometry. New York, NY: W.H. Freeman and Company; 1981. [Google Scholar]
- 39.Sakano K, Oikawa S, Murata M, Hiraku Y, Kojima N, Kawanishi S. Mechanisms of metal-mediated DNA damage induced by metabolites of carcinogenic 2-nitropropane. Mutat Res. 2001;479:101–111. doi: 10.1016/s0027-5107(01)00158-0. [DOI] [PubMed] [Google Scholar]
- 40.Andrae U, Homfeldt H, Vogl L, Lichtmannegger J, Summer KH. 2-Nitropropane induces DNA repair synthesis in rat hepatocytes in vitro and in vivo. Carcinogenesis. 1988;9:811–815. doi: 10.1093/carcin/9.5.811. [DOI] [PubMed] [Google Scholar]
- 41.Izumi T, Hazra TK, Boldogh I, Tomkinson AE, Park MS, Ikeda S, Mitra S. Requirement for human AP endonuclease 1 for repair of 3’-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis. 2000;21:1329–1334. [PubMed] [Google Scholar]
- 42.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Sabine G, kaina B. Transcriptional activation of Apurinic/Apyrimidinic endonuclease [Ape,Ref-1] by oxidative stress requires CREB. Biochem and Biophy Res Communications. 1999;261:859–863. doi: 10.1006/bbrc.1999.1125. [DOI] [PubMed] [Google Scholar]
- 44.Yau-Jan C, Rawson TY, Wilson SH. Cloning and characterization of a novel member of the human ATF/CREB family: ATF2 deletion, a potential regulator of the human DNA polymerase β promoter. Gene. 2003;312:117–124. doi: 10.1016/s0378-1119(03)00607-3. [DOI] [PubMed] [Google Scholar]
- 45.Fornance AJ, Jr, Zmudzka B, Hollander MC, Wilson SH. Induction of beta-polymerase mRNA by DNA-damaging agents in Chinese hamster ovary cells. Mol Cell Biol. 1989;9:851–853. doi: 10.1128/mcb.9.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nuc Acids Res. 1998;26:5116–5122. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersom S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Uracil-DNA Glycosylase [UNG]-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 48.Endres M, Biniszkiewicz D, Sobol RB, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. Increased potischemic brain injury in mice deficienct in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dizdaroglu M, Karakaya A, Jaruga P, Slupphaug G, Krokan HE. Novel activities of human uracil DNA N-glycosylase for cytosine derived products of oxidative DNA damage. Nuc Acids Res. 1996;24:418–422. doi: 10.1093/nar/24.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fritz G, Grosch S, Tomicic M, Kaina B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology. 2003;193:67–78. doi: 10.1016/s0300-483x(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 51.Hsieh MM, Hegde V, Kelley MR, Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nuc Acids Res. 2001;29:3116–3122. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F, Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nuc Acids Res. 2005;33:4379–4394. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pederson RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 54.Xanphoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huamani J, McMahan CA, Herbert DC, Reddick R, McCarrey JR, MacInnes MI, Chen DJ, Walter CA. Spontaneous mutagenesis is enhanced in Apex heterozygous mice. Mol Cell Biol. 2004;24:8145–8153. doi: 10.1128/MCB.24.18.8145-8153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage version 7, DNA repair 5. DNA repair. 2006;5:189–209. doi: 10.1016/j.dnarep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 57.Kohl C, Morgan P, Gescher A. Metabolism of the genotoxicant 2-nitropropane to a nitric oxide species. Chemico-Biological Interactions 97. Chemico-Biological Interactions. 1995;97:175–184. doi: 10.1016/0009-2797(95)03614-r. [DOI] [PubMed] [Google Scholar]
- 58.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS, Keefer LK. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 59.Carrero P, Okamoto K, Coumailleau P, O’Brien S, Tanaka H, Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziel KA, Campbell CC, Wilson GL, Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. FASEB J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 61.Raffoul JJ, Banerjee S, Singh-Gupta V, Knoll ZE, Fite A, Zhang H, Abrams J, Sarkar FH, Hillman GG. Down-regulation of apurinic/apyrimidinic endonuclease 1/redox-factor 1 expression by soy isoflavones enhances prostate cancer radiotherapy in vitro and in vivo. Cancer Res. 2007;67:2141–2149. doi: 10.1158/0008-5472.CAN-06-2147. [DOI] [PubMed] [Google Scholar]
- 62.Raffoul JJ, Wang Y, Kucuk O, Forman JD, Sarkar FH, Hillman GG. Genistein inhibits radiation-induced activation of NF-κB in prostate cancer cells promoting apopstosis and G2/M cell cycle arrest. BMC Cancer. 2006;6:107. doi: 10.1186/1471-2407-6-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baldwin AS., Jr The transcription factor NF-κB and human disease. The Journal of Clinical Investigation. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Srivastava DK, Vande Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Boil Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 65.Cabelof DC, Raffoul JJ, Yanamadala S, Ganir C, Guo Z, Heydari AR. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat Res. 2002;500:135–145. doi: 10.1016/s0027-5107(02)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cabelof DC, Guo Z, Raffoul JJ, Sobol RW, Wilson SH, Richardson A, Heydari AR. Base excision repair deficiency caused by polymerase beta haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003;63:5799–5807. [PubMed] [Google Scholar]
- 67.Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA Repair. 2003;2:295–307. doi: 10.1016/s1568-7864(02)00219-7. [DOI] [PubMed] [Google Scholar]
- 68.Suh D, Wilson DM, Povirk LF. 3’-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nuc Acids Res. 1997;25:2495–2500. doi: 10.1093/nar/25.12.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernstein C, Bernstein CH, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mut Res. 2002;511:145–178. doi: 10.1016/s1383-5742(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Ahn J, Wilson SH, Prives C. A role of P53 in base excision repair. EMBO J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saitoh T, Shinmura K, Yamaguchi S, Tani M, Seki S, Murakami H, Nojima Y, Yokota J. Enhancement of OGG1 protein AP lyase activity by increase of Apex protein. Mutation Research. 2001;486:31–40. doi: 10.1016/s0921-8777(01)00078-7. [DOI] [PubMed] [Google Scholar]
- 72.Huang D, Shenoy A, Cui J, Huang W, Liu PK. In situ detection of AP sites and DNA strand breaks bearing 3’-phosphate termini in ischemic mouse brain. FASEB J. 2000;14:407–417. doi: 10.1096/fasebj.14.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- 74.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J Biol Chem. 2005;280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]
- 75.Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Tao Q. The stress-responsive gene GADD45g is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clin Cancer Res. 2005;18:6442–6449. doi: 10.1158/1078-0432.CCR-05-0267. [DOI] [PubMed] [Google Scholar]
- 76.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4/MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 77.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Molecular aspects of Medicine. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]