

Abstract
Background
Limbic encephalitis (LE) frequently associates with antibodies to cell surface antigens. Characterization of these antigens is important because it facilitates the diagnosis of those disorders that are treatment-responsive. We report a novel antigen of LE and the effect of patients' antibodies on neuronal cultures.
Methods
Clinical analysis of 10 patients with LE. Immunoprecipitation and mass spectrometry were used to identify the antigens. HEK293 cells expressing the antigens were used in immunocytochemistry and ELISA. The effect of patients' antibodies on cultures of live rat hippocampal neurons was determined with confocal microscopy.
Results
Median age was 60 years (38-87); 9 were women. Seven had tumors of the lung, breast or thymus. Nine patients responded to immunotherapy or oncological therapy but neurologic relapses, without tumor recurrence, were frequent and influenced the long-term outcome. One untreated patient died of LE. All patients had antibodies against neuronal cell surface antigens that by immunoprecipitation were found to be the GluR1 and GluR2 subunits of the AMPA receptor (AMPAR). HEK293 cells expressing GluR1/2 reacted with all patients' sera or CSF, providing a diagnostic test for the disorder. Application of antibodies to cultures of neurons significantly decreased the number of GluR2-containing AMPAR clusters at synapses with a smaller decrease in overall AMPAR cluster density; these effects were reversed after antibody removal.
Conclusions
Antibodies to GluR1/2 associate with LE that is often paraneoplastic, treatment-responsive, and has a tendency to relapse. Our findings support an antibody-mediated pathogenesis in which patients' antibodies alter the synaptic localization and number of AMPAR.
Keywords: autoimmune, encephalitis, AMPAR, paraneoplastic, antibodies
INTRODUCTION
Limbic encephalitis (LE) is an inflammatory disorder that predominantly affects the grey matter of the medial temporal lobes, amygdala, and orbitofrontal cortex.1 As a result patients develop short-term memory deficits, emotional and behavioral disturbances, seizures, and sometimes dementia. Until recently, most cases of LE were thought to be paraneoplastic, mediated by immune responses against intracellular antigens, and poorly responsive to treatment.2 However, the clinical and immunological spectra of LE are far more extensive than initially considered and recent studies revealed a larger category of allied disorders in which the antigens are on the cell surface.3,4 For example, a series of 45 patients with LE demonstrated that 64% had antibodies against neuronal cell surface antigens; of these 45% were the voltage-gated potassium channels (VGKC) and 55% other antigens, most unknown.5 Given that disorders with antibodies to cell surface autoantigens are often treatment responsive, characterization of the antigens is important to facilitate a prompt and specific diagnosis.3,5 In addition, the immune response provides a link between the function of the antigens and neuronal events involved in memory, learning, cognition, and seizures. For example, antibodies to NR1/NR2 heteromers of the N-methyl-D-aspartate receptor (NMDAR) associate with a treatable encephalitis characterized by psychosis, memory deficits, seizures, dyskinesias, and autonomic instability.6
The current study describes a novel cell surface autoantigen in 10 patients with LE. Our findings are relevant for three reasons; 1) the antigen is the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), a subtype of glutamate receptor that underlies mechanisms of memory, learning, and seizures; 2) application of patients' antibodies into cultures of live rat hippocampal neurons altered the number of AMPAR clusters at synapses, supporting an antibody-mediated pathogenesis of the LE, and 3) we describe the associated syndrome and provide a diagnostic test for this disorder.
PATIENTS AND METHODS
The 10 patients of this study were identified among 109 cases who fulfilled strict criteria of limbic encephalitis2 seen at the Hospital of the University of Pennsylvania or whose sera, CSF or tissues were referred for immunological studies from July 2004 until July 2008. Thirty nine cases had antibodies to known antigens (16 VGKC, 7 NMDA receptor, 6 GAD, 4 Hu, 4 Ma2, 1 amphiphysin, 1 CV2/CRMP5), 43 had antibodies to unknown antigens, and 27 had no antibodies. Among the 43 patients with antibodies to unknown antigens, 10 were selected for having antibodies with a similar pattern of reactivity with the neuropil of brain and cerebellum. Nine of these 10 patients were seen by the authors (4 JD, 5 other authors) and the information of one was provided by the referring physician. The clinical features of 4 patients have been partially reported.4,5 In addition to the above cases, serum or CSF of 110 individuals served as controls. These included 20 patients with anti-NMDA receptor encephalitis, 20 LE and VGKC antibodies, 20 paraneoplastic encephalitis and antibodies to intracellular antigens (Hu, Ma2, CV2/CRMP5, or amphiphysin), 10 patients with Rasmussen's encephalitis, 10 patients with lupus erythematosus, 10 patients with small-cell lung cancer without paraneoplastic syndromes, 10 with thymoma without LE, and 10 blood donors.
Information on clinical antibody testing, animal tissue processing, IgG biotinylation, immunohistochemistry on tissue and cultures of live rat hippocampal neurons, immunocytochemistry on HEK293 cells, and enzyme-linked immunoabsorption assay (ELISA) is provided in Supplemental Material and Methods. Studies were approved by the University of Pennsylvania Institutional Review Board.
Immunoprecipitation and immunoblot
Rat hippocampal neuronal cultures were prepared as reported.7 Live neurons grown in 100 mm wells (density 106 neurons/well) were incubated at 37°C with filtered patient's serum (diluted 1:500) for 1 hour. Neurons were then washed with phosphate buffered saline (PBS), lysed with buffer (NaCl 150mM, EDTA 1mM, Tris-HCl 100mM, deoxycholate acid 0.5%, 1% Triton X-100, pH 7.5) containing protease inhibitors (Sigma P8340), and centrifuged at 16.1 × 103 g for 20 minutes at 4 °C. The supernatant was retained and incubated with protein A/G agarose beads (Pierce, 20423) overnight at 4 °C, centrifuged, and the pellet containing the beads with patients' antibodies bound to the target cell surface antigens was then washed with PBS, aliquoted and kept at -80 °C. An aliquot of this pellet was resuspended in Laemmli buffer, boiled for 10 minutes, separated in a 4-15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins visualized with EZBlue gel staining (Sigma G1041). Distinctive protein bands precipitated by patients' sera were excised from the gel and analyzed using mass spectrometry at the proteomic facility at the University of Pennsylvania.
After characterization of the antigens, frozen aliquots of the indicated pellets were separated in a SDS-PAGE as above, transferred to nitrocellulose (Bio-Rad 162-0115) and blotted with the indicated GluR1 (1:1000) or GluR2/3 (1:200) antibodies.
Quantitative analysis of AMPAR clusters using confocal microscopy
To determine the degree of immunolabeling of AMPAR by patients' antibodies, 14 days in vitro (div) live rat hippocampal neurons were exposed to patient's CSF and a rabbit polyclonal antibody against GluR1 or GluR2/3, washed, fixed, and incubated with the appropriate fluorescent-conjugated secondary antibodies (Supplemental Methods: Immunocytochemistry using live rat hippocampal neurons). Images were obtained using a laser-scanning confocal microscope (Leica TCS SP2). For each image, laser light levels and detector gain and offset were adjusted so that no pixel values were saturated. Images were thresholded, and the number of individual clusters along neuronal dendrites was determined using interactive software (MetaMorph; Universal Imaging, West Chester, PA or ImageJ).8
To determine the effects of patients' antibodies on the number and localization of AMPAR clusters, neurons were treated with patient or control CSF (1:15 dilution in NeuroBasal + B27 medium, GIBCO Carlsbad, CA) from 11 to 17 div. Every three days, 20 of the 300 μl of medium in each culture well were removed and replaced with 20 μl of fresh patient or control CSF. In another series of experiments, neurons were treated with patient CSF from 11 to 14 div followed by treatment with control CSF from 14 to 17 div. On 14 or 17 div, neurons were fixed in freshly made paraformaldehyde (4% paraformaldehyde, 4% sucrose in PBS) for 5 minutes, permeablized in 0.25% Triton X-100 for 10 minutes, and blocked in 5% normal goat serum for 1 hour. Neurons were then incubated with patient's CSF (1:15 dilution in Triton X-100), a rabbit polyclonal antibody against GluR1 or GluR2/3 (1:1000, Chemicon) or a mouse monoclonal antibody against PSD-95 (1:500, Affinity BioReagents, Golden, CO) or a guinea pig polyclonal antibody against VGlut (1:1000, polyclonal; Chemicon) for 2 hours followed by the appropriate fluorescent-conjugated secondary antibodies (Jackson Immunologicals, Inc.). Images were obtained and analyzed as above.
Statistics
Differences in antibody titers among groups were analyzed using the Mann Whitney test. The effects of IgG and CSF on neuronal cultures were analyzed using the Kruskal-Wallis non-parametric ANOVA followed by Dunn's pairwise comparison.
RESULTS
Clinical features
Demographic information, symptoms, diagnostic tests, treatment, and outcome are shown in Tables 1 and 2. Patients median age was 60 years (range 38-87); nine were female. Nine patients presented with subacute (< 8 weeks) confusion, disorientation and memory loss, classic of LE; 1 patient presented with a 4 month history of progressive memory loss, behavioral change and agitation that initially suggested a rapidly progressive dementia. Four patients had seizures, one of them at the relapse of the disorder 16 months after the first episode of encephalitis. In all patients the neurological examination was consistent with an encephalopathy predominantly involving the limbic system.
Table 1.
Clinical Features, CSF, EEG, and MRI Findings
| Case* | Sex/age | Symptom presentation | CSF | Initial EEG | Initial Brain MRI (FLAIR) | GluR1/2 antibodies (main antigen) |
|---|---|---|---|---|---|---|
| 14 | F/65 | Initial episode and relapses: short-term memory loss, confusion, agitation, aggressive behavior, confabulation. Transient downbeat nystagmus. | 30 WBC, protein 97, positive OB and ISAb | Normal, repeated twice | Mild increased signal in medial temporal lobes. | GluR1/2 positive (GluR1) |
| 24 | F/44 | Initial episode and relapses: short-term memory loss; confusion, combativeness. Focal motor seizures. Right beating nystagmus. | 44 WBC, protein 91, glucose 70, no OB, positive ISAb | Diffuse theta activity; episodes of epileptic activity in left temporal lobe. | Initial MRI normal. Followup 5 days later: mild temporal lobe increased signal (right> left). | GluR1/2 positive (GluR2) |
| 34 | M/38 | Initial episode and relapse: short-term memory loss, confusion, agitation, perseveration generalized tonic-clonic seizures | 7 WBC, protein 50, positive OB and ISAb, | NA | Increased signal in the right medial and lateral temporal lobe, right frontal, left insular and left occipital regions. | GluR1/2 positive (GluR2) |
| 4 | F/64 | Increased seizures, confusion, disorientation, lethargy, short-term memory loss. | 75 WBC, protein 79 | Slow activity in the right temporal region. No epileptic activity. | Post-operative changes in the right temporal lobe. New increased signal in the left medial temporal lobe. | GluR1/2 positive (GluR2) |
| 55 | F/44 | Confusion, behavioral change, hypersomnia, progressive unresponsiveness. Mild gait unsteadiness, low grade fever. | 15 WBC, normal protein | NA | NA** CT normal | GluR1/2 positive (GluR2>R1) |
| 6 | F/38 | Short-term memory loss, confabulation | 6 WBC, normal protein | Normal | Increased signal in medial temporal lobes, left septal nucleus, left cerebellum. | GluR1/2 positive (GluR2) |
| 7 | F/87 | Initial episode: short-term memory loss, disorientation. At relapse: memory loss, generalized tonic-clonic seizures | Initially normal; protein 50 at relapse | Diffuse slow activity (7-8 c/sec), delta activity in anterior frontotemporal areas | Increased signal in medial temporal lobes; mild transient contrast enhancement in the left hippocampus. | GluR1/2 positive (GluR1) |
| 8 | F/61 | Initial episode and relapse: Short-term memory loss, decreased level of consciousness. | 24 WBC, protein 420 | Theta activity in posterior temporal regions | Normal MRI | GluR1/2 positive (GluR2) |
| 9 | F/59 | Progressive memory loss, behavioral change, agitation, confabulation (4 months). Mild dysdiadochokinesia | 17 WBC, protein 51, positive OB | Bilateral sharp waves in temporal lobes; no seizures | Increased signal in medial temporal lobes, and medial orbitofrontal region. | GluR1/2 positive (GluR1) |
| 10 | F/67 | Confusion, combativeness, insomnia, hallucinations, short term memory loss | 32 WBC, normal protein, no OCB | Sharp waves in temporal lobes | Mild temporal lobe increased signal. | GluR1/2 positive (GluR2) |
OB: oligoclonal bands; ISAb: Intrathecal synthesis of antibodies; CSF normal values: WBC <4/μl; Protein 16-46 mg/dL. All patients had normal glucose levels in the CSF; ND: not done. NA: not available.
The initial clinical features of patients #1, 2 and 3, were described in detail in4 (cases 2,5 and 6); the long term follow-up is provided here. Patient #5 corresponds to case 4 of Tables 1 and 2 in5
Limbic encephalitis confirmed at autopsy.
Table 2.
Tumor Association, Other Autoimmune Features, Treatment and Outcome
| Case | Tumor (antigen) | Time from symptoms of LE to tumor diagnosis | Other autoimmune disorders or antibodies | Treatment | Number of relapses (#); Interval between presentation and last relapse | Outcome (follow-up in months) |
|---|---|---|---|---|---|---|
| 14 | - | - | At presentation: plasma exchange, corticosteroids. At relapse: IVIg, corticosteroids. Chronic treatment with azathioprine. | (3); 7 months | First episode: returned to baseline. Subsequent relapsing-remitting behavioral problem and memory deficit. Residual stable deficits after 3rd relapse (50) | |
| 24 | Thymic carcinoma (GluR1 & GluR2) | Concurrent with first episode of encephalitis | ANA, dsDNA, cardiolipin antibodies | Tumor removal. At presentation and relapses: IVIg, corticosteroids. Chronic treatment with azathioprine. | (3); 101 months | First episode: returned to baseline. Subsequent relapsing-remitting memory deficit. Residual short-term memory deficit after 3rd relapse (120) |
| 34 | Malignant thymoma (GluR2) | Concurrent with relapse of encephalitis | Stiff-person syndrome, diabetes mellitus, GAD antibodies | Tumor removal, radiation therapy; corticosteroids, plasma exchange, IVIg. | (1); 60 months | First episode: returned to baseline. Mild residual memory deficit after relapse; steroid dependant muscle spasms and rigidity (36) |
| 4 | Non-SCLC (N/A) | Concurrent with first episode of encephalitis | Chronic seizures due to cortical displasia (confirmed by surgery). | Tumor removal; corticosteroids | - | Returned to baseline (8) |
| 55 | Thymoma (N/A) | Concurrent with first episode of encephalitis | CV2/CRMP5 antibodies | - | (1 atypical)*; 24 months | Unexpected dead, cardiorespiratory arrest (0.5). Autopsy results in Supplemental material. |
| 6 | - | - | - | IVIg, corticosteroids | - | Returned to baseline (8) |
| 7 | - | - | ANA, hypothyroidism, | Corticosteroids | (1); 16 months | First episode: partial improvement followed by progressive deterioration. Died at relapse after status epilepticus (16) |
| 8 | Breast cancer GluR1 & GluR2) | Concurrent with relapse of encephalitis | Hypothyroidism | At presentation: corticosteroids At relapse: plasma exchange and corticosteroids | (1); 9 months | First episode and relapse responded to corticosteroids and plasma exchange; last follow-up: residual short-term memory loss and behavioral problems (28). |
| 9 | SCLC (GluR1) | 6 months | Raynaud's syndrome, +ANA speckled pattern (1:160); VGCC and SOX1 antibodies | Tumor removal, chemotherapy; corticosteroids, IVIg | - | Returned to baseline; died of myocardial infarction (15). Autopsy results in Supplemental material. |
| 10 | Breast cancer (N/A) | Concurrent with first episode of encephalitis | - | Tumor removal, radiation therapy, corticosteroids chemotherapy (including cyclophosphamide), IVIg | - | Rapid recovery of memory; mild persistent depression, apathy and reduced verbal fluency (3) |
Atypical: episode of confusion, hallucinations, of unclear etiology, attributed to a “psychotic break”; resolved spontaneously 2 years before the diagnosis of GluR1/2 associated LE.
The CSF showed lymphocytic pleocytosis in 9 patients (median white blood cell count 24/μl; range 6-75). The brain MRI demonstrated increased FLAIR signal involving the medial temporal lobes in 8 of 9 patients examined. Two patients had additional abnormalities in the anterior septal nuclei, one with several cortical areas of transiently increased signal on FLAIR imaging, and the other with transiently increased signal in the cerebellum.
Seven patients had an underlying neoplasm, 5 of them diagnosed by the time of the initial episode of LE and 2 at first relapse of LE. The other 3 patients had extensive studies (2 combined body CT/FDG-PET, and 1 CT of the chest, abdomen and pelvis along with serologic tumor markers, and ultrasound of the breast) without revealing a neoplasm (median follow-up 16 months; range 8-50 months). Five patients had a history or concurrent findings of systemic autoimmunity (Table 2; see in Supplemental Results: Concurrent autoimmune findings).
Nine patients received immunotherapy; 6 of them had an underlying tumor and also received oncological therapy. All 9 patients responded to the indicated treatments at the first episode of encephalitis. Five patients had 1 to 3 neurological relapses between 2 and 101 months (median 16) after the first episode of encephalitis. None of the 7 patients with tumors developed tumor recurrences.
At relapse of LE 4 of 5 patients responded to treatment, but in all cases the response was partial, and 1 patient died after a prolonged episode of status epilepticus. Among the 4 patients with a single episode of encephalitis, 2 returned to their baseline activities, 1 had partial improvement, and 1 returned to most of her baseline activities but had residual hypersomnia and depression for several months. This patient had small-cell lung cancer (SCLC) and died 7 months after neurological recovery; the cause of death was a cardiac dysrhythmia due to severe coronary atherosclerosis (Supplemental Results: Autopsy findings, case #9).
One patient was not treated and died of LE. She was a 44 year-old woman who developed a rapidly progressive encephalitis and sudden death. The final diagnosis was LE with thymoma and overlapping GluR1/2 and CV2/CRMP5 antibodies (Supplemental Results: Autopsy findings, case #5).
Identification of antibodies to cell surface antigens
Using immunohistochemistry of rat brain, all 10 patients' serum and CSF showed intense reactivity with the neuropil of the hippocampus, subiculum, caudate-striatum, and molecular layer of the cerebellum; there was substantially less reactivity with other areas of the brain, cerebellum, and brainstem (Figure 1). In cultures of live, non-permeabilized, rat hippocampal neurons, all patients' serum and CSF produced intense immunolabeling of the cell surface and neuronal processes (Figure 2 A). Further analysis of this reactivity using confocal microscopy demonstrated that the target antigens were distributed in a punctuate manner along dendrites (Figure 2 B).
Figure 1. Immunolabeling of rat brain by patients' antibodies.

Sagittal section of rat brain incubated with the CSF of a patient with limbic encephalitis and novel antibodies. Note the intense reactivity of the patient's antibodies with the neuropil of hippocampus (Hip), subiculum (S), molecular layer of the cerebellum and Purkinje cells (CB), caudate-putamen (CPu), and cerebral cortex (Ctx). Other regions of the brain (e.g., corpus callosum (cc) and brainstem (B)) do not show significant immunolabeling. The areas boxed in A correspond to hippocampus and cerebellum and are shown at high magnification in B and D. The box in B is located at the dentate gyrus and is shown amplified in C. In panels B-D the nuclei of the cells is demonstrated with 4',6-diamidino-2-phenylindole (DAPI).
Immunofluorescent technique, A × 2.5; B and D × 200; C × 400.
Figure 2. Patients antibodies react with extracellular epitopes and precipitate GluR1 and GluR2 subunits of the AMPAR.

Culture of rat hipppocampal neurons incubated (live, non-permeabilized) with the CSF of a patient with LE. Note the intense reactivity of patient's antibodies with cell surface antigens (A); scale bar = 10 μm. Confocal microscopy suggests that the antigens are concentrated in puncta along dendrites (B); scale bar = 5 μm. The precipitation of these antigens using patients' antibodies is shown in a gel in which proteins are visualized with EZBlue (C). Note that two patients' antibodies (P1 and P2) precipitated antigen(s) that produce a single band at ~100 kDa; this band is not seen in the precipitate using CSF from a control individual (N). The band ~50 kDa corresponds to patients' IgG. Sequencing of the 100 kDa band using mass spectrometry demonstrated the GluR1 and GluR2 subunits of the AMPAR (not shown). Subsequent transfer of the gel to nitrocellulose and immunoblotting with antibodies specific for GluR1 and GluR2 confirmed that the 100 kDa band contained both GluR1 and GluR2 subunits (Panels in D).
Three patients had additional antibodies to previously characterized antigens (Table 2): one had GAD antibodies, another had CV2/CRMP5 antibodies, and another had SOX1 and voltage-gated calcium channel (VGCC) antibodies without evidence of Lambert-Eaton myasthenic syndrome. In this patient, SOX1 antibodies predicted the presence of a SCLC.
The cell surface synaptic antigens are the GluR1 and GluR2 subunits of the AMPAR
In order to identify the cell surface antigens, live rat hippocampal neurons were incubated with patients' sera and the antigens immunoprecipitated. These studies produced a distinctive protein band of ~100 kDa (Figure 2 C) that when analyzed by mass spectrometry contained sequences derived from the GluR1 and GluR2 subunits of the AMPAR. Since GluR1 and GluR2 have similar molecular weight and share sequence homology, we further examined whether the precipitated band contained GluR1, GluR2, or both subunits, using western blot with antibodies specific for each subunit. These studies confirmed that the band precipitated by patients' antibodies contained both GluR1 and GluR2 subunits (Figure 2 D).
Since GluR1 and GluR2 co-assemble in neurons and immunoprecipitated together, the primary epitopes could be located on either or both subunits. Thus, we examined which of the two subunits contained the main epitopes recognized by patients' antibodies using HEK293 cells transfected with single subunits (GluR1 or GluR2). Non-transfected HEK cells and cells transfected with GluR3 were used as controls. Six patients had antibodies against GluR2, three against GluR1, and one against both GluR1 and GluR2. None of the patients' antibodies reacted with GluR3 (Figure S1). All patients' antibodies reacted with cells co-transfected with both GluR1 and GluR2 (GluR1/2) (Figure 3 A-C; Table 1). None of the 110 control patients' samples produced the neuropil reactivity shown in Figure 1 or reacted with cells transfected with GluR1 or GluR2 (Figure S1).
Figure 3. Antibody reactivity with HEK293 cells co-transfected with GluR1/2 and quantitative ELISA studies.
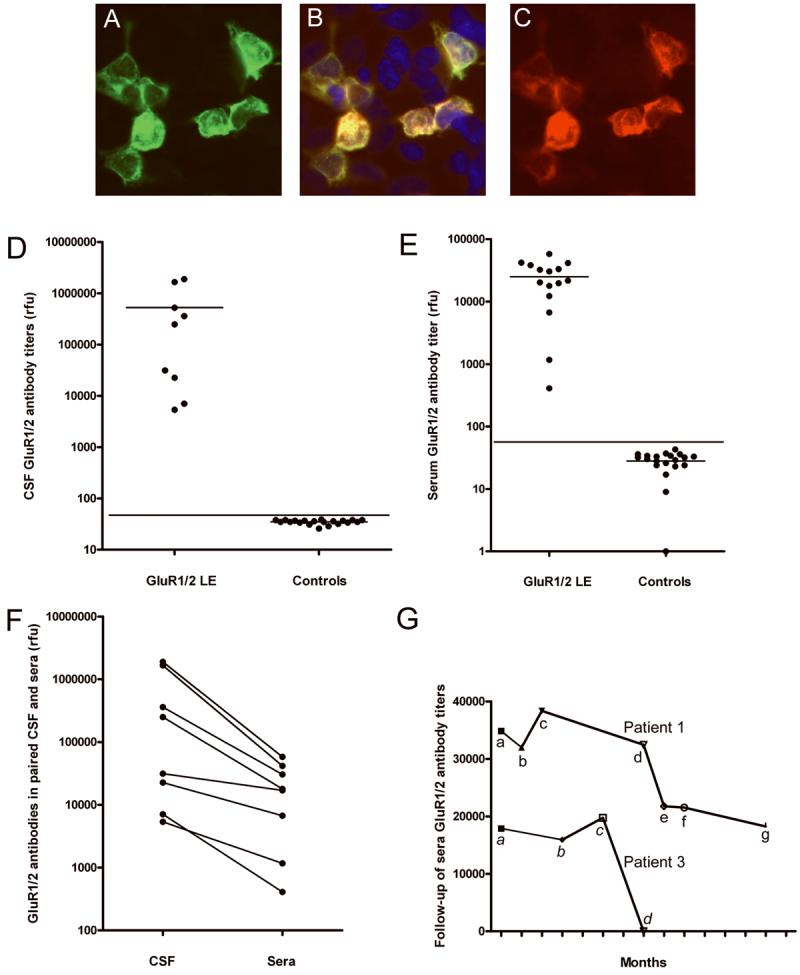
HEK293 cells co-transfected with GluR1/2 and immunostained with a patient's CSF (A) and an antibody specific for GluR2/3 (C). Note the co-localization of reactivities in (B). Protein extracts from HEK293 cells co-transfected with GluR1/2 were used to develop an ELISA (D-G).
Panel D shows the titers of GluR1/2 antibodies in the CSF of 9 patients with GluR1/2 associated LE and 20 CSF randomly selected from controls; horizontal lines in each subgroup indicate the mean; the line across subgroups indicates 3 standard deviations above the mean (p < 0.001).
Panel E shows the antibody titers of 8 patients' sera obtained during 15 episodes of LE (8 presentations and 7 relapses) and the titers of 20 controls (p < 0.001).
Panel F compares the titers of GluR1/2 antibodies in paired CSF and serum samples in which the IgG has been normalized; note that in all 8 patients the antibody titers are higher in the CSF, indicating intrathecal synthesis of antibodies.
Panel G shows the follow-up of serum antibody titers in two patients. Patient #1: a, titers at symptom presentation; b, 1 month after receiving 5-day treatment with intravenous methylprednisolone and plasma exchange (initially associated with substantial neurological improvement); c, first relapse of symptoms (after tapering corticosteroids), treated with intravenous methylprednisolone and IVIg; between c-d the patient had a second relapse partially treated with oral corticosteroids and anti-psychotic medication (she refused diagnostic tests and hospital admission); d, third relapse of symptoms, treated with IVIg and corticosteroids (partial neurological improvement); e-g, titers obtained while on azathioprine and stable neurological deficits. After the last time point (g) the patient was left with severe memory and behavioral deficits; for the last 3 years she has been living in a skill nursing facility with stable deficits (no follow-up titers available). Patient #3 had an episode of LE (that resolved spontaneously) 5 years before the current relapse; a, AMPAR antibody titers obtained at relapse of LE (which presented in association with anti-GAD related stiff-person syndrome); b-c, titers obtained during diagnostic studies and initial treatments (thymectomy, local radiation therapy); d, after intravenous corticosteroids (associated with dramatic improvement of LE). After the last time point (d), the patient remained only with symptoms of anti-GAD related stiff-person syndrome refractory to plasma exchange and IVIG; he eventually improved with chronic corticosteroids.
(rfu = relative fluorescence, ELISA reader, Biotek Instruments, Winooski, Vermont).
Taken together, these findings establish the presence of antibodies to GluR1/2 in this subgroup of patients with LE, and provide a diagnostic test for this disorder. We next developed an ELISA based on lysates of HEK293 cells expressing GluR1/2 and this proved to be useful for the quantification of antibodies (Figure 3 D-G). Analysis of GluR1/2 antibodies using normalized amounts of IgG from paired serum and CSF samples indicated that all tested patients had intrathecal synthesis of antibodies (Figure 3 G).
After identifying the GluR1/2 subunits as the target antigens, we next examined whether these subunits were present in patients' tumors (Supplemental Material and Methods: Immunohistochemistry on tissue). All tumors examined (4/4) expressed GluR1/2 subunits; 2 tumors predominantly expressed one subunit, which correlated with the patients' antibody specificity; the other two tumors expressed similar levels of GluR1/2 and both patients had GluR2 antibodies (Table 2, Figure 4).
Figure 4. Expression of GluR1 and GluR2 subunits by patients' tumors.

The tumor of a patient with GluR1 antibodies demonstrates high level of expression of GluR1 (A), mild expression of GluR2 (B), and reactivity with patient's antibodies (C). The tumor of a patient with GluR2 antibodies demonstrates high level of expression of GluR1 (D) and GluR2 (E), and reactivity with patient's antibodies (F).
Tissues immunostained with the indicated avidin-biotin peroxidase method, and mildly counterstained with hematoxylin. All panels x400.
Patients' antibodies decrease the number of AMPAR clusters and alter receptor localization at synapses
Given that the GluR2 subunit was the most frequent target of patients' antibodies and that most hippocampal AMPAR are composed of GluR1/2 heteromers, we next investigated whether patients' GluR2 antibodies affected the levels of GluR2 subunits in cultures of live hippocampal neurons. For these studies we first selected a representative patient's CSF containing antibodies that competed with the reactivity of other patients' GluR2 antibodies, indicating they target the same epitopes (data not shown). Then, the extent of specific GluR2 immunolabeling was quantified by confocal microscopy. These studies demonstrated that 91% of the clusters labeled by patient's antibodies corresponded to GluR2/3 (Figure 5 A).
Figure 5. Patient's antibodies selectively bind to GluR2 and alter the number and localization of AMPAR in live neurons.
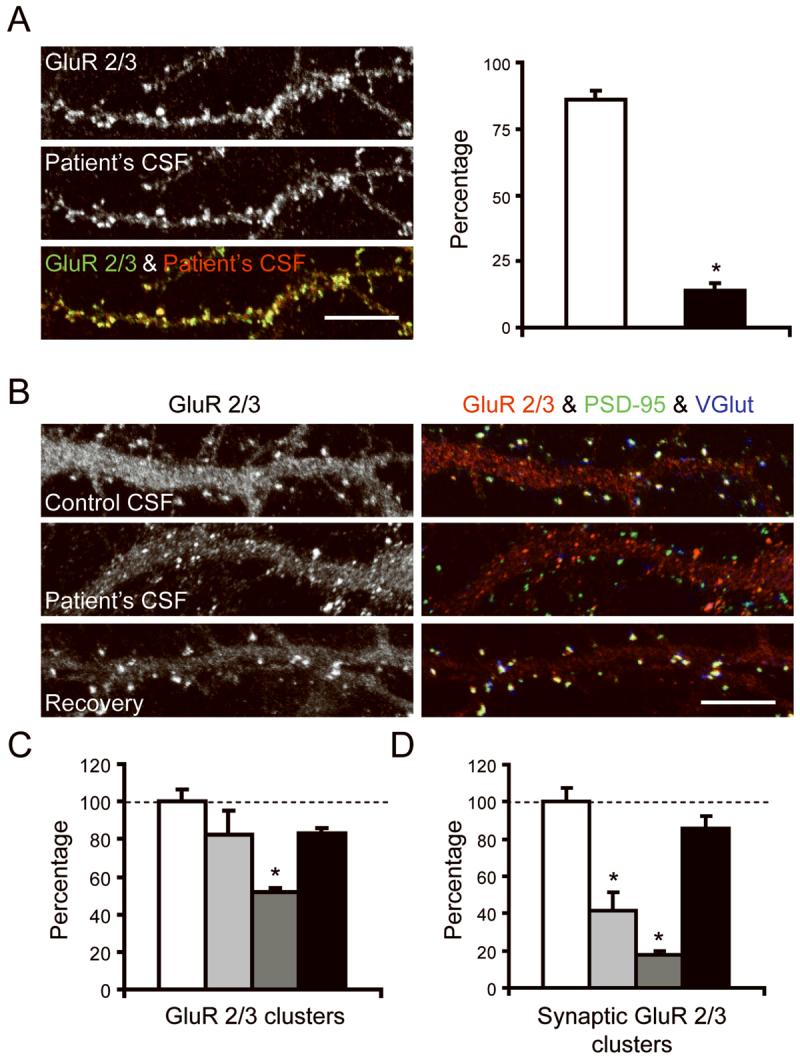
Panel A shows 17 div hippocampal neurons immunostained with patient's CSF (b+w, red) and with a commercial antibody specific for GluR 2/3 (b+w, green). White column indicates the number of GluR2/3-containing clusters labeled by patient's CSF; black column indicates the number of GluR2/3-containing clusters that are not labeled by patient's CSF. Since patient's antibodies react with GluR2, but not GluR3 (see text), the findings indicate that nearly all clusters labeled with patient's CSF correspond to GluR2 (91%, yellow puncta in overlay). Scale bar = 5 μm.
Panels B-D show hippocampal neurons cultured with control CSF or patient's CSF and subsequently immunostained for GluR 2/3 (b+w, red), the postsynaptic marker PSD-95 (green), and the pre-synaptic marker VGlut (blue). White columns indicate the number of GluR2/3-containing clusters in neurons cultured for 6 days with control CSF; Light grey columns indicate the number of clusters in neurons cultured for 3 days with patient's CSF; Dark grey columns indicates the number of clusters in neurons cultured for 6 days with patient's CSF; Black columns indicate the number of clusters in neurons cultured for 3 days with patient's CSF and subsequently cultured for 3 days with control CSF (3 day recovery). Note that patient's CSF, applied from 11-17 div (6 day treatment), reduces the number of GluR 2/3 labeled puncta compared with cultures exposed to control CSF (Panel B “GluR 2/3” and panel C; p < 0.001). Moreover, the patient's CSF applied for 3 or 6 days, but not the control CSF, reduces the number of GluR 2/3 clusters that colocalize with PSD-95 (yellow puncta = postsynaptic AMPAR), and the number of GluR 2/3 clusters that colocalize with VGlut (white puncta = presynaptic AMPAR) (Panel B “GluR 2/3 & PSD-95 & VGlut” and panel D; p < 0.01). These effects were reversed after removing the antibodies from the cultures and allowing the neurons to recover for 3 days (panels C and D). Scale bar = 5 μm.
For each condition, a minimum of 6-15 neurons was examined on each of 2-3 coverslips in 3 independent experiments. Neurons were randomly selected for analysis. The number of synaptic clusters was calculated per 20 μm and expressed as a percentage of control values. Results were analyzed using the Kruskal-Wallis non-parametric ANOVA followed by Dunn's pairwise comparison.
Having shown the high specificity of patients' antibodies for GluR2 in cultures of live hippocampal neurons, we next examined whether prolonged neuronal exposure to patient's antibodies changed the number or localization of GluR2-containing AMPAR clusters. For each condition, a minimum of 6-15 neurons was examined on each of 2-3 coverslips in 3 independent experiments. Neurons were randomly selected for analysis. The number of synaptic clusters was calculated per 20 μm and expressed as a percentage of control values. These studies demonstrated that patients' antibodies, but not controls, decreased the number of GluR2-containing AMPAR clusters after 6 days of antibody exposure (p < 0.001; Figure 5 B, C) but not after 3 days of antibody exposure (p > 0.05; Figure 5 B, C). In contrast, patients' antibodies, but not controls, decreased the co-localization of GluR2 with post-synaptic (PSD-95) and pre-synaptic (VGlut) markers after 3 and 6 days of antibody exposure (p < 0.01; Figure 5 B, D). The effects on receptor number and localization of AMPAR clusters at post- and presynaptic sites were reversed with removal of antibodies from the neuronal cultures (Figure 5 B-D). Moreover, the effects were AMPAR-selective as the normal co-localization of PSD-95 with VGlut at synapses and the localization of NMDA receptors at postsynaptic sites (data not shown) were not altered by patient's antibodies.
DISCUSSION
We report the clinical and immunological features of a new type of LE that associates with antibodies against GluR1/2 subunits of the AMPAR. The AMPAR are ionotropic glutamate receptors that are highly conserved among mammals, and mediate most of the fast excitatory neurotransmission in the brain.9 The majority of AMPAR are tetramers composed of GluR1, 2, 3 or 4 subunits that combine in a brain region-dependent manner.10 The regions with highest levels of GluR1/2 and GluR2/3 receptors are the synaptic CA3-CA1 areas of the hippocampus, followed by the subiculum, cerebellum, caudate-putamen, and cerebral cortex.11 This distribution is similar to the immunostaining of our patients' antibodies. Although GluR2 and GluR3 share sequence homology, our patients antibodies did not react with GluR3, a subunit that has been identified as an autoantigen in some patients with Rasmussen's encephalitis.12
In most respects the clinical presentation and MRI findings of anti-GluR1/2 associated encephalitis are typical of LE, and therefore this diagnosis was considered early in 9 patients. The other patient was suspected of having a rapidly progressive dementia but the MRI and antibody findings lead to the recognition of LE. Seven patients had tumors of the thymus, breast or lung, and an underlying neoplasm was excluded in the other 3 patients after extensive tumor screening. Although this follow-up may be too short to definitively exclude a neoplasm, the assessment of individual patients suggests that this disorder may occur as an autoimmune, non-paraneoplastic syndrome. For example, the patient with the shortest follow-up (8 months) is a 38 year-old woman that has fully recovered and has no risk factors for cancer. The only patient with risk factors for cancer (smoking) has been followed for 50 months without evidence for a neoplasm.
A frequent feature of anti-GluR1/2 associated LE is the tendency to relapse. A total of 14 episodes of LE (5 initial episodes and 9 relapses) occurred in 5 patients, ranging from 1 to 3 relapses in each patient. The presence of GluR1/2 antibodies in serum or CSF was demonstrated in 14/14 episodes studied. Except for two patients whose tumor was initially diagnosed at first relapse of LE, all other neurological relapses occurred in patients without tumor or without tumor recurrence. A compelling example is a patient who after successful treatment of a carcinoma of the thymus at first presentation of LE, subsequently developed 3 relapses over 101 months without evidence of tumor recurrence (total follow-up of 10 years). Taken together, the presence of a GluR1/2-expressing tumor and the susceptibility for autoimmune disorders (identified in 5 patients) either alone or combined may play a role in triggering this autoimmune disorder.
A propensity to relapse was also noted in a study of 100 patients with encephalitis and antibodies to NMDA receptor, a member of another category of ionotropic glutamate receptors.6 Although AMPAR and NMDAR are functionally related and play critical roles in synaptic plasticity,13 each autoimmunity associates with a different clinical phenotype. For example, none of the patients of the current study had an ovarian teratoma or developed dyskinesias, autonomic instability, and hypoventilation, which are common features of anti-NMDA receptor encephalitis.6,14
Neuronal plasticity can be studied through a series of models termed long-term potentiation (LTP) or depression (LTD).13 In these models synaptic strength increases or decreases in association with changes in trafficking of AMPAR.11 The results in our patients are analogous to those seen in some LTD paradigms.9 Our study shows that patients' antibodies specifically bind to AMPAR clusters causing a decrease in the number of the receptors at synapses, and to a lesser degree the total number of receptor clusters along dendrites. Since the major effect was on the synaptic location of the receptors, this finding suggests a mechanism whereby the antibodies disrupt receptor trafficking/turn over, relocating them from synaptic to extrasynaptic sites/intracellular pool. The reversibility of these effects provides an explanation for the improvement of patients' symptoms with plasma exchange, IVIg, or corticosteroids.
Nine patients dramatically improved after the first episode of LE. All nine patients received immunotherapy and when appropriate oncological therapy. However, the long-term outcome depended on the appropriate management of the relapses. In some patients treatment was challenging because in each episode they became agitated, belligerent, and unmanageable at home, yet refusing hospital admission and medication. In two patients, the recovery from each relapse was incomplete resulting in cumulative residual memory or behavioral deficits. After the third relapse both patients stabilized with prolonged use of azathioprine; one was left with moderate short-term memory deficits; the other required institutionalization in a skill nursing facility where she has been living for 3 years. Another patient, an 87-year old woman, died at the second relapse shortly after a prolonged episode of status epilepticus.
The neurological outcome was not influenced by the presence of a tumor (as long as this was well controlled), but was adversely influenced by the presence of overlapping immune responses. For example, after recovering from anti-GluR1/2 associated LE one patient suffered from prolonged residual symptoms of anti-GAD-associated stiff-person syndrome; the GluR1/2 antibodies had disappeared after treatment but the GAD antibodies remained elevated (data not shown). Another patient, with GluR1/2 and CRMP5 antibodies, had rapid neurological deterioration that resulted in death; the autopsy confirmed prominent cytotoxic T-cell infiltrates in the limbic system. A possible explanation for these outcomes is that the accompanying immune responses, particularly if associated with cytotoxic T-cell mechanisms, are more difficult to treat, or that the neuronal dysfunction is less reversible than that caused by the anti-GluR1/2 immune response, which appears to be directly mediated by antibodies.15,16
Anti-GluR1/2 associated LE represents a new category of immune-mediated encephalitis that may occur with or without systemic tumors, and shows a propensity to relapse. The disorder is treatable and can now be diagnosed serologically. Potential cases are patients who are currently categorized as “antibody negative LE” or “steroid-responsive LE”. Future studies should determine the extent of tumor association and overlapping autoimmunities, the role of chronic immune suppression in preventing relapses, and the molecular mechanisms whereby antibodies alter the synaptic localization of AMPAR.
Supplementary Material
Acknowledgments
We thank Drs. Alfredo Voloschin, Sergio Zanini, and Gian Carlo Muscas for providing clinical information. This work was supported in part by 2R56CA089054 and RO1CA107192 (JD); R21 MH057683 (RB-G); NSR56-45986, NSR01-45986, and Foederer Foundation of the Children's Hospital of Philadelphia (DL); NIH NIMH F31MH083395 (AJG); the McBean Family Foundation, the Larry L. Hillblom Foundation, and National Institute on Aging Training Grant AG023481 (HS).
Reference List
- 1.Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain. 1968;91:481–496. doi: 10.1093/brain/91.3.481. [DOI] [PubMed] [Google Scholar]
- 2.Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123(Pt 7):1481–1494. doi: 10.1093/brain/123.7.1481. [DOI] [PubMed] [Google Scholar]
- 3.Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. J Neurol Neurosurg Psychiatry. 2007;78:381–385. doi: 10.1136/jnnp.2006.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764–1777. doi: 10.1093/brain/awh526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graus F, Saiz A, Lai M, et al. Neuronal surface antigen antibodies in limbic encephalitis: clinical-immunologic associations. Neurology. 2008;71:930–936. doi: 10.1212/01.wnl.0000325917.48466.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008 doi: 10.1016/S1474-4422(08)70224-2. October 13, 2008. DOI:10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchhalter JR, Dichter MA. Electrophysiological comparison of pyramidal and stellate nonpyramidal neurons in dissociated cell culture of rat hippocampus. Brain Res Bull. 1991;26:333–338. doi: 10.1016/0361-9230(91)90003-3. [DOI] [PubMed] [Google Scholar]
- 8.Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 10.Palmer CL, Cotton L, Henley JM. The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev. 2005;57:253–277. doi: 10.1124/pr.57.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprengel R. Role of AMPA receptors in synaptic plasticity. Cell Tissue Res. 2006;326:447–455. doi: 10.1007/s00441-006-0275-4. [DOI] [PubMed] [Google Scholar]
- 12.Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science. 1994;265:648–651. doi: 10.1126/science.8036512. [DOI] [PubMed] [Google Scholar]
- 13.Genoux D, Montgomery JM. Glutamate receptor plasticity at excitatory synapses in the brain. Clin Exp Pharmacol Physiol. 2007;34:1058–1063. doi: 10.1111/j.1440-1681.2007.04722.x. [DOI] [PubMed] [Google Scholar]
- 14.Iizuka T, Sakai F, Ide T, et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology. 2008;70:504–511. doi: 10.1212/01.wnl.0000278388.90370.c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernal F, Graus F, Pifarre A, Saiz A, Benyahia B, Ribalta T. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis. Acta Neuropathol (Berl) 2002;103:509–515. doi: 10.1007/s00401-001-0498-0. [DOI] [PubMed] [Google Scholar]
- 16.Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7:327–340. doi: 10.1016/S1474-4422(08)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.