

Abstract
Niemann-Pick Type C (NPC) disease is a devastating developmental disorder with progressive and fatal neurodegeneration. Previous work has shown that a single injection of the neurosteroid allopregnanolone at postnatal day 7 significantly prolonged lifespan of Npc1-/- mice. However, the cellular/molecular basis for this beneficial effect remains undefined. Here, we further characterized the effect of allopregnanolone treatment on cholesterol accumulation, a pathological hallmark of NPC, as well as on autophagic/lysosomal dysfunction, myelination and inflammation in Npc1-/- mouse brains. At 1 month postnatal, accumulation of filipin-labeled unesterified cholesterol was clearly evident not only in neurons but also in microglia in untreated mutant mice, but was mostly absent in allopregnanolone-treated animals. Brain levels of the lysosomal enzymes cathepsins B and D were significantly higher in Npc1-/- than in wild-type mice. Levels of LC3-II, an autophagy marker, were also increased in mutant mouse brain as compared to wild-type mouse brain. Both changes were significantly reduced by allopregnanolone treatment. Injection of the neurosteroid also significantly reduced astrocyte proliferation and microglial activation. Furthermore, allopregnanolone treatment significantly enhanced myelination in mutant mice. Taken together, our results clearly show that allopregnanolone treatment not only reduces cholesterol accumulation and improves autophagic/lysosomal function but also enhances myelination and reduces inflammation. These results provide further support for the potential usefulness of allopregnanolone for treating NPC disease.
Keywords: cathepsin, glia, LC3, lipids, neurosteroid, myelination
1. Introduction
Niemann-Pick type C (NPC) disease is a rare but fatal lipid storage disease characterized by accumulation of cholesterol in the liver, spleen and brain (Vanier et al., 1988). In the majority of patients, the disease is caused by mutations in the NPC1 gene, which encodes the NPC1 protein (Carstea et al., 1997). NPC1 is an integral membrane protein participating in lipid trafficking from late endosomes/lysosomes into other subcellular compartments (Scott and Ioannou, 2004; Walkley and Suzuki, 2004). Loss of functional NPC1 proteins leads to lipid accumulation, especially unesterified cholesterol, in late endosomes/lysosomes, and subsequent decreased biosynthesis of downstream neurosteroids such as progesterone and allopregnanolone, which are important for neurogenesis, cell proliferation, survival, and migration [see (Mellon, 2007) for a recent review]. Cholesterol accumulation in neurons in Npc1-/- mutant mice was observed as early as postnatal day 9 (Ong et al., 2001; Reid et al., 2004). Other neuropathologic features include formation of meganeurites (spindle shaped swelling at the initial segments of axons) and axonal spheroids and progressive neuronal loss, especially of Purkinje cells (Higashi et al., 1993; March et al., 1997; Zervas et al., 2001a). Moreover, the neurofibrillary tangles found in NPC patients have linked NPC pathology to that of Alzheimer's disease (Auer et al., 1995; Suzuki et al., 1995). However, the association between cholesterol accumulation and neuronal death is not completely elucidated.
Currently there is no effective treatment for NPC disease. Introduction of functional npc1 gene in the Npc1-/- mouse brain prevents neurodegeneration, normalizes lifespan, and corrects sterility (Loftus et al., 2002). Another recent study has shown that restoring Npc1 function only in astrocytes triples Npc1-/- mice lifespan, indicating that astrocytes play a critical role in NPC disease (Zhang et al., 2008). N-butyldeoxynojirimycin (Miglustat), an inhibitor of glycosphingolipid biosynthesis, has also been shown to extend the average lifespan from 67 days to 89 days in NPC mouse model (Zervas et al., 2001b). To date, the most promising treatment is perhaps the neurosteroid, allopregnanolone (ALLO), as a single injection at postnatal day 7 almost doubled Npc1-/- mice lifespan (Griffin et al., 2004). Further in vitro experiments indicated that ALLO-mediated Purkinje cell survival was blocked by the GABAA receptor antagonist, bicuculline, suggesting that the effect of the drug might be mediated by GABAA receptors (Griffin et al., 2004). However, this hypothesis has been challenged by the finding that ent-ALLO, an ALLO stereoisomer without GABAA receptor agonist function has identical effect as natural ALLO, which strongly suggests that GABAA-independent mechanisms are involved in ALLO's effects (Langmade et al., 2006). On the other hand, T0901317, a synthetic oxysterol ligand, acts in concert with ALLO to promote survival and to delay the onset of neurological symptoms (Langmade et al., 2006). The effects of ALLO and T0901317 correlate with their ability to activate the pregnane X receptor (PXR), suggesting a role of this receptor. However, other researchers have reported that there is no detectable PXR activity in mouse cerebellum (Bookout et al., 2006; Gofflot et al., 2007; Repa et al., 2007). To complicate the issue further, a recent study reported that administration of β-cyclodextrin, the vehicle used in the ALLO studies, also rescued Npc1-/- mice (Liu et al., 2008). To better understand the molecular/cellular basis of ALLO-mediated protection, we assessed its effects on cholesterol accumulation, autophagic/lysosomal function, and microglia- and astrocyte-mediated inflammation in Npc1-/- mice.
2. Results
2.1. Reduction of cholesterol accumulation in Npc1-/- mice by ALLO treatment
In untreated 1-month-old Npc1-/- mice, filipin-positive granules corresponding to cholesterol accumulation in endosomes/lysosomes were clearly detected in several brain regions (Figs. 1-3). In cerebellum, filipin-labeled puncta (arrows in Fig.1A) were observed in calbindin immunopositive Purkinje cells. In ALLO-treated animals, the pattern of filipin staining was similar to that observed in wild-type animals, with the staining present mainly in plasma membranes (Fig. 1). At 2 months postnatal, cholesterol accumulation was observed in both treated and untreated animals, although much higher levels were observed in the latter (not shown). Treatment with β-cyclodextrin (with 5.6 degrees of substitution, Sigma, Cat. No. 33260-7) alone did not reduce cholesterol accumulation as efficiently as ALLO (Fig. 1C), although in some animals, filipin-stained clusters in Purkinje cells became smaller and distributed more evenly around nucleus in the soma.
Figure 1. Reduction in intraneuronal cholesterol accumulation in Npc1-/- mice by treatment with ALLO but not with β-cyclodextrin.

Brains of 1-month-old wild-type (Npc1+/+) or Npc1-/- mice injected at p7 with ALLO (Npc1-/-Al), water (Npc1-/-), or β-cyclodextrin (Npc1-/-CD) were processed for filipin staining followed by immunostaining with anti-calbindin antibodies as described in Materials and Methods.
A. Images of sagittal cerebellar sections stained with filipin (blue) and anti-calbindin antibodies (label Purkinje cells; red). gl, granular cell layer; ml, molecular layer; pl, Purkinje cell layer. B. Images of cortex (CX) stained with filipin. ALLO treatment markedly reduced filipin-stained puncta (arrows) in cerebellar Purkinje cells and in cortical neurons of Npc1-/- mice. C. Filipin staining in the cerebellum of a β-cyclodextrin-treated Npc1-/- mouse; there is no evident difference in cholesterol accumulation in Purkinje cells between β-cyclodextrin-treated and water-treated Npc1-/- mice. Scale bar = 10 μm.
Figure 3. Lack of cholesterol accumulation in oligodendrocytes in Npc1-/- mice.
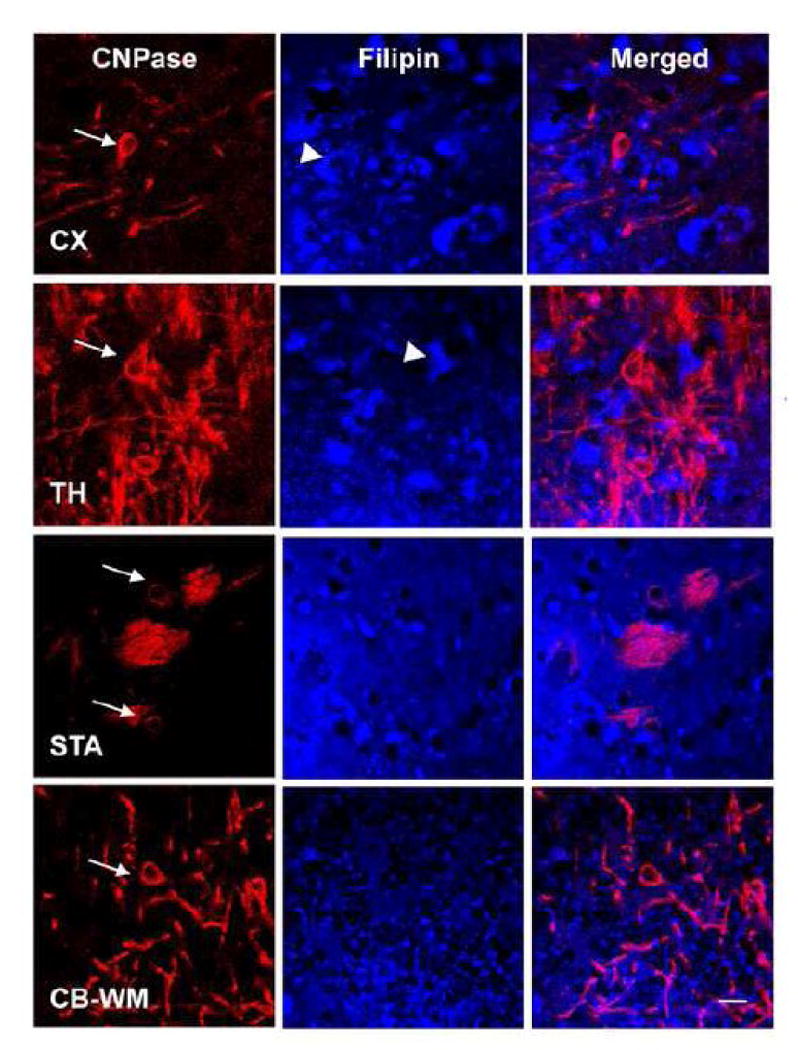
Images showing CNPase (red; an oligodendrocyte marker) immunoreactivity and filipin (blue) staining in cortex (CX), thalamus (TH), striatum (STA), and cerebellar white matter (CB-WM) of 1-month-old Npc1-/- mice. No clear filipin-positive puncta were found inside CNPase-immunopositive oligodendrocytes (arrows), although cholesterol accumulation was evident in adjacent neuron-like cells (arrowheads). Scale bar =10 μm.
Double immunofluorescent staining with F4/80 antibodies that label microglia showed that in 1-month-old Npc1-/- mice, filipin-labeled cholesterol aggregates were also found in reactive microglia that had large cell bodies and thick processes, especially in cortex (Fig. 2A) and cerebellum (Fig. 2B). Cholesterol aggregates were mainly found in cell bodies, and little filipin staining was found in processes. ALLO treatment not only reduced cholesterol accumulation in microglia, but also changed their morphology: the cell bodies became smaller and the processes thinner, and more similar to those found in wildtype mice (Fig. 2 and Fig. 6B). There was no obvious cholesterol accumulation in oligodendrocytes (Fig. 3) in 1-month-old Npc1-/- mice (Fig. 3) in all brain areas examined.
Figure 2. Reduced cholesterol accumulation in microglia in Npc1-/- mice by ALLO treatment.

Filipin- (blue) stained brain sections were further immunostained with anti-F4/80 (green; a marker of microglia) antibodies. Filipin-labeled cholesterol (blue) was found in cell bodies and processes of F4/80-immunopositive microglia in cortex (A, CX) and cerebellum (B, CB) of 1-month-old Npc1-/- mice. ALLO treatment (Npc1-/-Al) markedly reduced cholesterol accumulation and microglial activation. Scale bar =25 μm in A and 10 μm in B.
Figure 6. Effects of ALLO treatment on glial function in Npc1-/- mice.

A. Myelination is improved by ALLO treatment. Immunohistochemical analysis of CNPase in cortex (CX), striatum (STA), and cerebellum (CB) of 1-month-old Npc1+/+ and Npc1-/- mice treated with ALLO (Npc1-/-Al) or water (Npc1-/-) was carried out as described in Materials and Methods. Dashed lines in top panels (CX) indicate cortical layers containing CNPase-immunopositive elements. In Npc1-/- mice, only the deeper layer (layer 6) of cortex exhibit CNPase immunoreactive products, while in ALLO-treated mice, CNPase-immunoreactive oligodendrocytes and their processes expend up to the superficial layers. 2-6 indicate cortical layers. Similarly, more CNPase-immunoreactive processes were present in striatum and cerebellum of ALLO-treated compared to untreated Npc1-/- mice. gl, granular cell layer; ml, molecular layer; pl, Purkinje cell layer. Scale bar =50 μm.
B. Microglial reaction is diminished by ALLO treatment. Cerebellar sections from 1-month-old Npc1+/+ and Npc1-/- mice treated with ALLO (Npc1-/-Al) or water (Npc1-/-) were immunostained with anti-CD11b antibodies, Mac1, as described in Material and Methods. Larger reactive microglia (arrows) were found in Npc1-/- mice, but were absent in ALLO-treated Npc1-/- mice. gl, granular cell layer; ml, molecular layer; pl, Purkinje cell layer. Scale bar =50 μm.
C. Increased levels of GFAP in Npc1-/- mice are reduced by ALLO treatment. Shown are representative images of GFAP immunoblot processed with proteins from cortical homogenates from 1 (1M) and 2 (2M) month-old mice.
2.2. Reduction of brain autophagic/lysosomal dysfunction in Npc1-/- mice by ALLO treatment
To determine the effect of ALLO treatment on autophagic function in brains of Npc1-/- mice, homogenates from the cortex of 1 and 2-month-old Npc1-/- mice with or without ALLO treatment were subjected to immunoblotting analysis using antibodies against the microtubule-associated protein 1-light chain 3 (LC3) since during autophagy induction, LC3-I is converted to LC3-II, and this conversion is widely used as an index of autophagy (Kabeya et al., 2000). Immunoblot results showed that LC3-II levels were significantly higher in Npc1-/- mice (normalized to tubulin levels; 222 ± 21%; means ± SEM, expressed as % of values in wild-type mice; n =3-4, p <0.05) than in Npc1+/+ mice. The ratio of LC3-II/LC3-I, which reflects the conversion rate, was 198 ± 14% (Fig. 4A&C; n =3-4, p <0.05) at 1 month and 192% ± 16% at 2 months in Npc1-/- mice, of those found in age-matched wild-type mice (Fig. 4B&C; n =3-4, p <0.05). The increases in LC3-II levels and in LC3-II/LC3-I ratios were significantly reduced by ALLO treatment in both 1- and 2-month-old mutant mice, although a larger decrease was observed in younger animals (Fig. 4).
Figure 4. Reduced autophagic abnormality in Npc1-/- mice by ALLO treatment.
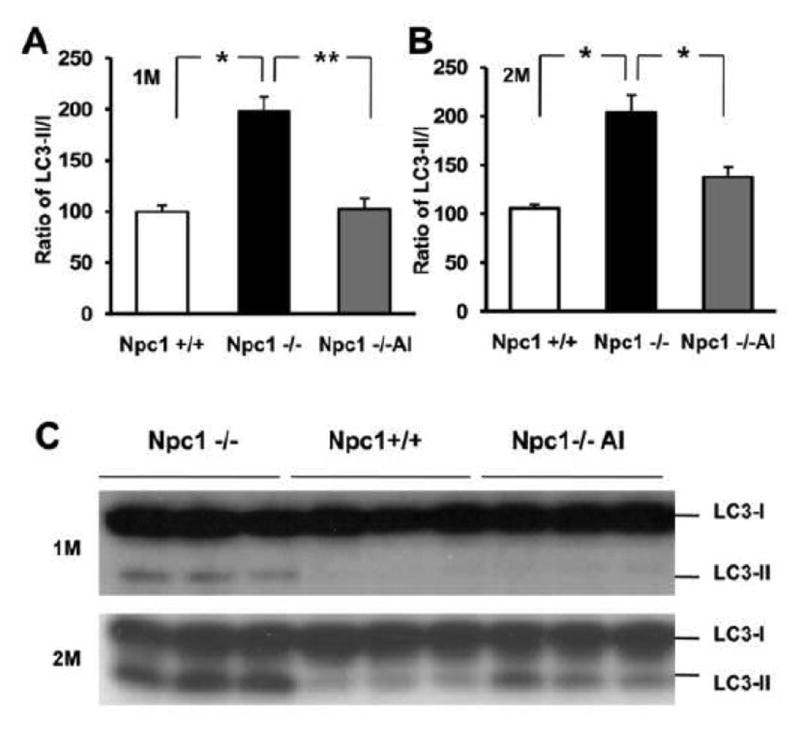
Proteins from cortical homogenates of 1 and 2-month-old wild-type (Npc1+/+) or Npc1-/- mice treated with ALLO (Npc1-/-Al) or water (Npc1-/-) were separated by SDS-PAGE (14 % resolving gel), transferred to PVDF membrane and probed with anti-LC3 antibodies. Representative blot images are shown in panel C, while quantitative data are presented in panels A&B. Levels of LC3I and LC3II were quantified using NIH Image J software and adjusted against loading control (GAPDH); LC3II/LC3I ratio was then calculated, normalized to values measured in NPC1+/+ mice and expressed as mean ± SEM. * p<0.05, ** p<0.01. The increase in LC3II/LC3I ratio in Npc1-/- mice, which represents abnormality in autophagic flux, was significantly reduced by ALLO treatment.
A previous study has shown that levels of beclin 1, another autophagy-related protein, are increased in Npc1-/- mouse brain (Pacheco et al., 2007). Our immunoblotting results revealed a small and not significant increase in beclin 1 levels in cortical homogenates of 1-month-old Npc1-/- mice (116 ± 2 %, p =0.054 compared to wild-type mice; Fig. S1). Nevertheless, ALLO-treatment induced a significant decrease in beclin 1 levels (p <0.01 compared to untreated mutant mice; Fig.S1). No significant change was observed in levels of phospho-mTOR and Atg5, two other proteins participating in autophagic activity (data no shown).
Autophagic/lysosomal pathways play important roles in the degradation and recycling of long-lived membrane proteins, especially under starvation stress. In agreement with our previous report (Liao et al., 2007), immunoblotting results indicated that levels of cathepsins B and D, two lysosomal proteases, were significantly increased in cortex of 1-month-old Npc1-/- mice. ALLO treatment significantly reversed the increase in levels of the 34 kDa mature isoform of cathepsin D, from 216 ±11% to 137 ± 8% (Fig. 5B&C; n =3-4, p <0.05) at 1 month, and from 249 ± 19% to 157 ± 16% (Fig. 5B&D; n=4, p <0.05) at 2 months of age. Increased cathepsin B levels in mutant mice were also significantly reduced by ALLO treatment (Fig. 5B&E). Immunofluorescent staining with anti-cathepsin D antibodies revealed cathepsin D-immunopositive puncta clustered in neuron-like cells in cortex of 1 month-old Npc1-/- mice; the neuronal identity of these cells was confirmed by double immunostaining with NeuN antibodies (Fig. S2). In some neurons, the puncta formed crescent-shaped caps at one pole (Fig. 5A, arrowheads). Levels of cathepsin D immunoreactivity in neurons further increased by 2 months of age and the polarized cathepsin D distribution was observed in most neurons. In mutant mice, but rarely in wild-type mice, cathepsin D immunoreactivity was also found in small cells that were immunopositive for the microglia marker F4/80 (Fig.5A, arrow). ALLO treatment reduced cathepsin D immunoreactivity to levels similar to those observed in wild-type mice (Fig. 5A). Interestingly, in cerebellum of Npc1-/- mice, increases in cathepsin D levels were mainly found in microglia in the molecular layer; little increase was found in the soma of Purkinje cells (Fig. S3). Changes in cathepsin D immunoreactivity in cerebellum were also markedly reduced by ALLO treatment.
Figure 5. Decreased lysosomal dysfunction in NPC1-/- mice by ALLO treatment.
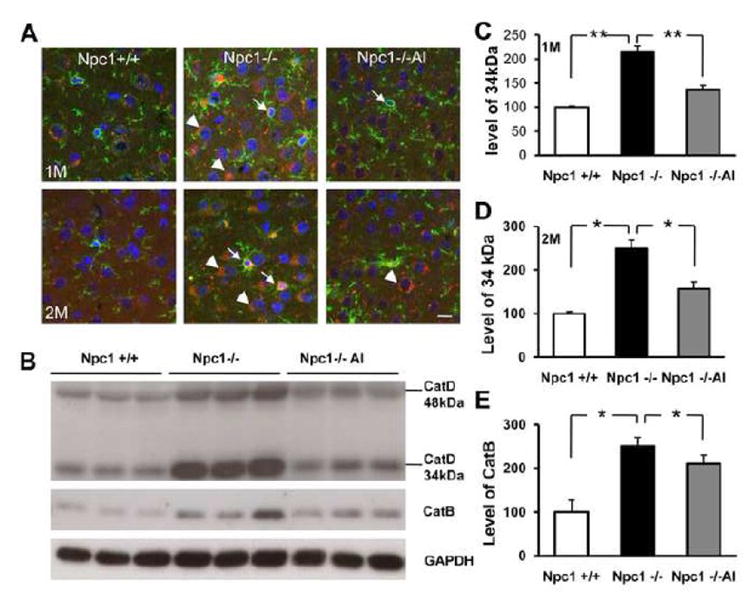
A. Immunostaining of cathepsin D (red) and F4/80 (green) in cortex of 1 and 2-month-old Npc1+/+ and Npc1-/- mice treated with ALLO (Npc1-/-Al) or water (Npc1-/-). In Npc1-/- mice, most cathepsin D immunoreactivity was localized in F4/80-positive microglia (arrows) and in neurons (arrowheads). Cathepsin D immunoreactivity was greatly reduced in ALLO-treated Npc1-/- mice. Note that in cortical neurons of Npc1-/- mice, but not of ALLO-treated, cathepsin D-immunoreactive products often clustered at one pole of the cell forming a “cap” like structure. Blue: DAPI stain to label nuclei. Scale bar =10 μm.
B. Representative images of cathepsin D and cathepsin B immunoblots performed with proteins from whole homogenates from cortex of 1-month-old Npc1+/+, Npc1-/- mice treated with ALLO (Npc1-/-Al) or water (Npc1-/-). GAPDH was used as loading control. Compared to Npc1+/+ mice, there was a clear increase in levels of both cathepsin B and D, which markedly eliminated by ALLO treatment. C&D. Quantification of levels of the 34 kDa band of cathepsin D in 1 (C) and 2 (D) month-old mice. E. Quantification of cathepsin B levels in 2-month-old mice. All data are presented as % of values found in Npc1+/+ mice. * p<0.05, ** p<0.01.
2.3. Enhanced myelination and suppression of microglial and astrocytic activation in brain of Npc1-/- mice by ALLO treatment
The effects of ALLO treatment on oligodendrocytes and myelination were determined by immunohistochemistry using an antibody against CNPase (2′, 3′-cyclic nucleotide 3′-phosphodiesterase [or -phosphohydrolase], EC 3.1.4.37), an enzyme found almost exclusively in oligodendrocytes in mammalian brain. In 1-month-old Npc1-/- mice, CNPase immunopositive areas in cortex and striatum were 37 ± 5% and 49 ± 4% of those found in Npc1+/+ mice, respectively (n =4-5, *p <0.01 compared to Npc1+/+ mice). However, in cortex of ALLO-treated Npc1-/-mice, CNPase-immunopositive area was 78 ± 2% of that in Npc1+/+ mice (Fig. 6A, p <0.01 compared to Npc1-/- mice). ALLO-treatment also increased CNPase-immunopositive area in striatum, although to a much smaller degree (63 ± 10%). Improvement in myelination by ALLO-treatment in both brain structures was still evident by 2-month of age, 73 ± 13% in treated compared to 27 ± 5% in untreated in cortex and 86 ± 7% compared to 56 ± 9% in striatum (n =4-5, p<0.01). ALLO-treatment also led to an enhancement in myelination in cerebellum, a result which is in agreement with a previously published study (Ahmad et al., 2005). On the other hand, β-cyclodextrin injection alone did not significantly improve myelination in 1-month old Npc1-/- mice (data no shown).
Early-onset inflammation mediated by microglia and astrocytes has been previously reported in Npc1-/- mice (Baudry et al., 2003). Immunohistochemistry carried out with the anti-CD11b antibody Mac-1, a commonly used microglia marker, revealed numerous Mac1-immunopositive microglia in cerebellum of 1-month-old Npc1-/- mice. The number of microglia further increased by 2 months of age and their cell bodies became larger and their processes shorter, suggesting that microglia were in a highly reactive state (not shown). In contrast, very few microglia were seen in cerebellum of ALLO-treated 1-month-old Npc1-/- mice (Fig. 6B). By 2 months of age, the number of Mac1-immunopositive microglia in cerebellum of ALLO treated Npc1-/- mice increased; however, their cell bodies were smaller and their processes longer compared with those observed in untreated Npc1-/- mice (not shown). ALLO treatment induced similar changes in microglia in other brain regions such as cortex, striatum, and thalamus (not shown).
Immunohistochemistry results indicated that ALLO treatment also significantly reduced the numbers of GFAP-immunopositive astrocytes in cortex of Npc1-/- mice (Fig. S4). Immunoblotting analysis confirmed that ALLO treatment significantly reduced GFAP levels in cortex of Npc1-/- mice (Fig. 6C). The effect of ALLO treatment on GFAP expression was smaller at 2 months of age compared with that observed at 1 month.
Previous studies from our laboratory provided evidence that the PI3K-Akt-GSK-3β signaling system was markedly disturbed in Npc1-/- mice (Bi et al., 2005). GSK-3β is constitutively active and its regulation is generally through inhibition by phosphorylation at Ser9 residue. As previously reported, high levels of phosphorylated GSK-3β (GSK-3β-p-Ser9) were observed in several brain regions of 1-month-old Npc1-/- mice (Fig. 7). However, in ALLO-treated 1-month-old Npc1-/- mice, GSK-3β-p-Ser9 immunoreactivity was almost completely abolished in cortex and striatum; ALLO treatment also significantly reduced the number of GSK-3β-p-Ser9-immunopositive Purkinje cells in cerebellum. These results suggest that ALLO treatment also affects GSK-3β signaling pathway.
Figure 7. Reduced abnormal levels of GSK-3βser9 immunoreactivity in brains of Npc1-/- mice by ALLO treatment.

Immunohistochemical analysis of inactive GSK-3β (phosphorylated at ser9 residue; GSK-3βser9) in cerebellum (CB), cortex (CX), and striatum (STA) of 1-month-old mice was performed with antibodies that specifically recognize GSK-3βser9. Note that numerous GSK-3βser9-immunoreactive cells are present in CB, CX, and STA of water-treated but not in ALLO-treated (Npc1-/-Al) Npc1-/- mice. gl, granular cell layer; ml, molecular layer; pl, Purkinje cell layer. Scale bar =50 μm.
3. Discussion
Our results show for the first time that a single injection of allopregnanolone (ALLO) at postnatal day 7 significantly reduced cholesterol accumulation, in both neurons and microglia, in several brains regions of Npc1-/- mice. In parallel with the improvement of cholesterol homeostasis, ALLO treatment also reduced abnormal autophagic activity as well as lysosomal dysfunction in neurons and in microglia and reduced microglial reaction. ALLO treatment also normalized brain levels of GSK-3β. Furthermore, our results show that the neurosteroid treatment improved myelination in brains of Npc1-/- mice in a cholesterol accumulation-independent manner, suggesting that ALLO treatment may activate multiple cell signaling pathways.
3.1. Delayed cholesterol accumulation in neurons and microglia of Npc1-/- mice by ALLO treatment
Our results indicated that a single ALLO injection at postnatal day 7 markedly reduced filipin-labeled unesterified cholesterol accumulation in neuronal endosome/lysosomes in several brain regions; a complete abrogation was detected at postnatal 1 month and a partial reduction at 2 months of age. The effect of ALLO was larger at 1 month than at 2 months, suggesting that ALLO treatment is delaying the onset of the disease rather than providing a real rescue.
A recent genetic/pharmacological study showed that deletion of liver X receptors accelerated the progression of NPC disease, while activators of the receptors increased cholesterol excretion from brain and slowed neurodegeneration in Npc1-/- mice (Repa et al., 2007). The authors of this study therefore proposed that ALLO-induced protective effects are mediated through the activation of liver X receptors, thereby reducing brain cholesterol levels. One caveat of this hypothesis is that cholesterol levels in NPC brain are not higher, if not lower, than in normal brain (Vanier, 1999; Xie et al., 1999). However, it is possible that ALLO-treatment induces cholesterol efflux from endosomes/lysosomes through the recruitment of cholesterol transporters, thereby decreasing intracellular cholesterol accumulation. In this regard, mRNA levels of NPC2, apoD, and Lip1, and protein level of apoD, which are all involved in lipoprotein and cholesterol transport, are increased in of Npc1-/- mice brain (Li et al., 2005; Ong et al., 2002; Suresh et al., 1998). These modifications might represent a spontaneous compensatory reaction to cholesterol modifications. Interestingly, results from a recent study using well powered microarray analyses in combination with immunohistochemistry and Morris spatial maze task suggest that, in rats, increases in cholesterol transport during brain aging may be associated with learning impairment (Kadish et al., 2009). Further investigation of potential effect of ALLO on cholesterol transport may shed insight on brain aging.
It is noteworthy that although intracellular cholesterol accumulation is present in both cortical neurons and Purkinje cells of Npc1-/- mice, the latter degenerate much earlier than the former. Such dissociation between cholesterol accumulation and neurodegeneration suggests that other factors may contribute to the selective vulnerability of Purkinje cells. Earlier in situ hybridization studies have shown that cerebellar Purkinje neurons express higher levels of Npc1 than neurons from other brain regions, which may contribute to the vulnerability of these cells to the lack of Npc1 proteins (Prasad et al., 2000). More importantly, recent research has revealed that Purkinje cells are the major site of neurosteroidogenesis in brain (Tsutsui et al., 2003; Tsutsui, 2008): in particular, the steroid hormones, estradiol and progesterone, and the neurosteroids allopregnanolone and pregnenolone are synthesized in Purkinje neurons (Tsutsui et al., 2003). High levels of allopregnanolone and pregnenolone are associated with Purkinje cell development in neonatal rats. In vitro experiments have also shown that steroid products play neuroprotective roles in Purkinje cell survival and maturation. It is thus conceivable that lack of Npc1 proteins leads to reduced steroid synthesis, which then leads to neurodegeneration.
In addition to neurons, neurosteroids may also change the function of microglia and oligodendrocytes. Despite the severe impairment in myelination in Npc1-/- mice, no obvious cholesterol accumulation was observed in cell bodies of CNPase-expressing oligodendrocytes, and ALLO-treatment did not induce evident alterations in cholesterol distribution in this cell type, although it markedly improved myelination in mutant mice. It is tempting to speculate that demyelination and ALLO-induced remyelination in Npc1-/- mice are most likely NOT due to intracellular cholesterol accumulation. One potential mechanism may be related to changes in cell adhesion function (Liao et al. unpublished observation).
3.2. Reduced lysosomal dysfunction and abnormal autophagy in Npc1-/- mice by ALLO treatment
Cholesterol accumulation in NPC is associated with lysosomal dysfunction and abnormal autophagy. Lysosomal dysfunction can further disrupt autophagic function since recycling of autophagosomal cargos depends on protein degradation carried out in lysosomes; accordingly, cholesterol accumulation could create an adverse feedback loop in NPC pathology involving both autophagy and lysosomal dysfunction. Results from previous and the present studies demonstrated that brain levels of the lysosomal hydrolases cathepsins B and D were increased in Npc1-/- compared to wild-type mice. Increases in these lysosomal enzymes could be a consequence of lysosomal dysfunction. In addition, recent studies have shown that cathepsin D plays critical roles in the pathogenesis of several degenerative disorders. For instance, cathepsin D cleaves microtubule-associated protein tau, which may potentially contribute to tauopathies and Alzheimer's disease (Bi et al., 2000). Several lines of evidence suggest that autophagic dysfunction may contribute to neurodegeneration in Npc1-/- mice (Bi and Liao, 2007; Ko et al., 2005; Liao et al., 2007; Pacheco et al., 2007). Surprisingly, autophagy alterations were not associated with changes in mTOR, but with one of its downstream targets, GSK3β, which suggests an mTOR-independent autophagy induction. Results from the present study confirmed our previous finding that increased autophagy is closely associated with the conversion of LC3-I to LC3-II. The normalization effect of ALLO treatment on the ratio of LC3-II/LC3-I, as well as on levels of cathepsins B and D could therefore be due to improved lysosomal function resulting from reversal of cholesterol accumulation, although we cannot exclude the possibility that ALLO treatment could directly regulate elements of the autophagic machinery. Abnormal autophagic activities have also been implicated in other neurodegenerative diseases such as Alzheimer's disease (Nixon et al., 2005), Parkinson's disease (Anglade et al., 1997), and lysosomal storage diseases (Koike et al., 2005). It would be therefore interesting to determine whether allopregnanolone could be beneficial for these diseases as well.
3.3. Decreased inflammatory glial reaction in Npc1-/- mice by ALLO treatment
Activation of inflammation is an early neuropathological event in Npc1-/- mice (German et al., 2002); microglial activation precedes while astroglia alterations follow neuronal degeneration in Npc1-/- mice (Baudry et al., 2003). However, whether glial activation represents a reactive response to neuronal damage, or results from intrinsic alterations remains undetermined. Our results indicated that cholesterol accumulation resulting from lack of functional Npc1 proteins occurs not only in neurons but also in glia, especially in microglia. Therefore, it is conceivable that, instead of being elicited by neuronal damage, microglia and astrocyte activation could be a consequence of cholesterol accumulation in these glial cells. ALLO treatment dramatically reduced microglia and astrocyte activation in Npc1-/- mice and diminished cholesterol accumulation in microglia, suggesting that increases in cholesterol efflux from endosomes may improve glial function. The recent finding that restoring NPC1 function in astrocytes led to dramatic increase in lifespan suggests that improving cholesterol movement in glia could be a potential target for treating NPC disease (Zhang et al., 2008). It would be of interest to investigate whether ALLO could suppress inflammatory responses in other neurodegenerative diseases, especially in age-related neurodegenerative diseases in which lysosomal dysfunction and inflammation have been implicated (Block et al., 2007; Koprich et al., 2008; Sivaprakasam, 2006).
A few studies have recently reported different degrees of protective effects in Npc1-/- mice with β-cyclodextrin, the vehicle used in the present study. However, most of these studies used lifespan as a readout while our study has focused on the molecular/cellular basis of the disease. Results from our study clearly indicated that, at least considering the prevention of intracellular cholesterol accumulation and demyelination, the beneficial effects are mostly due to the neurosteroid allopregnanolone, as β-cyclodextrin alone did not show comparable effects. Liu et al. (2008) have shown that treatment with β-cyclodextrin alone not only extended lifespan but also increased the numbers of cerebellar Purkinje cells. Whether the difference between their study and ours is due to different genetic background is an interesting question that will need to be addressed in the future.
In conclusion, results from the present study for the first time demonstrated that a single injection of ALLO at postnatal day 7 significantly delayed cholesterol accumulation in both neurons and microglia, a process that might be related to improved cholesterol efflux from endosomes/lysosomes. Decreases in cholesterol accumulation were associated with improved function in the autophagic/lysosomal system and with diminished microglial and astrocytic reactions, both of which may contribute to decreased neuropathology and increased lifespan. Improvement in autophagic/lysosomal function could result in improved cell function and lead to neuroprotection and diminished inflammation. On the other hand, the reversal of demyelination was not associated with changes in cholesterol redistribution, suggesting that other mechanism may be involved. Further investigation of mechanisms underlying cholesterol efflux and remyelination may open up novel therapeutic targets and better treatment for NPC.
Experimental Procedures
3.1. Animals
Npc1NIH heterozygous mice on BALB/c background were purchased from Jackson Laboratory (Bar Harbor, MN) and housed in the vivarium facility under protocols approved by the local IACUC. Npc1-/- mice were produced by in-house breeding from heterozygous mice. Npc1 genotype was determined with PCR method as previous described (Baudry et al., 2003). Both male and female mice were used in this study because there was no gender-specific difference in onset and progression of the disease.
3.2. Drug treatment
Allopregnanolone (EMD Biosciences, La Jolla, CA) was dissolved in 20% hydroxypropyl-β-cyclodextrin (Cat. No. 33260-7, Sigma, St. Louis, MO) in water and subcutaneously injected at 25 mg/kg body weight to postnatal day 7 pups once as previously described (Ahmad et al., 2005; Griffin et al., 2004). Pups treated with the same amount of β-cyclodextrin served as vehicle controls (Npc1-/-CD mice), and those received distilled water injection served as untreated controls.
3.3. Immunohistochemistry
One and two month-old mice were killed by deep anesthesia with pentobarbital, and were perfused with 4% paraformaldehyde (PFA); brains were cut into 25 μm coronal or sagittal sections for forebrain and cerebellum, respectively. Brain sections were kept in a cryoprotectant solution at -20°C before being processed for immunohistochemistry. Briefly, sections were washed in phosphate buffer (PB) once, blocked with 10% horse serum and 3% goat serum; sections were then incubated with primary antibodies in blocking buffer overnight at 4 °C. Sections were then washed with PB before being incubated with secondary antibodies (conjugated with Alexa-594 or Alexa-488). For signaling developed with avidin-biotin horseradish peroxidase complex method, after primary antibodies, sections were incubated with biotinylated secondary antibodies (1:400, Vector Laboratories, Burlingame, CA), washed with PB, incubated in avidin-biotin horseradish peroxidase complex solution (Vector Laboratories) for 45 min, and the reaction product visualized with diaminobenzidine (DAB Vector Laboratory) as the final chromogen. Antibodies used were: anti-LC3 (1:2000, Abgent, San Diego, CA), anti-cathepsin D (1:200, EMD Biosciences, La Jolla, CA), anti-F4/80 (1:500, Serotec, Oxford, UK), anti-CD11b (1:500, Chemicon, Temecula, CA), anti-CNPase (1:500, Chemicon), anti-GFAP (1:7500, Sigma, St. Louis, MO), and anti-calbindin (1:1000, Abcam, Cambridge, MA). The number of animals used for each group was: n =5 for Npc1+/+ mice, n =4 for Npc1-/- mice, n =5 for ALLO treated Npc1-/-, and n =4 for β-cyclodextrin treated Npc1-/- mice.
To quantify myelination in cortex, images of primary somatosensory cortex were taken with a 2× objective with acquisition parameters kept consistent across different experimental groups. In the area that covers all layers of cortex, CNPase immunolabeled portions of each image were measured with NIH Image J software, then compared to that in Npc1+/+ mice. For quantification of CNPase-immunopositive areas in striatum, images were also taken with a 2× objective and the whole striatum (at the level of medial septal nucleus) was manually selected and CNPase-immunopositive areas were measured using Image J software. Image analysis was done by investigators who were blind to the identity of the animals. Generally data from 4-6 animals were used for statistical analysis.
3.4. Filipin staining
Filipin staining was performed as previous described (Bornig and Geyer, 1974; Butler et al., 1987; Liao et al., 2007).
3.5. Immunoblotting
One and two month-old mice were decapitated and tissues from different brain regions (cortex, CX; hippocampus, HP; cerebellum, CB; brainstem, BS) were collected. Total protein was extracted with Tris lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, Protease inhibitor cocktail, Phosphatase inhibitor Cocktails 1 and 2). Forty μg of total proteins were separated on 8-14% SDS gels and transferred onto PVDF membranes. Membranes were probed with primary antibodies in 5% milk followed by incubation with peroxidase-conjugated secondary antibodies (1:10,000; Amersham Biosciences). Peroxidase reaction was developed with ECL-plus detection kit. The following primary antibodies were used: Anti-cathepsin D (1:1000, EMD Biosciences, La Jolla, CA), anti-cathepsin B (1:500, EMD Biosciences, La Jolla, CA), anti-LC3 (1:500, Abgent, San Diego, CA), anti-beclin 1 (1:1000, Santa Cruz), and anti-GFAP (1:7500, Sigma). Images were scanned and levels of different antigens were analyzed by using NIH Image J software. All blots were stripped and re-incubated with anti-GAPDH (1:10,000, Chemicon, CA) antibodies as loading control and all results were adjusted by GAPDH signal except the LC3 blots were adjusted by tubulin (1:10,000, Sigma).
3.6. Statistics analysis
Data were presented as mean ± S.E.M. One-way ANOVA was used to assess statistical differences between two groups. P<0.05 was considered as statistically significant.
Acknowledgments
The authors would like to thank Ms. Yueqin Yao, Mr. Ang Xie and Mr. Kevin Lee for their participation in some of the drug treatment experiments. This work was supported by a grant from NINDS (NS048423 to XB) and by funds from Western University to X.B. Xiaoning Bi was also supported by funds from the Daljit and Elaine Sarkaria Chair.
Abbreviations
- ALLO
allopregnanolone
- GABA
gamma-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- LC3
microtubule-associated protein 1 light chain 3
- PXR
pregnane X receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad I, Lope-Piedrafita S, Bi X, Hicks C, Yao Y, Yu C, Chaitkin E, Howison CM, Weberg L, Trouard TP, Erickson RP. Allopregnanolone treatment, both as a single injection or repetitively, delays demyelination and enhances survival of Niemann-Pick C mice. J Neurosci Res. 2005;82:811–21. doi: 10.1002/jnr.20685. [DOI] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, Pentchev PG, Carstea ED, Trojanowski JQ. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. 1995;90:547–51. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Bi X, Haque TS, Zhou J, Skillman AG, Lin B, Lee CE, Kuntz ID, Ellman JA, Lynch G. Novel cathepsin D inhibitors block the formation of hyperphosphorylated tau fragments in hippocampus. J Neurochem. 2000;74:1469–77. doi: 10.1046/j.1471-4159.2000.0741469.x. [DOI] [PubMed] [Google Scholar]
- Bi X, Liu J, Yao Y, Baudry M, Lynch G. Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in Npc1-/- mouse brain. Am J Pathol. 2005;167:1081–92. doi: 10.1016/S0002-9440(10)61197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Liao G. Autophagic-lysosomal dysfunction and neurodegeneration in Niemann-Pick Type C mice: lipid starvation or indigestion? Autophagy. 2007;3:646–8. doi: 10.4161/auto.5074. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–99. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornig H, Geyer G. Staining of cholesterol with the fluorescent antibiotic “filipin”. Acta Histochem. 1974;50:110–5. [PubMed] [Google Scholar]
- Butler JD, Comly ME, Kruth HS, Vanier M, Filling-Katz M, Fink J, Barton N, Weintroub H, Quirk JM, Tokoro T, et al. Niemann-pick variant disorders: comparison of errors of cellular cholesterol homeostasis in group D and group C fibroblasts. Proc Natl Acad Sci U S A. 1987;84:556–60. doi: 10.1073/pnas.84.2.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, 3rd, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O'Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- German DC, Liang CL, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–50. doi: 10.1016/s0306-4522(01)00517-6. [DOI] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, Auwerx J. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;131:405–18. doi: 10.1016/j.cell.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–11. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993;85:175–84. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–16. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, von Figura K, Mizushima N, Saftig P, Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflammation. 2008:5–8. doi: 10.1186/1742-2094-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci U S A. 2006;103:13807–12. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, Dietschy JM. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J Neuropathol Exp Neurol. 2005;64:323–33. doi: 10.1093/jnen/64.4.323. [DOI] [PubMed] [Google Scholar]
- Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A, Liang X, Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol. 2007;171:962–75. doi: 10.2353/ajpath.2007.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li H, Repa JJ, Turley SD, Dietschy JM. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J Lipid Res. 2008;49:663–9. doi: 10.1194/jlr.M700525-JLR200. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Erickson RP, Walkley SU, Bryant MA, Incao A, Heidenreich RA, Pavan WJ. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum Mol Genet. 2002;11:3107–14. doi: 10.1093/hmg/11.24.3107. [DOI] [PubMed] [Google Scholar]
- March PA, Thrall MA, Brown DE, Mitchell TW, Lowenthal AC, Walkley SU. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type. C Acta Neuropathol. 1997;94:164–72. doi: 10.1007/s004010050689. [DOI] [PubMed] [Google Scholar]
- Mellon SH. Neurosteroid regulation of central nervous system development. Pharmacol Ther. 2007;116:107–24. doi: 10.1016/j.pharmthera.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Ong WY, Kumar U, Switzer RC, Sidhu A, Suresh G, Hu CY, Patel SC. Neurodegeneration in Niemann-Pick type C disease mice. Exp Brain Res. 2001;141:218–31. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- Ong WY, Hu CY, Patel SC. Apolipoprotein D in the Niemann-Pick type C disease mouse brain: an ultrastructural immunocytochemical analysis. J Neurocytol. 2002;31:121–9. doi: 10.1023/a:1023993405851. [DOI] [PubMed] [Google Scholar]
- Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- Prasad A, Fischer WA, Maue RA, Henderson LP. Regional and developmental expression of the Npc1 mRNA in the mouse brain. J Neurochem. 2000;75:1250–7. doi: 10.1046/j.1471-4159.2000.0751250.x. [DOI] [PubMed] [Google Scholar]
- Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang TY. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res. 2004;45:582–91. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Li H, Frank-Cannon TC, Valasek MA, Turley SD, Tansey MG, Dietschy JM. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J Neurosci. 2007;27:14470–80. doi: 10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C, Ioannou YA. The NPC1 protein: structure implies function. Biochim Biophys Acta. 2004;1685:8–13. doi: 10.1016/j.bbalip.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sivaprakasam K. Towards a unifying hypothesis of Alzheimer's disease: cholinergic system linked to plaques, tangles and neuroinflammation. Curr Med Chem. 2006;13:2179–88. doi: 10.2174/092986706777935203. [DOI] [PubMed] [Google Scholar]
- Suresh S, Yan Z, Patel RC, Patel YC, Patel SC. Cellular cholesterol storage in the Niemann-Pick disease type C mouse is associated with increased expression and defective processing of apolipoprotein. D J Neurochem. 1998;70:242–51. doi: 10.1046/j.1471-4159.1998.70010242.x. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D'Agostino AN, Carstea ED. Neurofibrillary tangles in Niemann-Pick disease type. C Acta Neuropathol. 1995;89:227–38. doi: 10.1007/BF00309338. [DOI] [PubMed] [Google Scholar]
- Tsutsui K, Sakamoto H, Ukena K. A novel aspect of the cerebellum: biosynthesis of neurosteroids in the Purkinje cell. Cerebellum. 2003;2:215–22. doi: 10.1080/14734220310016169. [DOI] [PubMed] [Google Scholar]
- Tsutsui K. Neurosteroids in the Purkinje cell: biosynthesis, mode of action and functional significance. Mol Neurobiol. 2008;37:116–25. doi: 10.1007/s12035-008-8024-1. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet. 1988;33:331–48. doi: 10.1111/j.1399-0004.1988.tb03460.x. [DOI] [PubMed] [Google Scholar]
- Vanier MT. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem Res. 1999;24:481–9. doi: 10.1023/a:1022575511354. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–44. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001a;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type. C Curr Biol. 2001b;11:1283–7. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- Zhang M, Strnatka D, Donohue C, Hallows JL, Vincent I, Erickson RP. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1(-/-) mice. J Neurosci Res. 2008 doi: 10.1002/jnr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]