

Abstract
Th1 and Th17 T cells are often co-localized in pathological environments, yet Th1 derived-interferon (IFN)-γ inhibits Th17 cell development in vitro. We explored the physiologic basis of this paradox in humans. Here we demonstrate increased numbers of CD4+IL-17+ and CD8+IL-17+ T cells in skin lesions of psoriasis. Furthermore, we show that myeloid antigen presenting cells (APCs) potently support induction of IL-17+ T cells, and that this activity is greatly increased in psoriasis. We tested stimuli which might account for this activity. Th1 cells and IFN-γ are increased in psoriatic blood and lesional skin. We show that IFN-γ programs myeloid APCs to induce human IL-17+ T cells via IL-1 and IL-23. IFN-γ also stimulates APC production of CCL20, supporting migration of IL-17+ T cells, and synergizes with IL-17 in the production of β-defensin 2 (HBD-2), an antimicrobial and chemotactic protein highly overexpressed by psoriatic keratinocytes. This study reveals a novel mechanistic interaction between Th1 and IL-17+ T cells, challenges the view that Th1 cells suppress Th17 development through IFN-γ, and suggests that Th1 and IL-17+ T cells may collaboratively contribute to human autoimmune diseases.
Keywords: Th17, IFN-γ, APC, Chemokine receptor, T cell, Autoimmune disease, Psoriasis
IL-17+CD4+ T cells (Th17) and IL-17 play an active role in inflammation and autoimmune diseases in murine systems (1-9). While Th17 and IL-17+CD8+ T cells were found in both mouse and human tumors (10, 11), IL-17+CD8+ T cells have not been described as increased in human autoimmune diseases. In mice, the cytokine cocktail of TGFβ and IL-6 promotes differentiation of Th17 (12-14) and IL-17+CD8+ T cells (10, 11), but in humans, TGFβ or this combination of cytokines is thought to suppress Th17 cell differentiation from naïve T cells (15, 16). The cytokines IL-1 and IL-23 are important for amplifying and stabilizing Th17 cell differentiation from naïve T cells in mice (2, 4-6, 11, 17-21) and in humans (16). However, it remains poorly understood which factors promote IL-23 production in humans and control the development and trafficking of human IL-17+ T cells (22). Furthermore, Th1 cells are reported to inhibit Th17 development through IFN-γ (12-14). Why, then, are Th1 and Th17 cells often colocalized in pathological environments (1-9, 23, 24), and what is the mechanism and pathological relevance of this colocalization?
To address these questions, we studied psoriasis, a common, chronic disease of the skin and joints mediated by T cell interactions with keratinocytes and other skin cells. Previous studies implicated Th1 cells and the Th1 promoting cytokine, IL-12, in psoriasis pathogenesis and maintenance (23-26). However, polymorphisms in IL-12p40 and the IL-23 receptor, but not the IL-12 receptor, have been associated with psoriasis (27, 28). Furthermore, an antibody which neutralizes IL-12p40, and thereby both IL-23 and IL-12, has potent effects in treating psoriasis (29). Recently, IL-17+CD4+ (Th17) T cells have been isolated from psoriatic plaques (16, 24, 30). However, the relative numbers of Th17 cells in these lesions remains unquantified, and the proportion of IL-17+CD8+ cells is unknown. Further, it has been reported that blocking IL-23 has no effects on Th17 cell development mediated by myeloid APCs in humans (15). Whereas another study suggested that APC-derived IL-23 was associated with Th17 cell development in psoriasis (16). However, it has not been experimentally demonstrated whether psoriatic APC subsets induce IL-17+ T cells, and whether psoriatic APC-induced Th17 cells are mechanistically due to IL-23 or/and other cytokines.
In the current investigation, we explored the phenotype and function of IL-17 secreting T cells in psoriatic and healthy skin, and the factors supporting their trafficking to and induction in lesional skin. Specifically, we show that IFNγ is demonstrated herein as a potent promoter of IL-17+ T cell trafficking, induction, and function. Our observations support a model wherein Th1 and IL-17+ T cells mechanistically interact and collaboratively contribute to human autoimmune disease pathogenesis.
Materials and methods
Human subjects
All subjects provided written, informed consent. Peripheral blood mononuclear cells (PBMCs) and skin biopsies were obtained from healthy donors and patients with psoriasis. Prior to biopsy, cases were required to be off all systemic therapies for at least 2 weeks, and off topical antipsoriatic medications for at least 1 week. Controls were identified from respondents to advertisements in the Ann Arbor area, had no personal or family history of psoriasis, and were free of inflammatory skin disease at the time of biopsy. The study was approved by the Institutional Review Board of the University of Michigan Medical School.
Immune cell isolation
Single-cell suspensions were prepared from PBMCs and skin tissues. Skin biopsies were incubated in 50 U/ml dispase (BD Biosciences, Bedford, MA) at 37°C for 90 minutes, and then epidermis and dermis were teased apart with fine forceps. The dermal portions were then cut into 1 mm pieces and digested in collagenase for 2 hours at room temperature. Single cell suspensions of epidermal portions were generated by incubation in Cell Dissociation buffer (Invitrogen, Grand Island, NY) at 37°C for 2 hours. Skin explant cultures of T cells from skin biopsies were prepared as described (31).
Immune cells including T cells and CD14+ or/and CD11c+ myeloid APCs were enriched using paramagnetic beads (StemCell Technology, Vancouver, Canada) and sorted from stained single cell suspensions using a high speed cell sorter (FACSaria, Becton Dickinson Immunocytometry Systems, San Jose, CA) as we described (32, 33). Cell purity was > 98% as confirmed by flow cytometry (LSR II, BD). CD14+ or/and CD11c+ myeloid APCs were used to stimulate T cells as indicated.
APC activation and cytokine production
Fresh peripheral blood CD11c+ APCs (0.5 × 106/ml) were incubated for 72 hours with or without recombinant human IFN-γ (200 ng/ml, R&D Systems, Minneapolis, MN). These cells were washed and used for T cell stimulation or activated for 12 hours with LPS (1 μg/ml, Sigma Aldrich) or incubated 3 days (1 × 106 cells/ml) in the presence of LPS to detect cytokine levels in supernatants (all antibodies from R&D Systems, except IL-23p19 from E-Biosciences, San Diego, CA).
T cell culture system
Myeloid APCs were co-cultured with peripheral blood T cells in ratios of 1:3 to 1:10 (0.5 × 106 T cells/ml) for 4 days in the presence of anti-CD3 (2.5 - 5 μg/ml) and anti-CD28 (1.2–2.5 μg/ml) mAbs (Becton-Dickinson Biosciences, San Jose, CA). Different cytokines and neutralizing antibodies or their combinations including IL-1α (2.5 ng/ml), IL-1β (2.5 ng/ml), IL-23 (10 ng/ml), anti-IL-4 (1 μg/ml), anti-IFN-γ (2 μg/ml), anti-IL-1 (1 μg/ml anti-IL-1R + 1 μg/ml anti-IL-1α) were used as indicated (all from R&D). Cells were subjected to flow cytometric phenotyping, intracellular cytokine staining and transcript detection by real-time PCR. Culture supernatants were collected for detection of IL-17 by ELISA (R&D). For flow cytometry analysis, cells were first stained extracellularly with specific antibodies, then were fixed and permeabilized with Perm/Fix solution (E-Biosciences) and finally were stained intracellularly with specific antibodies against the indicated cytokines (BD Biosciences). Samples were acquired on a LSR II (BD) and data were analyzed with DIVA software (BD).
siRNA knockdown of human IL-23 gene expression
HEK293 cells were transfected with a Flag-tagged IL-23 expression plasmid and either a nonfunctional scrambled control siRNA or IL-23-specific siRNA using Lipofectamine 2000 (Invitrogen). After the siRNA treatment, the hIL-23 silencing efficiency was measured by Western-blot using anti-Flag tag (not shown). Peripheral blood myeloid APCs were transfected with the siRNA or pmaxGFP vector using Nucleofector technology (Macrophages Nucleofector Kit, Amaxa, Köln, Germany) as we described (34). The transfection efficiency reached 60-80% as confirmed by pmaxGFP vector transfection.
Analysis of HBD-2 induction in keratinocytes
Normal human keratinocytes (NHK) were grown in serum-free medium optimized for high - density keratinocyte growth (Medium 154; Cascade Biologics, Portland, OR) and used for experiments in the second or third passage at subconfluence. All cells were plated at 5,000 cells / cm2, and switched to basal Medium 154 lacking growth factors 24 hours prior to assay. In some experiments, the calcium concentration was increased from 0.1 to 1.4 mM to enhance production of HBD-2 (35, 36). HBD-2 in supernatants was measured by ELISA using antibodies and recombinant protein from Peprotech (Rocky Hill, NJ) as described (37).
Migration assays
Migration assays were performed in a Transwell system with a polycarbonate membrane of 6.5-mm diameter with a 3-μm pore size as we described (32). Purified T-cell subsets were added to the upper chamber and CCL-20 (5 ng/ml, R&D) was added to the lower chamber. After 4h incubation at 37°C, the phenotype and number of T cells in the upper and lower chambers was determined by FACS.
Quantification of gene expression
Quantitative real-time PCR was performed on control, lesional and nonlesional skin samples from 10 psoriatic patients and 10 normal controls. Primers for the genes IL-1α (Cat no. PPH00690A), IL-1β (cat no. PPH00171A), DEFB4 (cat no. PPH11010A), IL12A (p35, Cat no PPH00544B) and IL12B (p40, Cat no PPH00545A) were obtained from Superarray Biosciences (Frederick, MD). Results were normalized to the expression of the housekeeping gene, ribosomal protein, large, P0 (RPLPO, 36B4, cat no. PPH21138E). Quantification of gene expression in the cultured myeloid APCs were performed as we described (10, 11). The gene transcripts were quantified in a MasterCycler RealPlex system (Eppendorf, Westbury, N.Y.) and expressed as mRNA quantities normalized to GAPDH levels.
Immunohistochemistry
Skin biopsies were stained with anti-CD4 or anti-CD8 antibody as described (32). The staining was detected using the one-step avidin-biotin complex (ABC) technique (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA).
Statistical calculations
The Wilcoxon rank-sum test was used to determine pairwise differences and the X2 test used to determine differences between groups. P < 0.05 was considered as significant. Differences in phenotype of T cell subsets were tested with the paired Student's t-test. Correlation was tested by Spearman's test. All statistical analysis was done on Statistica software (StatSoft Inc., Tulsa, OK).
Results
IL-17+ T cells are increased in psoriatic plaques
Th17 cells are thought to play a role in psoriatic pathogenesis (16, 30); however, the phenotype and distribution of IL-17+ T cells in patients with psoriasis remain poorly defined. We investigated the numbers and distribution of IL-17+CD4+ and IL-17+CD8+ T cells in peripheral blood (Fig. 1A) and skin (Fig. 1A-C) of healthy donors and patients with psoriasis. The highest percentages (Fig. 1A, B) and absolute numbers (Fig. 1C) of IL-17+CD4+ and IL-17+CD8+ T cells were observed in psoriatic skin lesions (plaques). Notably, we observed increased numbers of IL-17+CD8+ T cells in psoriatic skin for the first time (Fig. 1C). We further analyzed the expression of IFNγ and IL-17 per single cell level. Interestingly, 30 to 50% IL-17+ T cells coexpressed IFNγ in the skin from psoriatic plaque (Fig. 1 D). After enzymatic separation of epidermis and dermis, we found increased numbers of both IL-17+CD4+ and IL-17+CD8+ T cells in lesional psoriatic dermis, relative to uninvolved skin from persons with psoriasis or healthy donors. However, in plaque epidermis most of the IL-17+ T cells were CD8+ (Fig. 1E), while IL-17+CD8+ T cells were virtually absent from healthy donor epidermis (Fig. 1 E). The data demonstrate the prevalence, phenotype, and distribution of IL-17+ T cells in patients with psoriasis, and indicate that psoriatic skin is an environment containing abundant IL-17+ T cells including Th17 cells and IL-17+CD8+ T cells.
FIGURE 1.

IL-17+ T cells in psoriasis lesions. Skin biopsies and peripheral blood from healthy donors and patients with psoriasis were stained with specific antibodies as described in Materials and Methods. A-E, IL-17+ T cells were analyzed with FACS. A, IL-17+ T cells. Results were expressed as the mean percent of IL-17+ T cells in T cells, error bars indicate SEM, n = 8-10. B, Representative dot-plots are shown. C, Total number of IL-17+ T cells in the skin. Total number of IL-17+ T cells was calculated by multiplying the percentage of IL-17+ T cells by the absolute number of T cells per mm2 of skin, as determined previously (26). Results are expressed as mean number ± SEM, n = 10. D, Coexpression of IL-17 and IFNγ on IL-17+ T cells. Single cell suspensions were made from skin tissues in healthy donors and patients with psoriasis. The expression of IFNγ and IL-17 was analyzed by intracellular cytokine staining gated on CD3+ T cells. E, Total number of IL-17+ T cell subsets in dermis and epidermis. Total number of IL-17+CD4+ and IL-17+CD8+ T cell subsets was calculated by multiplying the percentage of each IL-17+ T cell subset by the absolute number of T cell subsets per mm2 skin (26) in the separated dermis and epidermis. Results are expressed as mean number ± SEM, n = 5. *P < 0.05 (A, C, E) compared to uninvolved and healthy donor skin.
IFN-γ+ T cells in patients with psoriasis
As a control for relative IL-17+ T cell quantification, we examined the distribution of CD4+, CD8+, and IFN-γ+ T cells in psoriatic skin in patients with psoriasis. Consistent with previous reports (23, 26, 38), we observed large numbers of CD4+ and CD8+ T cells in psoriatic dermis and increased numbers of primarily CD8+ T cells in psoriatic epidermis (Fig. 2A). The epidermal-dermal distribution of IFNγ+CD4+ and CD8+ T cells is similar for T-cells expressing IFN-γ and IL-17 in psoriatic skin (Fig. 1). It has been hypothesized that Th1 cells promote psoriatic pathogenesis (23, 26, 38). Consistent with these observations, high levels of IFN-γ+ T cells (Fig. 2B) and IFN-γ transcripts (Fig. 2C) were also detected in psoriatic plaques. In the following studies, we therefore analyzed the relationship between IFN-γ and IL-17+ T cells in psoriatic pathogenesis.
FIGURE 2.
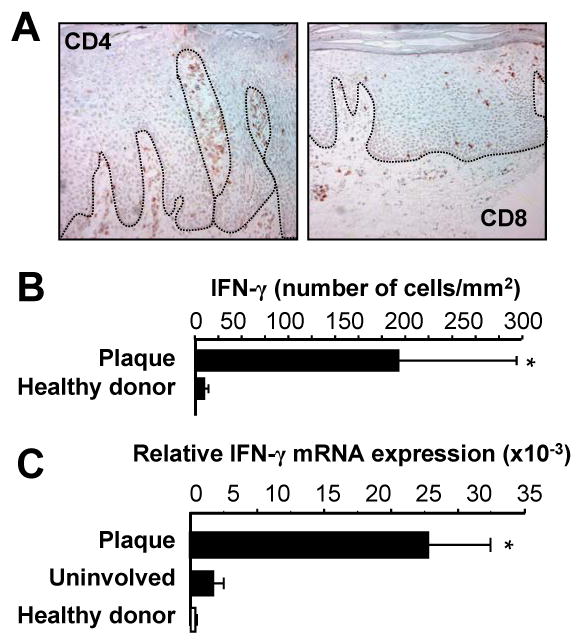
IFNγ+ T cells in psoriasis lesions. A, T cells in psoriasis lesions. Psoriasis plaques were stained with anti-CD4 and anti-CD8. One of 5 representative individuals is shown. B, C, IFNγ+ T cells in psoriasis lesions. IFN-γ+ T cells in psoriatic lesions: T cells (CD45+CD3+) were analyzed by FACS in cell suspensions obtained from healthy skin and psoriatic plaque. B, IFN-γ+ T cells were analyzed by FACS and quantified in the skin. Results were expressed as the mean number per mm2 skin ± SEM. (n = 3). C, Expression of IFN-γ in skin from healthy donors and patients with psoriasis. IFN-γ was quantified by real-time PCR. Results are expressed as the mean values of relative expression ± SEM. (n = 5, *P < 0.03 compared to uninvolved and healthy skin).
Psoriatic myeloid APCs induce IL-17+ T cells
APCs contribute to T cell polarization. It is thought that myeloid derived gene products contribute to pathogenic inflammation in psoriasis (39). We therefore investigated a potential role for myeloid APCs in inducing IL-17+ T cells. Myeloid APCs were found in psoriatic skin (Fig. 3A, B). Although numerically CD14+ cells are the major APCs in the skin, CD11c+ cells may be functionally the most powerful APCs in the skin. Therefore, we initially sorted both CD14+ and CD11c+ myeloid APCs from peripheral blood and skin plaques of people with psoriasis and healthy donors. We observed that CD14+ and CD11c+ myeloid APCs induced similar levels of Th17 cells (Fig. 3C, and not shown). Interestingly, APCs from psoriatic peripheral blood and skin lesions were significantly more efficient than those from healthy donors in inducing IL-17 production (Fig. 3C) and IL-17+ T cells (Fig. 3D). Myeloid APCs isolated from psoriatic plaques were superior to those isolated from blood in IL-17+ T cell induction (Fig. 3C, D) although these APCs induced comparable levels of IFNγ production. Myeloid APCs induced both IL-17+CD4+ T cells and IL-17+CD8+ T cells (Fig. 3D). We performed similar experiments with responder T cells from normal donors and psoriatic patients. We observed that APCs from psoriatic skin lesions induced comparable Th17 cell development in responder T cells from normal donors and psoriatic patients (Fig. 3C, D and data not shown). Altogether, these data indicate that psoriatic myeloid APCs potently induce IL-17+ T cells, and may thereby stimulate and maintain the IL-17+ T cell pool in patients with psoriasis.
FIGURE 3.

Psoriatic myeloid APCs stimulate memory IL-17+ T cell expansion. A, B, Myeloid APCs (CD45+CD14+) were analyzed by FACS in cell suspensions obtained from healthy skin and psoriatic plaque. A, One representative dot-plot of CD45+ leukocytes from the skin of a healthy donor and a patient with psoriasis is shown. Total number of CD3+ or CD14+ cells is indicated. B, Myeloid APCs were analyzed by FACS and quantified in the skin. Results were expressed as the mean number per mm2 skin ± SEM (n = 3). C, D, Psoriatic myeloid APCs induce IL-17+ T cells. Normal peripheral blood T cells were stimulated for 5 days with myeloid APCs derived from skin and blood of healthy donors or patients with psoriasis in the presence of anti-CD3 and anti-CD28. C, IL-17 was detected in the culture supernatants. Results are expressed as mean ± SEM, n = 5, *P<0.05 compared to healthy donor. D, One representative dot-plot of IL-17+ T cells induced by myeloid APCs isolated from psoriatic skin or blood of one patient is shown.
IFN-γ programs myeloid APCs to stimulate IL-17+ T cells
We next studied why psoriatic myeloid APCs are potent inducers of IL-17+ T cells. The contribution of IFN-γ was investigated, as IFN-γ is increased in serum of persons with psoriasis (40). IFN-γ+ T cells are enriched in psoriatic skin (Fig. 2), and previous studies implicated IFN-γ and Th1 cells in psoriasis pathogenesis (23, 25, 26). To test the hypothesis that IFN-γ may program myeloid APCs to stimulate IL-17+ T cells, CD11+ cells from the blood of non-psoriatic donors were conditioned with IFN-γ and tested for their capacity to induce IL-17+ T cells. IFN-γ profoundly increased the capacity of these CD11c+ cells to elicit IL-17+ T cells (Fig. 4A, B). We also conditioned peripheral blood myeloid APCs from psoriatic patients with exogenous IFN-γ, and observed that IFN-γ was able to further enhance the ability of normal and psoriatic myeloid APCs to induce IL-17-secreting T cells (Fig. 4C, D). The data suggest that IFN-γ released by psoriatic T-cells (Fig. 2) may condition myeloid APCs to induce IL-17+ T cells.
FIGURE 4.

IFN-γ programs myeloid APCs to stimulate IL-17+ T cells. Normal (A, B) or psoriatic (C, D) blood-derived ex vivo CD11c+ cells were conditioned for 72 hours with or without IFN-γ, and then cultured with normal T cells for 5 days in the presence of anti-CD3 and anti-CD28. A-C, IL-17+ T cells were detected by FACS. Results are expressed as the mean percentage of IL-17+ T cells in T cells ± SEM. D, IL-17 was measured in the supernatants by ELISA. Results are expressed as the mean value of IL-17 ± SEM. *P < 0.05 compared to control. (A, n = 11; B, one of 11; c, d, n = 2).
IL-1 and IL-23 mediate IFN-γ-stimulated induction of IL-17+ T cells by myeloid APCs
We further investigated the mechanism by which IFN-γ programs myeloid APCs to induce IL-17+ T cells. We showed that exogenous IL-1 and IL-23 were each capable of enhancing the induction of IL-17+ T cells by blood CD11c+ cells (Fig. 5A). We next analyzed the expression of IL-1 and IL-23 in CD11c+ cells treated with IFN-γ. IFN-γ moderately increased the expression of IL-12p35. However, IFN-γ dramatically increased the expression of IL-1, IL-12p40 and IL-23p19 (Fig. 5B). Similar effects of IFN-γ were observed on CD11c+ and CD14+ APCs. The data indicate that the effects of IFN-γ are more pronounced on IL-23 than IL-12p70.
FIGURE 5.

Myeloid APCs stimulate IL-17+ T cell expansion through IL-1 and IL-23. A, IL-1 and IL-23 enhance the induction of IL-17+ T cells by CD11c+ cells. Normal peripheral blood T cells were stimulated for 5 days with normal CD11c+ myeloid APCs with or without IL-1 and IL-23 in the presence of anti-CD3 and anti-CD28. IL-17+ T cells were determined by FACS. Results are expressed as mean ± SEM, n = 7, * P < 0.03 compared to control. B, IFN-γ stimulates IL-1 and IL-23 expression by CD11c+ myeloid APCs. CD11c+ myeloid APCs were conditioned with or without IFN-γ. IL-1, IL-12 and IL-23 transcripts were quantified by QRT-PCR. Results are expressed as the mean of relative expression ± SEM, n = 7, *P < 0.01 compared to control. C, Blockade of IL-1 and IL-23 abrogates CD11c+ cell-mediated IL-17+ T cell induction. Blood-derived CD11c+ cells were transfected with IL-23-specific siRNA or control siRNA, then conditioned with IFN-γ. T cells were stimulated for 5 days with these myeloid APCs with or without neutralizing IL-1R antibodies. IL-17+ T cells were detected by FACS. Results are expressed as the mean of IL-17+ T cells in T cells ± SEM, n = 5, *P < 0.05 compared to control. D, E, Expression of IL-23, IL-12 and IL-1 were quantified by real-time PCR in skin from healthy donors and patients with psoriasis. Results are expressed as the mean of relative expression ± SEM, n = 5, *P < 0.03 compared to uninvolved and healthy skin.
We next examined the role of CD11c+ cell-derived IL-1 and IL-23 in inducing memory IL-17+ T cells. Reliable and specific neutralizing antibodies against IL-23p19 are not commercially available, and anti-IL-12p40 neutralizing antibody blocks both IL-12 and IL-23. Therefore, to conclusively block IL-1 and IL-23, we used neutralizing antibodies against IL-1α and IL-1 receptor and designed three siRNA sequences which specifically block human IL-23 (not shown). Blockade of IL-1 and IL-23 separately each reduced CD11c+ cell-mediated induction of IL-17+ T cells, and simultaneous blockade of both cytokines abrogated the IL-17 response (Fig. 5C). We extended these observations to patients with psoriasis. Consistent with the high levels of IL-17+ T cells (Fig. 1) and of IL-17+ inducing myeloid APCs (Fig. 3), we detected high levels of IL-1β and IL-23 transcripts in psoriatic plaques (Fig. 5D, E). Taken together, our data suggest that IFN-γ programs myeloid APCs to induce IL-17+ T cells through IL-1 and IL-23 in patients with psoriasis.
CCR6+IL-17+ T cells and CCL20 in the psoriatic environment
We next examined how IL-17+ T cells traffic to the psoriatic environment. We found that IL-17+CD4+ and IL-17+CD8+ T cells derived from psoriatic plaque highly expressed CCR6 (Fig. 6A, B). We therefore asked whether IL-17+ T cells could migrate toward CCL20, the ligand for CCR6. We observed that T cells efficiently migrated in response to CCL20, and that the migrating cells were enriched for IL-17+ T cells (from 0.2% IL-17- cells in the upper chamber to 11% IL-17+ T cells in the lower chamber) (Fig. 6C). We further tested the role of IFN-γ in CCL20 production. We observed that IFN-γ stimulated CCL20 production from CD11c+ APCs (Fig. 6D). High levels of CCL20 mRNA were detected in lesional psoriatic skin (39, 41) (Fig. 6E). The data suggest that IFN-γ derived from psoriatic T cells induces CCL20 and promotes homing of IL-17+ T cells to the psoriatic environment.
FIGURE 6.

CCR6+IL-17+ T cells and CCL20 in the psoriatic environment. A, B, Psoriatic IL-17+ T cells highly express CCR6. Expression of CCR6 was determined by FACS on IL-17+ and IL-17- T cells in psoriatic skin. A, Results are expressed as the mean of CCR6+ T cells in IL-17+ T cells or IL-17- T cells ± SEM. (n = 6, *P < 0.05 compared to IL-17- T cells). B, One representative dot-plot of 6 is shown. Gated on CD4+ and CD8+ T cells. C, IL-17+ T cells migrate in response to CCL20. Migration assay was performed as described in Methods. The migrated T cells were subjected to intracellular staining for IL-17. The percent of IL-17+ T cells in the upper and lower chambers are shown. D, IFN-γ induces CCL-20 production. Blood CD11c+ cells were stimulated for 3 days with or without IFN-γ. CCL20 was detected by ELISA in the supernatants. Results are expressed as the mean value ± SEM (n = 7, *P < 0.01 compared to control). E, High expression of CCL20 in psoriatic skin. CCL20 transcript was quantified by real-time PCR. Results are expressed as the mean values of relative expression ± SEM. (n = 5, *P < 0.03 compared to uninvolved and healthy skin). F, High expression of CD103 on psoriatic IL-17+ T cells. CD103 expression was determined by FACS on IL-17+ and IL-17- T cell subsets. Results are expressed as the mean of CD103+ T cells in each T cell subset ± SEM. (n = 5, *P < 0.05 compared to IL-17- T cells). Right panel: One representative dot-plot of psoriatic CD103+IL-17+ T cells. Gated on CD4+ and CD8+ T cells.
In addition to CCR6, we observed that psoriatic IL-17+ T cells highly expressed CD103 as compared to IL-17- T cells (Fig. 6F). CD103 has been implicated in localization of T cells to psoriatic epidermis (42, 43), and may play a specific role in IL-17+ T cell trafficking.
Psoriatic IL-17+ T cells are functional
We next asked whether myeloid APC-induced IL-17+ T cells are functional. HBD-2 is an important component of skin innate immunity (35, 44). Increased HBD-2 copy number has recently been shown to be a risk factor for development of psoriasis (45). We confirmed that high levels of HBD-2 transcripts are present in psoriatic plaques (Fig. 7A). Further, IL-17 induced a dose dependent increase in secreted HBD-2 protein in keratinocytes, which could be blocked by a neutralizing antibody specific for the human IL-17 receptor (anti-IL-17R) (Fig. 7B). We next generated IL-17+ T cells with psoriatic myeloid APCs, collected the conditioned medium from these cultures, assayed them for IL-17 protein, and treated normal human keratinocytes with a 1:5 dilution of conditioned medium. The T cell supernatants strongly induced human HBD-2 secretion by keratinocytes (Fig 7C). Moreover, anti-IL-17R blocked the induced HBD2 expression (Fig. 7C). Furthermore, the concentrations of IL-17 in the T cell supernatants were positively correlated with the levels of HBD-2 secreted by keratinocytes (Fig 7D). We next examined if IFN-γ has an effect on HBD-2 production. IFN-γ alone had no detectable effect on HBD-2 secretion by keratinocytes. However, IFN-γ synergistically increased HBD-2 production induced by IL-17 (Fig. 7E). The data indicate that IL-17+ T cells generated by psoriatic myeloid APCs are functional, and that IFN-γ and IL-17 synergistically promote the innate immune function of keratinocytes. Based on all our findings, we propose a novel mode of interaction among Th1, IL-17+ T cells and APCs in human autoimmune disease (Fig. 7F): IFN-γ derived from Th1 and other cells promotes tissue homing of IL-17+ T cell by stimulating local CCL20 production, and stimulates IL-17+ T cells by programming IL-17-inducing myeloid APCs.
FIGURE 7.

Function of IL-17+ T cells in psoriasis. A, High levels of HBD-2 transcript in psoriatic plaque. Expression of HBD-2 was quantified by real-time PCR in the skin from healthy donors and patients with psoriasis. Results are expressed as the mean values of relative expression ± SEM. (n = 5, *P < 0.03 compared to uninvolved and healthy skin). B, IL-17 induces HBD-2 production by NHK. NHKs were cultured for 48 hours with variable concentrations of IL-17 with or without anti-IL-17 receptor. HBD-2 in the supernatants was detected by ELISA. One representative of 3 is shown. C, D, IL-17+ T cells stimulate NHK HBD-2 production through IL-17. T cells were stimulated with psoriatic myeloid APCs for 5 days to generate IL-17+ T cells. C, NHKs were cultured for 48 hours in the presence of 20% concentration of these T cell supernatants in the absence (control) or presence of anti-IL-17 receptor. HBD-2 in the NHK supernatants was detected by ELISA. (n = 10, *P < 0.01 compared to control). D, Strong correlation was observed between the amount of T cell derived IL-17 in the supernatants and NHK HBD-2 production (n = 10, R2 = 0.8, P < 0.01). E, Synergistic effect of IFN-γ on IL-17 mediated HBD-2 production. NHK cells were cultured with IL-17 (10 ng/ml) in the presence of variable concentrations of IFN-γ. HBD-2 was detected in the NHK cell supernatants by ELISA. Results are expressed as the mean values of ± SEM. (n = 3, P < 0.05). F, Proposed schematic model of interaction between Th1, IL-17+ and myeloid APCs. IFN-γ derived from Th1 cells induces CCL-20 production by local APCs, which in turn promotes IL-17+ T cell trafficking to the psoriatic environment. IFN-γ also programs APCs and expands memory IL-17+ T cells through APC-derived IL-1 and IL-23. Additionally, IFN-γ derived from Th1 acts synergistically with IL-17 on keratinocytes. Thus, Th1 and IL-17+ T cells collaboratively contribute to psoriasis pathogenesis.
Discussion
Th17 cells are thought to play a role in psoriatic pathogenesis (16, 30). In this report we have investigated the phenotype, distribution, trafficking, development and function of IL-17+ T cells in healthy human beings and people with psoriasis. We show that both CD4+ and CD8+ T cells express IL-17 in normal donors as well as in people with psoriasis. While the phenotype of human Th17 cells has not yet been fully defined, IL-17+CD4+ T cells (Th17) are postulated to play a role in psoriasis pathogenesis (16, 30). Entry of CD8+ T cells into the epidermis is necessary for the epidermal hyperproliferative response in psoriasis (43, 46). We observed high levels of IL-17+CD8+ T cells in the epidermis of psoriatic lesions. IL-17+CD8+ T cells are thus ideally positioned to respond to potential keratinocyte autoantigens on HLA class I molecules, which have been genetically implicated in psoriasis (47). Our observations support the hypothesis that IL-17+CD8+ T cells are critical mediators of the persistently altered epidermal growth and differentiation and local inflammation that is characteristic of psoriasis. We have also observed IL-17+CD8+ T cells in cancers (10, 11) and colitis (W. Zou, et al, unpublished observation). Our data provide the first evidence that IL-17+CD8+ T cells are at least as important as Th17 cells (16, 30) in autoimmune diseases, including but not limited to psoriasis.
We further characterize the phenotype of human psoriatic IL-17+ T cells: CD103+CCR6+. IL-17+ T cells are effector T cells often found in environments with chronic inflammation (1, 3). It is therefore not surprising that human IL-17+ T cells highly express CD103, which may facilitate trafficking of IL-17+ cells into inflammatory tissues (42, 43). We confirm that psoriatic skin is an environment enriched with CCL20, and show that CCL20 is triggered by IFN-γ in myeloid APCs. Further, we show that psoriatic Th1 cells are one of the major sources of IFN-γ, and that IL-17+ T cells efficiently migrate toward a CCL20 enriched psoriatic environment via CCR6. Our data lead us to propose a first mode of potential interaction between Th1 and IL17+ T cells in psoriasis: IFN-γ derived from Th1 cells (and other cells) promotes trafficking of IL-17+ T cells to the psoriatic environment through the induction and maintenance of local CCL20 production.
There are < 1% IL-17+CD4+ T cells and < 0.5% IL-17+CD8+ T cells in peripheral blood of healthy human beings (10, 11). In contrast, high levels of Th17 and IL-17+CD8+ T cells are observed in psoriatic plaques. How does this dramatic induction occur? In addition to migration from peripheral blood, IL-17+ T cells may be induced within the psoriatic environment. It has been reported that myeloid cell derived genes contribute to pathogenic inflammation in psoriasis (39). We demonstrate that myeloid APCs including macrophages and myeloid DCs potently induce human IL-17+ T cells. Our observation may explain why IL-17+ T cells are often found in inflammatory tissues and organs. In support of this concept, we show that myeloid APCs isolated from psoriatic plaques potently stimulate IL-17+ T cells. Additionally, our observation that circulating myeloid APCs from people with psoriasis promote IL-17+ T cell development may partially explain why persons with psoriasis experience inflammation at distinct anatomical sites, including skin plaques, arthritis, and atherosclerosis. Increased circulating IFN-γ (48) may activate circulating APCs, allowing them to enter tissue and promote expansion of IL-17+ T cells. Interestingly, recent studies in mouse models of chronic psoriasiform skin inflammation support pathogenic involvement of activated macrophages, although IL-17+ T cells have not been specifically investigated in these models (49, 50).
How and why do activated myeloid APCs contribute to psoriatic skin inflammation? Th1 cells can suppress Th17 cell differentiation through IFN-γ (1-3, 5, 51). However, this presents the paradox of why Th17 and Th1 cells often coexist in autoimmune diseases (17, 18, 20) including psoriasis (24). We demonstrate that psoriatic T cells produce IFN-γ, IFN-γ triggers myeloid APCs to produce IL-1 and IL-23, and in turn induce IL-17+ T cells. Our data therefore demonstrate a second mode of potential interaction between Th1 and IL17+ T cells in psoriasis: IFN-γ derived from Th1 cells (and other cells) activates psoriatic myeloid APCs, and the latter promote IL-17+ T cell development through IL-1 and IL-23 in the psoriatic environment. Therefore, IFN-γ may possibly play dual roles in regulating the IL-17+ T cell pool: IFN-γ targets APCs to initiate and promote Th17 polarization (this study) whereas it targets naïve T cells (1-3, 5, 51) to suppress Th17 polarization.
In addition to promoting the trafficking and development of IL-17+ T cells, IFN-γ also regulates the function of IL-17+ T cells. Human β-defensins are greatly enriched in the psoriatic environment (35) and play a role in increasing genetic susceptibility to psoriasis (45). Interestingly, we demonstrate that HBD-2 is induced by IL-17 and its expression is synergistically enhanced by IFN-γ, similar to synergistic effects of IFN-γ and IL-17 on induction of IL-6, IL-8, GRO-α, and GM-CSF (52, 53). Our data thereby illustrate a third mode of potential interaction between Th1 and IL17+ T cells in psoriasis: IFN-γ derived from Th1 (and other cells), and IL-17 derived from IL-17+ T cells, may synergistically promote keratinocyte function. The three modes of interaction described in this study suggests a novel, collaborative contribution of IL-17+ T cells and IFN-γ+ cells including Th1 cells to inflammatory reactions and autoimmune diseases, including psoriasis.
In summary, we show that IFN-γ may promote trafficking, induction and function of IL-17+ T cells in people with psoriasis. This study suggests a novel mechanistic interaction between Th1 and Th17 cells, challenges the view that Th1 cells suppress Th17 cell development, and suggests a collaborative contribution of Th1 and Th17 to human autoimmune diseases. Improved mechanistic insight into the collaborative interactions between these central regulatory players promises novel approaches to management of these diseases.
Acknowledgments
The authors thank the volunteers who provided blood and skin samples for this study, and acknowledge the skilled technical assistance of Linda Hodges, Kathleen McCarthy, Suzan Rehbine, and Thy Thy Do.
This work was supported by the Dermatology Foundation (AB, JEG), the National Psoriasis Foundation (AB), the American Skin Association (JEG), the National Institute of Arthritis, Musculoskeletal, and Skin Diseases (AR052889) (JTE) and the National Cancer Institute (CA99985) (WZ).
Footnotes
Disclosures: The authors have no financial conflict of interest.
References
- 1.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 2.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 4.Wynn TA. T(H)-17: a giant step from T(H)1 and T(H)2. Nat Immunol. 2005;6:1069–1070. doi: 10.1038/ni1105-1069. [DOI] [PubMed] [Google Scholar]
- 5.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 6.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 8.Tato CM, O'Shea JJ. Immunology: what does it mean to be just 17? Nature. 2006;441:166–168. doi: 10.1038/441166a. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 10.Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W. Cutting Edge: Th17 and Regulatory T Cell Dynamics and the Regulation by IL-2 in the Tumor Microenvironment. J Immunol. 2007;178:6730–6733. doi: 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- 11.Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, Zou W. Cutting Edge: Opposite Effects of IL-1 and IL-2 on the Regulation of IL-17+ T Cell Pool IL-1 Subverts IL-2-Mediated Suppression. J Immunol. 2007;179:1423–1426. doi: 10.4049/jimmunol.179.3.1423. [DOI] [PubMed] [Google Scholar]
- 12.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 14.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 15.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 16.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 17.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 19.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 20.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A. 2003;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang ST. Mechanisms of T-cell homing to skin. Adv Dermatol. 2001;17:211–241. [PubMed] [Google Scholar]
- 23.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 24.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, Krueger JG. Psoriasis Vulgaris Lesions Contain Discrete Populations of Th1 and Th17 T Cells. J Invest Dermatol. 2008 doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 25.Uyemura K, Yamamura M, Fivenson DF, Modlin RL, Nickoloff BJ. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701–705. doi: 10.1111/1523-1747.ep12371679. [DOI] [PubMed] [Google Scholar]
- 26.Szabo SK, Hammerberg C, Yoshida Y, Bata-Csorgo Z, Cooper KD. Identification and quantitation of interferon-gamma producing T cells in psoriatic lesions: localization to both CD4+ and CD8+ subsets. J Invest Dermatol. 1998;111:1072–1078. doi: 10.1046/j.1523-1747.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- 27.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nair RP, Ruether A, Stuart PE, Jenisch S, Tejasvi T, Hiremagalore R, Schreiber S, Kabelitz D, Lim HW, Voorhees JJ, Christophers E, Elder JT, Weichenthal M. Polymorphisms of the IL12B and IL23R Genes Are Associated With Psoriasis. J Invest Dermatol. doi: 10.1038/sj.jid.5701255. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley LT, Lebwohl M. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–592. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- 30.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Farinas MS, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–3194. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark RA, Chong BF, Mirchandani N, Yamanaka K, Murphy GF, Dowgiert RK, Kupper TS. A novel method for the isolation of skin resident T cells from normal and diseased human skin. J Invest Dermatol. 2006;126:1059–1070. doi: 10.1038/sj.jid.5700199. [DOI] [PubMed] [Google Scholar]
- 32.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 33.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 34.Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L, Lackner A, Alvarez X, Ochoa A, Chen L, Zou W. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;387:861–861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 36.Pernet I, Reymermier C, Guezennec A, Branka JE, Guesnet J, Perrier E, Dezutter-Dambuyant C, Schmitt D, Viac J. Calcium triggers beta-defensin (hBD-2 and hBD-3) and chemokine macrophage inflammatory protein-3 alpha (MIP-3alpha/CCL20) expression in monolayers of activated human keratinocytes. Exp Dermatol. 2003;12:755–760. doi: 10.1111/j.0906-6705.2003.00086.x. [DOI] [PubMed] [Google Scholar]
- 37.Voss E, Wehkamp J, Wehkamp K, Stange EF, Schroder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem. 2006;281:2005–2011. doi: 10.1074/jbc.M511044200. [DOI] [PubMed] [Google Scholar]
- 38.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 39.Haider AS, Lowes MA, Suarez-Farinas M, Zaba LC, Cardinale I, Khatcherian A, Novitskaya I, Wittkowski KM, Krueger JG. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol. 2008;180:1913–1920. doi: 10.4049/jimmunol.180.3.1913. [DOI] [PubMed] [Google Scholar]
- 40.Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273–279. doi: 10.1155/MI.2005.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, Catron D, Buchanan ME, Muller A, de Waal Malefyt R, Deng G, Orozco R, Ruzicka T, Lehmann P, Lebecque S, Caux C, Zlotnik A. Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol. 2000;164:6621–6632. doi: 10.4049/jimmunol.164.12.6621. [DOI] [PubMed] [Google Scholar]
- 42.Pauls K, Schon M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, Ruzicka T, Parker CM, Schon MP. Role of integrin alphaE(CD103)beta7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol. 2001;117:569–575. doi: 10.1046/j.0022-202x.2001.01481.x. [DOI] [PubMed] [Google Scholar]
- 43.Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, Kotelianski V, Gardner H, Nestle FO. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med. 2007;13:836–842. doi: 10.1038/nm1605. [DOI] [PubMed] [Google Scholar]
- 44.Buchau AS, Gallo RL. Innate immunity and antimicrobial defense systems in psoriasis. Clin Dermatol. 2007;25:616–624. doi: 10.1016/j.clindermatol.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, van de Kerkhof PC, Traupe H, de Jongh G, Heijer MD, Reis A, Armour JA, Schalkwijk J. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2007;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gudjonsson JE, Johnston A, Sigmundsdottir H, Valdimarsson H. Immunopathogenic mechanisms in psoriasis. Clin Exp Immunol. 2004;135:1–8. doi: 10.1111/j.1365-2249.2004.02310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, Jenisch S, Weichenthal M, Abecasis GR, Lim HW, Christophers E, Voorhees JJ, Elder JT. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78:827–851. doi: 10.1086/503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szegedi A, Aleksza M, Gonda A, Irinyi B, Sipka S, Hunyadi J, Antal-Szalmas P. Elevated rate of Thelper1 (T(H)1) lymphocytes and serum IFN-gamma levels in psoriatic patients. Immunol Lett. 2003;86:277–280. doi: 10.1016/s0165-2478(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N, Stratis A, Renkl AC, Sunderkotter C, Wlaschek M, Haase I, Scharffetter-Kochanek K. Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest. 2006;116:2105–2114. doi: 10.1172/JCI27180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stratis A, Pasparakis M, Rupec RA, Markur D, Hartmann K, Scharffetter-Kochanek K, Peters T, van Rooijen N, Krieg T, Haase I. Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J Clin Invest. 2006;116:2094–2104. doi: 10.1172/JCI27179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–649. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 53.Albanesi C, Scarponi C, Cavani A, Federici M, Nasorri F, Girolomoni G. Interleukin-17 is produced by both Th1 and Th2 lymphocytes, and modulates interferon-gamma- and interleukin-4-induced activation of human keratinocytes. J Invest Dermatol. 2000;115:81–87. doi: 10.1046/j.1523-1747.2000.00041.x. [DOI] [PubMed] [Google Scholar]