

Abstract
 An efficient synthetic strategy for installation of the two vicinal quaternary carbon centers of the communesins is reported. Key steps include the O-allylation/Claisen rearrangement of spirolactone systems, which are formed by tandem intramolecular Heck cyclization/carbonylation. Substituent and solvent effects on the stereochemical outcome of the Claisen rearrangements have been examined. The stereochemical assignment of the allyl spirolactone previously reported as 17 has now been revised to 32, which has the communesin relative configuration at the quaternary carbons. Key C-allyl spirolactone 59 bearing functional handles required for the communesin core has been constructed with a 9.8:1 diastereomer ratio.
An efficient synthetic strategy for installation of the two vicinal quaternary carbon centers of the communesins is reported. Key steps include the O-allylation/Claisen rearrangement of spirolactone systems, which are formed by tandem intramolecular Heck cyclization/carbonylation. Substituent and solvent effects on the stereochemical outcome of the Claisen rearrangements have been examined. The stereochemical assignment of the allyl spirolactone previously reported as 17 has now been revised to 32, which has the communesin relative configuration at the quaternary carbons. Key C-allyl spirolactone 59 bearing functional handles required for the communesin core has been constructed with a 9.8:1 diastereomer ratio.
Introduction and Background
About a decade ago, Numata, et al. reported two unusual natural products isolated from a Penicillium mold found growing on the marine alga Enteromorpha intestinalis.1 The unique structures of these two compounds, communesin A (1) and communesin B (5), were established by spectroscopic analysis (Figure 1). An interesting feature of these complex, highly functionalized polycyclic compounds is the two contiguous quaternary centers at C-7,8. Communesins A and B were found to have in vitro cytotoxic activity against P-388 lymphoid leukemia cells. In 2001, Hemscheidt and coworkers described a metabolite called nomofungin, which was isolated from an unidentified fungus growing on the bark of Ficus microcarpa in Hawaii.2 This material was found to have cytotoxic activity against LoVo and KB cells, which was shown to be due to the ability of the metabolite to cause microfilament disruption. The metabolite was initially proposed to have structure 9, but it was later found that this assignment was in error, and that nomofungin is actually communesin B (5). It should be noted, however, that the Hemscheidt work did serve to establish both the configuration at C-21 of 5 as well as the absolute configuration of the molecule, which were not originally determined by the Numata group. Recently, several other modified communesin derivatives have been isolated3 including communesin C (6),4 D (7), E (2), F (8), G (3) and H (4) (Figure 1). Some of these new compounds were found to have significant biological activity. For example, communesins D, E and F are insecticidal,3b and communesins C and D are moderately active against various leukemia cell lines.3a
Figure 1.

Structures of the Communesins and Perophoramidine
In 2002, Ireland reported the isolation of a congeneric compound, perophoramidine, from the marine ascidian Perophora namei collected in the Philippines.5 Based mainly upon NMR spectral analysis, this metabolite was assigned the hexacyclic structure 10, which contains some unique features including two amidine units, two chlorines and a bromine. Like the communesins, perphoramidine has two adjacent quaternary carbons at C-4,20, but interestingly, the natural products have the opposite relative stereochemistry at these centers. Perphoramidine has cytotoxicity against the HCT116 colon carcinoma cell line, and also induces apoptosis via PARP cleavage.
Perophoramidine and the communesins have closely related structures and presumably arise via a common biogenetic pathway.6 Some model studies on biogenetically-patterned synthesis of the communesin/perophoramidine ring system by the Stoltz,7 and Qin8 groups have appeared, as well as a model hetero Diels-Alder-based approach by Crawley and Funk.9 Moreover, Fuchs and Funk have completed an elegant, biogenetically-inspired total synthesis of racemic perophoramidine.10 In addition, Rainier and coworkers recently described a nice total synthesis of dehaloperophoramidine.11
In 2003, we disclosed some preliminary results on a Heck-based strategy for total synthesis of these alkaloids.12 In this work, it was found that acrylamides 11 and 12 underwent tandem intramolecular Heck cyclization/carbonylation13 to afford E/F-lactam esters 13 and 14, respectively, in good yields (Scheme 1).These compounds could then be cleanly cyclized to lactones 15 and 16 after removal of the TBS protecting groups. As part of these studies, we reported that lactone 16 could be deprotonated and alkylated with allyl bromide to afford a single product to which we assigned the perophoramidine relative stereochemistry at the quaternary centers as shown in 17 via 1H NMR NOESY experiments. Based upon the subsequent studies discussed in this paper, we have found that the configurational assignment of this alkylation product is in fact incorrect (vide infra).
Scheme 1.

Results and Discussion
Because we believed that we had been able to efficiently access the requisite perophoramidine C-4,20 quaternary carbon stereochemistry, work was undertaken on synthesis of a Heck substrate bearing the appropriate C-ring substituents. Using a two-step procedure developed by Coe14 heating commercially available 4-bromo-2-nitrotoluene (18) in the presence of N,N-dimethylformamide dimethyl acetal and pyrrolidine in DMF generated the known enamine 19,15 which was oxidatively cleaved using NaIO4 in THF/H2O to afford the desired benzaldehyde 20 in 93 % yield over two steps (Scheme 2). Wittig olefination of the aldehyde 20 with readily available ylide 2116 in refluxing toluene generated the benzylidene lactone 22 in excellent yield. Ring opening of this unsaturated lactone with the aluminium amide reagent17 derived from the commercially available aniline 23 produced the nitro acrylamide 24 in quantitative yield. Subsequent protection of the primary alcohol moiety of 24 as its TBS ether and alkylation of the amide nitrogen using NaH/MOMCl resulted in the N-MOM acrylamide 25 in 84 % yield over the two steps.
Scheme 2.

To continue the synthesis, the Heck reaction/carbonylation sequence with substrate 25 was examined, leading to the desired lactam ester 28 as a mixture of diastereomers along with the reductive Heck product 2718 and ester 26 derived from uncyclized starting iodide (Scheme 3). The ratios of these products varied widely depending upon the specific reaction conditions (Pd source, ligand, solvent, base, etc) used for the Heck/carbonylation step. However, the formation of the desired cyclization products 28 was optimal under the conditions shown in the scheme. Although compounds 26-28 could be separated by chromatography and characterized spectrally (see Experimental Section), it was found best to simply expose the crude reaction mixture to TBAF, leading to lactone 29 (4:1 diastereomeric mixture) in 51% isolated overall yield from substrate 25.
Scheme 3.

This mixture of lactones was deprotonated with sodium hydride in DMF at room temperature, upon which allyl iodide was added, and the mixture was heated at in an oil bath at 90 °C. This reaction produced a single crystalline alkylation product in excellent yield whose structure was established unambiguously by X-ray analysis. We were quite surprised to find, however, that the allylated lactone has the communesin stereochemistry shown in 30 rather than the expected perophoramidine configuration at the quaternary centers.
In view of this unexpected stereochemical result, we decided to reexamine the model system shown in Scheme 1.12 Thus, the lactone product from C-allylation of 1619 was exposed to the reagent derived from ammonium chloride and trimethylaluminum20 to afford hydroxy amide 32 (Scheme 4). This material provided crystals suitable for X-ray analysis, which established that the allylation product of 16 in fact has the communesin stereochemistry shown in 31, and is not the perophoramidine diastereomer 17 which had originally been asssigned.
Scheme 4.

Since the experiments outlined above suggested that we might be able to access the quaternary carbons of the communesins stereoselectively, we turned to an investigation of systems bearing useful C and F aromatic ring substitutents for synthesis of this group of metabolites. Using methodology similar to that described above, Heck substrate 33 was prepared in a few steps. It was found, however, that under most conditions, the Heck/carbonylation sequence led to the reduced compound 35 as the major or sole product rather than the desired ester 34 (Scheme 5). After substantial experimentation, it was possible at best to produce equal amounts of 34 and 35 along with a small quantity of ester 36 using the conditions shown in the scheme. It should be noted that a similar tendency for intramolecular reductive Heck reactions has been observed by Denmark and Schnute when using nitroolefins.18a The rationale presented for these results would also apply to our cases involving nitroarenes 25 and 33. Since the desired transformation of Heck substrate 33 to ester 34 was not synthetically useful, we turned to an alternative system lacking the nitro group which was expected to be more amenable to the requisite Heck/carbonylation process.
Scheme 5.

One solution we considered was to install the required C-ring nitrogen via a late-stage Curtius rearrangement of a system bearing a suitable carbon substituent. Thus, Heck substrate 43 was constructed via the chemistry outlined in Scheme 6. Known benzyl alcohol 3721 was first converted to the benzyl ether 38, and the nitro group was subsequently reduced to afford aniline 39. Conversion of this amine to the corresponding aluminum amide17 followed by reaction with benzylidene lactone 4022 then led to amide 41 in high yield. The alcohol functionality of 41 was protected as the TBS ether 42, and the amide nitrogen was methylated to yield the desired substrate 43. This compound underwent smooth tandem Heck cyclization/carbonylation to produce the desired lactam ester 44 as a single stereoisomer whose configuration was not proven, but by analogy with 13/1412 is assumed to be as shown.
Scheme 6.

Treatment of silyl ether ester 44 with TBAF led to desilylation and in situ cyclization to lactone 45, which was produced as a 2:1 mixture of diastereomers (Scheme 7). During work with spirolactone 45 it was discovered that allylation of the corresponding enolate does not actually involve a direct C-alkylation. Thus, treatment of 45 with sodium hydride in DMF along with allyl iodide at room temperature affords the chromatographically isolable ketene acetal 46 in 57% yield (71% based on recovered lactone, formed via hydrolysis of 46 during purification). Ketene acetal 46 appears to exist as a 2:1 mixture of atropisomers, as can be seen by 1H NMR. It was found that heating ketene acetal 46 at 130 °C in either DMF or toluene led to a Claisen rearrangement producing a separable mixture of diastereomeric C-allylation products 47 and 48, with a slightly better stereoselectivity in the former solvent. The stereochemistry of the minor isomer was established by 1H NMR NOESY analysis to have the configuration shown in 48 (see Supporting Information). It should be noted that although we cannot isolate the ketene acetals in the two alkylations described above, we believe these processes also involve an initial O-allylation followed by a Claisen rearrangement (vide infra).
Scheme 7.

In view of the disappointing level of stereoselectivity in the alkylation of lactone 45, we decided to explore an alternative system with different substitution in the F-ring with the hope of improving this key step. Known, easily prepared phenol 4923 was therefore first converted to the carbamate 50 and the nitro group was reduced to yield the aniline 51 (Scheme 8). Condensation of this intermediate with benzylidene lactone 40 then produced amide 52 in excellent overall yield. The alcohol functionality of 52 was protected as the MOM ether 53, and the amide nitrogen was methylated to afford 54. The tandem Heck cyclization/carbonylation of this substrate proceeded cleanly to generate the requisite lactam ester 55 as a single stereoisomer assumed to be as indicated.
Scheme 8.

The MOM protecting group of 55 was next removed with methanolic HCl to give the desired spirolactone 57 along with some of the uncyclized hydroxy ester 56 (Scheme 9). O-Allylation of the spirolactone as was done with 45 gave the ketene acetal 58, which in this case was again a 2:1 mixture of atropisomers. Heating 58 in DMF at 110°C did not show improved stereoslectivity over the system in Scheme 7, leading to a 2.3:1 mixture of stereoisomers 59 and 60 in good yield. We have been unable to definitively establish the configurations of these diastereomers, but by analogy with the three cases described above we believe the major isomer has the communesin configuration shown in 59. We were pleased to find, however, that heating ketene acetal 58 in toluene induced a Claisen rearrangement, which led to a synthetically useful 9.8:1 mixture of 59:60.
Scheme 9.

To rationalize the high stereoselectivity shown in the first two lactone allylations (cf. Schemes 3 and 4), we considered four possible half-chair transition state conformations for the Claisen rearrangement of the corresponding ketene acetal intermediates (Figure 2). The carbonyl group of the oxy-indole moiety (EF rings) could occupy either pseudo-axial (A) or pseudo-equatorial (E) positions. In addition, each transition state has two possible atropisomers (α and β) derived from the position of the substituent (R) at C(10) in the C-ring aryl moiety. It seems reasonable that the allyl group approaches the ketene acetal double bond from the face opposite to this substituent in order to avoid severe steric interactions.24 Since axial attack of the allyl group is more favorable stereoelectronically,25 transition states A-β and E-α are expected to be preferable to conformations A-α and E-β. In addition, it appears from inspection of models that the aryl group of the oxy-indole moiety blocks attack of the allyl substituent more effectively than does the carbonyl group, and therefore transition state E-α would seem to be favored over A-β. Steric interactions and/or electronic repulsions between the carbonyl group and the substituent R (OMe or NO2) may additionally destabilize conformation A-β.
Figure 2.

Models for Allylation of Lactones 16 and 29
On the other hand, allylation of the lactones bearing substituents (R”) at the C-12a position of the oxy-indole moiety showed lower stereoselectivity than did the above two cases (cf. Schemes 7 and 9). As shown in Figure 3, substituent R” causes substantial steric interactions not only with substituent R in E-α but also with the allyl group in A-β. As noted above, NMR analysis shows that there is little preference for one atropisomer in ketene acetals 46 and 58 at room temperature. These steric interactions presumably slow the Claisen rearrangements of the latter two ketene acetals, making these species more easily isolable, and also lead to generally low stereoselectivities. At this point, however, we are unable to explain why we observe high stereoselectivity in the rearrangement of ketene acetal 58 in toluene.
Figure 3.

Transition State Models for Claisen Rearrangement of Ketene Acetals 46 and 58
In conclusion, we have developed a concise strategy to construct systems bearing the two quaternary centers of the communesins. Key steps in this approach involve an intramolecular tandem Heck cyclization/carbonylation, followed by a stereoselective O-allylation/Claisen rearrangement of a spirolactone enolate. We are currently attempting to utilize this methodology in a total synthesis of the communesins.
Experimental Section
3-(4-Bromo-2-nitrophenyl)-2-[2-(tert-butyldimethylsilanyloxy)ethyl]-N-(2,4-dichloro-6-iodophenyl)-N-methoxymethylacrylamide (25)
N,N-Dimethylformamide dimethyl acetal (7.25 mL, 54.6 mmol) and pyrrolidine (4.50 mL, 53.9 mmol) were added to 4-bromo-2-nitrotoluene (18, 9.66 g, 44.7 mmol, TCI) in DMF (25 mL). The reaction mixture was heated at 110 °C for 4 h to give a dark red solution. The reaction mixture was cooled to rt and the volatile organics were removed under reduced pressure resulting in the known enamine 1915 as a mass of red crystals.
The above enamine was dissolved in THF (30 mL) and added slowly to NaIO4 (28.9 g, 135 mmol) in 50% aqueous THF (200 mL) while maintaining the temperature below 20 °C. The reaction mixture was rapidly stirred for an additional 2 h and the insoluble salts were removed by filtration and washed with EtOAc. The combined organic filtrate was washed with water, dried (MgSO4) and concentrated. The crude product was purified by flash silica gel chromatography (1:9 EtOAc: hexanes) to afford the desired 4-bromo-2-nitrobenzaldehyde (20, 9.58 g, 93%) as a yellow solid (mp 93-95 °C; lit. mp 95 °C26). IR (KBr) 3085, 2912, 2863, 1812, 1691, 1525 cm−1; 1H NMR (300 MHz, CDCl3) δ 10.38 (d, J = 0.6 Hz, 1H), 8.26 (d, J = 1.7 Hz, 1H), 7.93 (ddd, J = 8.3, 1.8, 0.6 Hz, 1H), 7.84 (d, J = 8.3 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 187.2, 150.1, 137.4, 131.1, 129.9, 128.4, 127.8.
3-(Triphenyl-λ5-phosphanylidene)dihydrofuran-2-one (21, 4.62 g, 13.3 mmol)16 was added to nitrobenzaldehyde 20 (3.07 g, 13.3 mmol) in toluene (70 mL). The reaction mixture was heated at reflux for 2 h and cooled to rt. The volatile organics were removed under reduced pressure. The crude product was absorbed onto silica gel and purified by flash silica gel column chromatography (1:2 EtOAc: hexanes) to afford the benzylidene lactone 22 (3.96 g, 100%) as a light brown solid (mp 129-131 °C). IR (KBr) 2929, 2867, 1762, 1523 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 1.8 Hz, 1H), 7.82 (dd, J = 8.3, 1.8 Hz, 1H), 7.78 (t, J = 2.9 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 4.46 (t, J = 7.2 Hz, 2H), 3.07 (dd, J = 7.2, 3.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 170.8, 148.8, 136.7, 131.3, 129.3, 129.0, 128.4, 123.5, 65.7, 26.9.
2,4-Dichloro-6-iodoaniline (23, 11.1 g, 39 mmol, TCI) was dissolved in benzene (60 mL) and slowly added via syringe to AlMe3 (2.0 M in hexanes, 21 mL, 42 mmol) in benzene (10 mL) at 0 °C. After stirring the mixture for an additional 45 min, the benzylidine lactone 22 (10.4 g, 35 mmol) dissolved in CH2Cl2 (60 mL) was slowly added via syringe. The reaction mixture was heated at 65 °C for 15 h then cooled to 0 °C. 1 N HCl (50 mL) was slowly added dropwise and the reaction mixture was stirred for an additional 30 min. The organic layer was removed and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over MgSO4 and concentrated in vacuo. The crude oil was absorbed onto silica gel and purified by flash silica gel chromatography (1:2 EtOAc:hexanes) to afford the bromo dichloro acrylamide 24 (16.5 g, 81%) as a foam. 1H NMR (300 MHz, CDCl3) δ 8.33 (d, J = 2.0 Hz, 1H), 8.05 (s, 1H), 7.80-7.83 (m, 2H), 7.67 (s, 1H), 7.49 (d, J = 2.2 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 3.73-3.80 (br m, 2H), 2.63 (t, J = 5.8 Hz, 2H), 2.48 (br s, 1H); 13C NMR (75 MHz, CDCl3) δ 167.9, 148.2, 137.6, 137.3, 137.0, 135.1, 134.8, 133.2, 132.8, 132.4, 130.3, 128.4, 122.8, 100.2, 61.5, 31.4.
TBSCl (5.11 g, 33.9 mmol) was added in a single portion to acrylamide 24 (16.3 g, 27.8 mmol) and imidazole (4.20 g, 61.7 mmol) in DMF (30 mL) and the mixture was stirred at rt for 60 min. Water was added and the aqueous phase was extracted with ether. The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure to afford the desired TBS ether (19.5 g, 100%) as an oil, which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 8.28 (d, J = 2.0 Hz, 1H), 7.74-7.77 (m, 2H), 7.70 (s, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.45 (d, J = 2.2 Hz, 1H), 3.75 (t, J = 5.6 Hz, 2H), 2.67 (t, J = 5.6 Hz, 2H), 0.82 (s, 9H), 0.02 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 167.3, 148.4, 137.9, 137.7, 136.9, 135.9, 134.7, 133.6, 133.4, 132.8, 130.7, 130.4, 128.4, 122.7, 100.8, 62.8, 31.8, 26.3, 18.7, −5.0; HRMS-APCI: [M+H]+ calcd for C23H26BrCl2IN2O4Si 698.9340; found, 698.9366.
NaH (60 % dispersion in mineral oil, 1.33 g, 33.2 mmol) was added to the TBS ether (19.5 g, 27.8 mmol) in THF (140 mL) at 0 °C. The solution was stirred for an additional 20 min at this temperature before MOMCl (3.10 mL, 41.6 mmol) was introduced. The reaction solution was warmed to rt and stirred for 12 h. Saturated aqueous NaHCO3 (100 mL) was slowly added and the organic layer was removed. The aqueous phase was extracted with EtOAc. The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure. The crude product was purified by flash silica gel column chromatography (1:4 EtOAc: hexanes) to afford the N-MOM amide 25 (17.4 g, 84%) as an oil, which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.32 (m, 1H), 7.87 (d, J = 2.1 Hz, 1H), 7.75−7.78 (m, 2H), 7.65 (s, 1H), 7.54 (d, J = 2.1 Hz, 1H), 5.10 (ABq, J = 67.1, 10.5 Hz, 2H), 3.81−3.85 (m, 2H), 3.28 (s, 3H), 2.71 (br. m, 2H), 0.92 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 179.4, 150.8, 137.4, 135.6, 135.1, 133.8, 130.7, 129.3, 128.9, 128.1, 123.3, 121.8, 116.9, 72.0, 59.5, 56.6, 52.8, 40.2, 39.6, 26.1, −5.4; HRMS-APCI: [M+H]+ calcd for C25H30BrCl2IN2O5Si 742.9602; found, 742.9572.
(4-Bromo-2-nitrophenyl)-{3-[2-(tert-butyldimethylsilanyloxy)-ethyl]-5,7-dichloro-1-methoxymethyl-2-oxo-2,3-dihydro-1H-indol-3-yl}-acetic Acid Methyl Ester (28)
To a solution of N-MOM amide 25 (27.4 mg, 0.037 mmol) in DMA (2.0 mL) and MeOH (0.4 mL) were added Pd2(dba)3 (3.4 mg, 0.004 mmol), P(o-Tol)3 (4.5 mg, 0.015 mmol), n-Bu4NBr (23.7 mg, 0.074 mmol) and NEt3 (26 μL, 0.187 mmol). The mixture was stirred at 85 °C under a CO atmosphere (1 atm) for 21 h. The catalyst was removed by filtration and washed with EtOAc. The filtrate was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The crude mixture was used for the next step without further purification. For analytical purposes the compounds were separated by preparative TLC (1:5 EtOAc:hexanes). More polar major diastereomer of 28 (pale yellow oil): IR (film) 2946, 2860, 1735, 1536, 1461, 1095 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 2.1 Hz, 1H), 7.70 (dd, J = 8.5, 2.1 Hz, 1H), 7.45 (d, J = 8.5 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H), 6.76 (d, J = 2.0 Hz, 1H), 5.37, 5.30 (ABq, J = 10.5 Hz, 2H), 5.09 (s, 1H), 3.65 (s, 3H), 3.42 (s, 3H), 3.24 (td, J = 6.2, 2.6 Hz, 2H), 2.25 (td, J = 6.2. 3.7 Hz, 2H), 0.72 (s, 9H), −0.17 (d, J = 2.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 178.4, 170.2, 151.3, 138.3, 135.6, 133.5, 133.0, 130.9, 128.5, 128.4, 126.8, 124.0, 123.2, 117.2, 72.6, 59.4, 57.1, 53.1, 52.8, 50.4, 37.2, 26.1, 18.6, −5.3, −5.4; HRMS-ES: [M+H]+ calcd for C27H34BrCl2N2O7Si 675.0696; found, 675.0712. Less polar minor diastereomer of 28 (pale yellow oil): IR (film) 2946, 2860, 1735, 1536, 1461 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 2.1 Hz, 1H), 7.56 (dd, J = 8.5, 2.1 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 5.24, 5.19 (ABq, J = 10.5 Hz, 2H), 5.16 (s, 1H), 3.64 (s, 3H), 3.29 (t, J = 6.2 Hz, 2H), 3.22 (s, 3H), 2.22−2.12 (m, 2H), 0.72 (s, 9H), −0.17 (d, J = 2.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 178.6, 170.4, 151.4, 138.3, 135.7, 133.7, 133.5, 131.2, 129.0, 128.1, 127.0, 124.3, 123.0, 116.9, 72.3, 59.3, 56.8, 53.3, 53.2, 51.0, 39.3, 26.1, 18.6, −5.3, −5.4; HRMS-ES: [M+H]+ calcd for C27H34BrCl2N2O7Si 675.0696; found, 675.0672.
3-(4-Bromo-2-nitrobenzyl)-3-[2-(tert-butyldimethylsilanyloxy)-ethyl]-5,7-dichloro-1-methoxymethyl-1,3-dihydroindol-2-one (27)
Pale yellow oil. IR (film) 2935, 2860, 1730, 1536, 1461, 1348 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 2.0 Hz, 1H), 7.52 (dd, J = 8.3, 2.0 Hz, 1H), 7.21 (d, J = 2.0 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H), 5.19 (s, 2H), 3.80, 3.41 (ABq, J = 13.7 Hz, 2H), 3.37 (m, 2H), 3.32 (s, 3H), 2.32 (dt, J = 13.8, 6.9 Hz, 1H), 2.08 (dt, J = 13.8, 5.5 Hz, 1H), 0.75 (s, 9H), − 0.14 (d, J = 3.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 179.4, 150.8, 137.3, 135.7, 135.1, 133.8, 130.6, 129.3, 128.9, 128.1, 123.4, 121.8, 116.9, 72.0, 59.5, 56.5, 52.8, 40.3, 39.6, 26.1, 18.5, −5.36, −5.37; HRMSES: [M+H]+ calcd for C25H32BrCl2N2O5Si 617.0641; found, 617.0659.
2-{[2-(4-Bromo-2-nitrobenzylidene)-4-(tert-butyldimethylsilanyloxy)-butyryl]-methoxymethylamino}-3,5-dichlorobenzoic Acid Methyl Ester (26)
Yellow solid (mp 125-126 °C). IR (film) 2946, 2860, 1730, 1665, 1531, 1278 cm−1; 1H NMR (360 MHz, CDCl3, 5:1 atropisomer mixture) δ 8.30 (d, J = 1.8 Hz, 1H, major), 8.09 (d, J = 1.7 Hz, 1H, minor), 7.89 (d, J = 2.4 Hz, 1H, major), 7.85 (d, J = 2.4 Hz, 1H, minor), 7.77 (d, J = 1.8 Hz, 1H, minor), 7.75−7.69 (m, 3H, major), 7.67 (m, 1H, minor), 7.46 (s, 1H, major), 7.22 (d, J = 8.2 Hz, 1H, minor), 7.00 (s, 1H, minor), 5.23, 4.85 (ABq, J = 10.5 Hz, 2H, minor), 5.20, 4.79 (ABq, J = 10.2 Hz, 2H, major), 3.92 (s, 3H, minor), 3.89 (s, 3H, major), 3.84 (t, J = 6.0 Hz, 2H, major), 3.74 (m, 2H, minor), 3.46 (s, 3H, minor), 3.21 (s, 3H, major), 2.68 (m, 2H, major), 2.41 (m, 2H, minor), 0.90 (s, 9H, major), 0.83 (s, 9H, minor), 0.07 (s, 6H, major), -0.02 (s, 6H, minor); 13C NMR (75 MHz, CDCl3) δ 171.7, 164.7, 148.5, 137.6, 136.6, 136.4, 136.2, 134.9, 134.1, 133.8, 133.1, 130.9, 130.5, 129.4, 128.2, 122.4, 83.3, 61.0, 56.8, 53.3, 33.4, 26.3, 18.7, −5.0; HRMS-ES: [M+H]+ calcd for C27H34BrCl2N2O7Si 675.0696; found, 675.0665.
Synthesis of Spirolactone 29
To a stirred solution of the above crude mixture of Heck/carbonylation adducts in THF (3.0 mL) was added TBAF (0.072 mL, 1.0 M in THF, 0.072 mmol) at 0 °C. The reaction mixture was warmed to rt and stirred overnight. The mixture was diluted with EtOAc and saturated aqueous NH4Cl. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:4 EtOAc:hexanes) to give the spiro lactone 29 (9.9 mg, 51% over 2 steps) as a pale yellow oil. IR (film) 2924, 1725, 1531, 1455, 1348 cm−1; 1H NMR (400 MHz, CDCl3, 4:1 diastereomeric mixture) δ 7.95 (s, 1H, minor), 7.79 (d, J = 1.9 Hz, 1H, major), 7.61 (dd, J = 8.5, 1.9 Hz, 1H, major), 7.45 (dd, J = 8.6, 1.8 Hz, 1H, minor), 7.41 (d, J = 8.5 Hz, 1H, major), 7.34 (s, 1H, minor), 7.25 (d, J = 1.8 Hz, 1H, major), 7.22 (s, 1H, minor), 7.12 (d, J = 1.7 Hz, 1H, major), 6.68 (d, J = 8.5 Hz, 1H, minor), 5.50 (br s, 1H, minor), 5.44 (s, 1H, major), 5.30 (s, 1H, minor), 5.21, 5.18 (ABq, J = 10.3 Hz, 2H, major), 5.13 (s, 1H, minor), 5.06 (dt, J = 11.5, 4.2 Hz, 1H, major), 4.85 (m, 1H, minor), 4.72 (m, 1H, minor), 4.64 (dt, J = 11.7, 4.7 Hz, 1H, major), 3.23 (s, 3H, major), 2.99 (s, 3H, minor), 2.59-2.52 (m, 1H, major and minor), 2.26 (m, 1H, minor), 2.19 (dt, J = 14.6, 4.0 Hz, 1H, major); 13C NMR (75 MHz, CDCl3) δ 177.4, 168.8, 150.6, 136.3, 136.2, 134.5, 132.5, 131.8, 130.5, 127.9, 126.6, 123.1, 122.7, 117.6, 72.0, 65.2, 56.8, 52.7, 46.8, 33.2; HRMS-ES: [M+Na]+ calcd for C20H15BrCl2N2NaO6 550.9388; found, 550.9388.
Allylation of Spirolactone 29
To a stirred suspension of spiro lactone 29 (102 mg, 0.191 mmol) and NaH (9.2 mg, 60% dispersion in mineral oil, 0.230 mmol) in DMF (2.0 mL) was added allyl iodide (0.026 mL, 0.287 mmol) at 0 °C. The mixture was stirred at rt for 1 h and heated at 90 °C for 7 h. The reaction mixture was cooled to rt, diluted with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:2 EtOAc:hexanes) to give the allyl lactone 30 (95 mg, 87%) as a yellow solid (mp 157-158 °C). Recrystallization from EtOAc provided X-ray quality crystals of 30. IR (film) 2924, 1725, 1536, 1461, 1359 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.6, 2.0 Hz, 1H), 7.10 (d, J = 1.9 Hz, 1H), 6.81 (d, J = 1.8 Hz, 1H), 6.68 (d, J = 8.7 Hz, 1H), 5.51-5.34 (m, 4H), 4.99 (d, J = 17.2 Hz, 1H), 4.94 (d, J = 10.4 Hz, 1H), 4.61 (dt, J = 11.6, 4.0 Hz, 1H), 3.44 (s, 3H), 3.35 (dd, J = 15.2, 6.3 Hz, 1H), 2.99 (ddd, J = 15.2, 10.9, 4.3 Hz, 1H), 2.86 (dd, J = 15.4, 7.1 Hz, 1H), 2.16 (dt, J = 14.7, 3.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 178.3, 168.0, 150.8, 136.1, 134.8, 134.3, 132.7, 132.6, 132.4, 131.1, 129.0, 128.0, 125.1, 122.1, 120.3, 116.9, 72.3, 65.0, 57.1, 54.4, 54.2, 44.1, 31.5; HRMS-ES: [M+Na]+ calcd for C23H19BrCl2N2NaO6 590.9701; found, 590.9697.
2-[5,7-Dichloro-3-(2-hydroxyethyl)-1-methoxymethyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-2-(2-methoxyphenyl)-pent-4-enoic Acid Amide (32)
To a stirred suspension of NH4Cl (229 mg, 4.28 mmol) in toluene (5 mL) was slowly added Me3Al (2.30 mL, 2.0 M in hexanes, 4.60 mmol) at 0 °C. After the addition was complete, the reaction mixture was allowed to warm to rt and was stirred for 1.5 h. To a solution of lactone 31 (291 mg, 0.61 mmol) in toluene (15 mL) was added the above aluminum amide reagent at rt, and the mixture was heated at 80 °C overnight. The reaction mixture was cooled to rt and was carefully quenched with 1N HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (10:10:1 CH2Cl2:EtOAc:MeOH) to give the amide 32 (286 mg, 95%) as a yellow solid (mp 179-180 °C). Recrystalization from EtOAc provided X-ray quality crystals of 32. IR (film) 3342, 1708, 1672, 1461 cm−1; 1H NMR (300 MHz, CDCl3+CD3OD) δ 7.85 (br s, 1H), 7.14 (t, J = 7.7 Hz, 1H), 7.06 (d, J = 1.6 Hz, 1H), 6.78 (t, J = 7.6 Hz, 1H), 6.54 (br s, 1H), 5.87 (m, 1H), 5.51 (br s, 1H), 5.17 (d, J = 16.8 Hz, 1H), 5.03 (d, J = 10.0 Hz, 1H), 4.65 (br s, 2H), 3.55-2.89 (m, 10H), 2.42 (br s, 1H); 13C NMR (75 MHz, CDCl3+CD3OD) δ 180.2, 174.7, 156.9, 136.9, 134.6, 134.0, 131.1, 129.6, 129.1, 128.2, 127.3, 124.7, 119.4, 118.4, 114.6, 110.7, 71.3, 58.8, 58.4, 57.0, 56.3, 54.1, 37.3, 34.7; HRMS-ES: [M+H]+ calcd for C24H27Cl2N2O5 493.1297; found, 493.1285.
1-Benzyloxymethyl-2-iodo-3-nitrobenzene (38)
To a stirred solution of (2-iodo-3-nitrophenyl)-methanol (37)21 (0.86 g, 3.08 mmol) in CH2Cl2 (6 mL) and cyclohexane (10 mL) was added benzyl 2,2,2-trichloroacetimidate (1.14 mL, 6.13 mmol), followed by TfOH (0.03 mL, 0.34 mmol). The mixture was stirred at rt for 9 h and then diluted with saturated aqueous NaHCO3 and EtOAc. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:15 EtOAc:hexanes) to give benzyl ether 38 (1.06 g, 93%) as a yellow oil. IR (film) 2871, 1712, 1526, 1350 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.6 Hz, 1H), 7.58 (dd, J = 7.9, 1.4 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.44−7.32 (m, 5H), 4.71 (s, 2H), 4.63 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 144.6, 137.9, 131.6, 129.4, 129.0, 128.4, 128.2, 123.9, 89.1, 76.9, 73.5; HRMS-EI: [M]+ calcd for C14H12INO3 368.9862; found, 368.9866.
3-Benzyloxymethyl-2-iodophenylamine (39)
To a solution of the nitrobenzene 38 (1.05 g, 2.83 mmol) in EtOH (10 mL) and glacial acetic acid (10 mL) was added iron powder (0.79 g, 14.15 mmol). The mixture was heated at 60 °C for 3 h and then cooled to rt. The mixture was diluted with iced water and carefully neutralized with solid Na2CO3. The resulting solution was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:5 EtOAc:hexanes) to give the aniline 39 (0.83 g, 97%) as a colorless oil. IR (film) 3447, 3357, 2859, 1610, 1463 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.45-7.29 (m, 5H), 7.14 (t, J = 7.7 Hz, 1H), 6.89 (d, J = 6.9 Hz, 1H), 6.72 (dd, J =7.9, 0.9 Hz, 1H), 4.65 (s, 2H), 4.56 (s, 2H), 4.00 (bs, 2H); 13C NMR (75 MHz, CDCl3) δ 147.4, 141.8, 138.6, 129.2, 128.9, 128.3, 128.1, 119.4, 114.5, 88.6, 77.2, 73.0; HRMS-ES: [M+H]+ calcd for C14H15INO 340.0198; found, 340.0195.
N-(3-Benzyloxymethyl-2-iodophenyl)-4-hydroxy-2-[2-(4-methoxyphenoxymethyl)-benzylidene]-butyramide (41)
To a stirred solution of the aniline 39 (0.69 g, 2.03 mmol) and lactone 4022 (0.57 g, 1.84 mmol) in CH2Cl2 (30 mL) was added AlMe3 (1.80 mL, 2.0 M in hexane, 3.60 mmol) at 0 °C. The mixture was warmed to rt, stirred overnight and then poured into ice cold aqueous NH4Cl. The aqueous layer was extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the amide 41 (1.16 g, 97%) as a white solid (mp 126.5-127 °C). IR (film) 3368, 2859, 1661, 1509, 1226 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 1H), 8.11 (dd, J = 8.0, 1.5 Hz, 1H), 7.68 (s, 1H), 7.54 (m, 1H), 7.51−7.28 (m, 10H), 6.94−6.89 (m, 2H), 6.84−6.80 (m, 2H), 5.03 (s, 2H), 4.64 (s, 2H), 4.55 (s, 2H), 3.79 (t, J = 5.8 Hz, 2H), 3.74 (s, 3H), 2.77 (t, J = 5.8 Hz, 2H), 2.70 (br s, 1H); 13C NMR (75 MHz, CDCl3) δ 168.9, 154.6, 153.1, 141.9, 138.64, 138.61, 138.3, 135.5, 135.3, 134.3, 129.9, 129.6, 129.3, 129.00, 128.95, 128.9, 128.3, 125.9, 122.5, 116.3, 115.2, 96.0, 77.1, 73.1, 69.7, 62.5, 56.2, 31.9; HRMS-ES: [M+H]+ calcd for C33H33INO5 650.1404; found, 650.1404.
N-(3-Benzyloxymethyl-2-iodophenyl)-4-(tert-butyldimethylsilanyloxy)-2-[2-(4-methoxyphenoxymethyl)-benzylidene]-butyramide (42)
To a solution of the amide 41 (568 mg, 0.87 mmol) and imidazole (178 mg, 2.62 mmol) in DMF (8.7 mL) was added TBSCl (316 mg, 2.09 mmol). The reaction mixture was stirred at rt overnight and then diluted with EtOAc. The solution was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:3 EtOAc:hexanes) to give the TBS ether 42 (641 mg, 96%) as a colorless oil. IR (film) 2950, 2848, 1678, 1509, 1226, 1090 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.2 (dd, J = 8.0, 1.4 Hz, 1H), 7.74 (s, 1H), 7.56 (m, 1H), 7.44−7.29 (m, 9H), 6.94−6.91 (m, 2H), 6.85−6.82 (m, 2H), 5.03 (s, 2H), 4.65 (s, 2H), 4.59 (s, 2H), 3.9 (t, J = 6.1 Hz, 2H), 3.76 (s, 3H), 2.84 (t, J = 6.1 Hz, 2H), 0.87 (s, 9H), 0.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 167.7, 154.5, 153.3, 141.8, 139.0, 138.5, 138.3, 135.7, 135.2, 134.1, 129.9, 129.3, 129.2, 128.9, 128.8, 128.5, 128.23, 128.21, 125.6, 122.4, 116.4, 115.1, 95.9, 77.2, 73.1, 69.4, 62.5, 56.1, 31.8, 26.4, 18.8, −4.9; HRMS-ES: [M+H]+ calcd for C39H47INO5Si 764.2268; found, 764.2271.
N-(3-Benzyloxymethyl-2-iodophenyl)-4-(tert-butyldimethylsilanyloxy)-2-[2-(4-methoxyphenoxymethyl)-benzylidene]-N-methylbutyramide (43)
To a stirred suspension of NaH (40 mg, 60% dispersion in mineral oil, 1.01 mmol) in THF (8 mL) was added a solution of the amide 42 (641 mg, 0.84 mmol) in THF (3 mL) at 0 °C. The mixture was stirred at rt for 30 min and recooled to 0 °C. To the solution was added MeI (0.08 mL, 1.29 mmol) and the reaction mixture was warmed to rt and stirred for 12 h. The reaction mixture was diluted with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:3 EtOAc:hexanes) to give the N-methyl amide 43 (630 mg, 96%) as a colorless oil. IR (film) 2950, 2860, 1650, 1509, 1226, 1090 cm−1; 1H NMR (300 MHz, toluene-d8, 90 °C) δ 7.38 (m, 1H), 7.28−7.01 (m, 12H), 6.78 (dt, J = 12.8, 3.8 Hz, 2H), 6.72 (dt, J = 12.8, 3.8 Hz, 2H), 4.59 (d, J = 5.5 Hz, 2H), 4.42 (d, J = 7.1 Hz, 2H), 4.38 (s, 2H), 3.94 (m, 2H), 3.45 (s, 3H), 3.22 (s, 3H), 2.75 (m, 1H), 2.57 (m, 1H), 0.95 (s, 9H), 0.07 (s, 6H); 13C NMR (75 MHz, toluene-d8, 90 °C) δ 170.7, 155.1, 153.6, 148.3, 144.3, 137.5, 129.2, 136.3, 135.1, 131.9, 129.2, 129.1, 128.7, 128.5, 128.1, 127.84, 127.78, 127.7, 127.53, 127.48, 116.9, 115.3, 103.3, 77.1, 73.1, 69.1, 62.5, 55.4, 37.6, 34.0, 26.2, 18.5, −5.2; HRMS-ES: [M+H]+ calcd for C40H49INO5Si 778.2425; found, 778.2397.
{4-Benzyloxymethyl-3-[2-(tert-butyldimethylsilanyloxy)-ethyl]-1-methyl-2-oxo-2,3-dihydro-1Hindol-3-yl}-[2-(4-methoxyphenoxymethyl)-phenyl]-acetic Acid Methyl Ester (44)
To a solution of N-methyl amide 43 (222 mg, 0.286 mmol) in DMA (3.0 mL) and MeOH (1.5 mL) were added Pd(OAc)2 (6.4 mg, 0.029 mmol), biphenyldicyclohexylphosphine (30.1 mg, 0.086 mmol) and NaOAc (46.9 mg, 0.572 mmol). The mixture was stirred at 85 °C under a CO atmosphere (1 atm) for 24 h. The catalyst was removed by filtration and washed with EtOAc. The filtrate was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:5 EtOAc:hexanes) to give the methyl ester 44 (145 mg, 71%) as a colorless oil. IR (film) 2950, 2859, 1740, 1712, 1509, 1226 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35−7.17 (m, 9H), 7.12 (dd, J = 14.8, 7.5 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.87 (d, J = 9.1 Hz, 2H), 6.82 (d, J = 9.2 Hz, 2H), 6.65 (d, J = 7.7 Hz, 1H), 4.93, 4.69 (ABq, J = 11.6 Hz, 2H), 4.63 (s, 1H), 4.41, 4.29 (ABq, J = 11.9 Hz, 2H), 3.89, 3.77 (ABq, J = 11.1 Hz, 2H), 3.78 (s, 3H), 3.59 (s, 3H), 3.19 (m, 2H), 3.10 (s, 3H), 2.56 (dt, J = 14.4, 7.2 Hz, 1H), 2.13 (dt, J = 13.4, 4.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 178.2, 171.6, 154.5, 153.1, 145.4, 138.2, 136.5, 136.0, 133.4, 130.6, 129.7, 129.0, 128.8, 128.4, 128.3, 128.2, 128.1, 126.1, 123.5, 116.2, 115.1, 107.6, 73.4, 69.2, 68.9, 60.0, 56.1, 55.1, 52.5, 38.0, 26.9, 26.4, 26.2, 18.6, −5.3, −5.4; HRMS-ES: [M+H]+ calcd for C42H52NO7Si 710.3513; found, 710.3521.
Synthesis of Spirolactone 45
To a stirred solution of methyl ester 44 (172 mg, 0.24 mmol) in THF (10 mL) was added TBAF (0.27 mL, 1.0 M in THF, 0.27 mmol) at 0 °C. The reaction mixture was warmed to rt and stirred for 4 h. The mixture was diluted with EtOAc and saturated aqueous NH4Cl. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the lactone 45 (116 mg, 85%) as a separable mixture of diastereomers (2:1). For analytical purposes the diastereomers were separated by preparative TLC (1:1 EtOAc:hexanes). More polar major diastereomer of 45 (colorless oil): IR (film) 2927, 1752, 1712, 1605, 1509, 1226 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.38−7.01 (m, 11H), 6.83−6.74 (m, 4H), 6.54 (d, J = 7.7 Hz, 1H), 5.03 (s, 1H), 4.83 (ddd, J = 11.7, 6.7, 4.9 Hz, 1H), 4.67−4.43 (m, 6H), 4.30 (ddd, J = 11.8, 7.2, 4.6 Hz, 1H), 3.77 (s, 3H), 2.96 (s, 3H), 2.61 (ddd, J = 14.7, 6.8, 4.8 Hz, 1H), 2.27 (ddd, J = 14.6, 7.4, 5.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 178.2, 171.1, 154.5, 153.0, 144.1, 137.3, 136.5, 134.4, 132.3, 130.8, 129.7, 129.2, 129.1, 128.79, 128.75, 128.7, 128.2, 127.8, 125.1, 116.3, 115.1, 108.6, 73.8, 70.0, 69.7, 64.8, 56.2, 52.6, 47.2, 30.3, 26.5; HRMS-ES: [M+H]+ calcd for C35H34NO6 564.2386; found, 564.2391. Less polar minor diastereomer of 45 (colorless oil): IR (film) 2916, 1746, 1706, 1610, 1509, 1232 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.44−7.30 (m, 5H), 7.27−7.23 (m, 3H), 7.09 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 9.0 Hz, 2H), 6.88−6.84 (m, 3H), 6.63 (d, J = 7.8 Hz, 1H), 6.54 (d, J = 7.4 Hz, 1H), 5.32 (d, J = 11.7 Hz, 1H), 4.98 (s, 1H), 4.80−4.55 (m, 6H), 4.38 (dd, J = 11.7, 5.6 Hz, 1H), 3.79 (s, 3H), 2.91 (s, 3H), 2.73 (td, J = 14.5, 5.8 Hz, 1H), 2.23 (d, J = 14.9 Hz, 1H); 13C NMR (75 MHz, CDCl3), δ 179.4, 172.6, 154.6, 153.1, 144.2, 138.2, 136.7, 136.0, 131.5, 130.8, 129.8, 129.7, 129.0, 128.9, 128.6, 128.3, 128.2, 127.5, 125.4, 116.6, 115.1, 108.5, 73.6, 70.2, 69.2, 66.1, 56.1, 52.5, 47.1, 31.6, 26.8; HRMS-ES: [M+Na]+ calcd for C35H33NNaO6 586.2206; found, 586.2210.
O-Allylation of Spirolactone 45
To a stirred suspension of spirolactone 45 (20.6 mg, 0.037 mmol) and NaH (2.0 mg, 60% dispersion in mineral oil, 0.050 mmol) in DMF (2 mL) was added allyl iodide (0.010 mL, 0.109 mmol) at 0 °C. The mixture was stirred for 1 h at rt. The reaction mixture was diluted with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:2 EtOAc:hexanes) to give the O-allyl ketene acetal 46 (12.7 mg, 57%, 71% based on recovered starting material) as a coloress oil, and unreacted spiro lactone 45 (3.9 mg). IR (film) 3470, 2927, 1706, 1509, 1226 cm−1; 1H NMR (400 MHz, CDCl3, 2:1 atropisomeric mixture) δ 7.44-7.22 (m, 7H, major and minor), 7.13 (t, J = 7.7 Hz, 1H, minor), 7.08−7.01 (m, 1H, major and minor), 6.98−6.96 (m, 2H, major and minor), 6.91 (d, J = 6.7 Hz, 1H, minor), 6.86−6.82 (m, 3H, major and minor), 6.77 (t, J = 7.6 Hz, 1H, major), 6.58 (d, J = 8.1 Hz, 1H, major and minor), 6.35 (d, J = 7.8 Hz, 1H, minor), 5.78 (m, 1H, major and minor), 5.27 (d, J = 13.3 Hz, 1H, major), 5.17−5.06 (m, 3H, major, 2H, minor), 5.01−4.84 (m, 2H, major and minor), 4.77−4.65 (m, 2H, major, 3H, minor), 4.44−4.24 (m, 4H, major and minor), 3.78 (s, 3H, major and minor), 3.16 (s, 3H, minor), 2.95 (s, 3H, major), 2.58 (m, 1H, major and minor), 1.98 (d, J = 14.6 Hz, 1H, major), 1.85 (d, J = 14.3 Hz, 1H, minor). 13C NMR (300 MHz, CDCl3) δ 180.7, 179.4, 157.6, 156.3, 154.0, 153.9, 153.8, 153.6, 144.0, 143.6, 138.32, 138.27, 137.2, 135.2, 134.7, 134.0, 133.7, 133.4, 132.7, 130.9, 130.2, 129.8, 129.6, 129.0, 128.9, 128.81, 128.76, 128.4, 128.34, 128.26, 128.1, 127.5, 127.4, 127.1, 127.0, 126.7, 126.4, 125.2, 124.4, 118.22, 118.16, 116.2, 115.9, 115.0, 114.9, 108.0, 107.9, 87.1, 84.4, 73.8, 73.5, 69.30, 69.26, 68.33, 68.26, 65.5, 64.6, 56.1, 50.5, 50.4, 32.6, 31.4, 26.8, 26.6; HRMS-ES: [M+H]+ calcd for C38H37NO6 604.2699; found, 604.2684.
Thermal Claisen Rearrangement of Ketene Acetal 46
A solution of ketene acetal 46 in DMF or toluene was heated at 130 °C and stirred for 15 h. The solvent was removed under reduced pressure. The residue was purified by preparative TLC (1:1 EtOAc:hexanes) to give the two allyl lactone diastereomers 47 and 48. More polar major diastereomer 47 (oil): IR (film) 2929, 1704, 1508, 1228 cm−1; 1H NMR (300 MHz, toluene-d8, 90°C) δ 7.56 (d, J = 7.9 Hz, 1H), 7.26−6.98 (m, 9H), 6.90 (t, J = 7.2 Hz, 1H), 6.80−6.67 (m, 4H), 6.38 (d, J = 6.8 Hz, 1H), 6.10 (m, 1H), 5.21 (br. s, 1H), 5.13 (td, J = 12.1, 4.4 Hz, 1H), 4.77 (s, 1H), 4.72 (d, J = 8.8 Hz, 1H), 4.52 (br. s, 1H), 4.20, 4.13 (ABq, J = 12.1 Hz, 2H), 4.06 (dd, J = 11.0, 7.0 Hz, 1H), 3.47 (s, 3H), 3.16 (m, 1H), 3.15 (s, 2H), 2.85 (m, 1H), 2.81 (s, 3H), 2.53 (dd, J = 15.4, 6.8 Hz, 1H), 1.35 (dd, J = 14.7, 4.6 Hz, 1H); 13C NMR (75 MHz, toluene-d8, 90 °C) δ 176.5, 172.4, 155.3, 154.0, 145.1, 139.6, 139.1, 138.6, 137.8, 137.3, 134.1, 132.3, 129.4, 128.7, 128.2, 127.9, 127.5, 126.7, 127.5, 126.7, 125.8, 116.1, 115.7, 107.5, 73.0, 69.6, 69.0, 64.6, 59.1, 57.0, 55.7, 41.2, 27.2, 25.7; HRMS-ES: [M+H]+ calcd for C38H38NO6 604.2699; found, 604.2714. Less polar minor diastereomer 48 (oil): IR (film) 2918, 1704, 1508, 1228 cm−1; 1H NMR (300 MHz, toluene-d8, 90°C) δ 7.83 (d, J = 7.6 Hz, 1H), 7.29 (d, J = 7.6 Hz, 2H), 7.22−6.96 (m, 8H), 6.77 (d, J = 8.8 Hz, 2H), 6.60 (t, J = 7.8 Hz, 1H), 6.36 (br s, 1H), 5.99 (d, J = 7.7 Hz, 1H), 5.77 (m, 1H), 5.52 (td, J = 12.3, 4.1 Hz, 1H), 5.33 (d, J = 11.8 Hz, 1H), 4.98−4.85 (m, 3H), 4.60, 4.57 (ABq, J = 10.3 Hz, 2H), 4.50, 4.44 (ABq, J = 11.9 Hz, 2H), 4.0 (dd, J = 11.1, 6.9 Hz, 1H), 3.47 (s, 3H), 3.39 (dd, J = 14.7, 5.6 Hz, 1H), 2.95 (dd, J = 14.3, 6.7 Hz, 1H), 2.74 (td, J = 14.4, 6.9 Hz, 1H), 2.32 (s, 3H), 1.42 (dd, J = 14.7, 3.7 Hz, 1H); 13C NMR (75 MHz, toluene-d8, 90 °C) δ 177.2, 169.9, 155.1, 154.8, 145.9, 139.7, 138.1, 137.4, 137.2, 134.5, 133.9, 129.5, 129.4, 129.3, 128.9, 128.4, 127.5, 127.3, 125.9, 125.6, 117.7, 117.1, 115.6, 107.8, 73.6, 71.2, 70.4, 64.7, 57.9, 57.8, 55.7, 42.0, 26.9, 25.4; HRMS-ES: [M+H]+ calcd for C38H38NO6 604.2699; found, 604.2722.
Dimethylcarbamic Acid 2-Iodo-3-nitrophenyl Ester (50)
To a stirred solution of 2-iodo-3-nitrophenol (49, 422 mg, 1.59 mmol), NEt3 (0.33 ml, 2.35 mmol)23 and DMAP (19.4 mg, 0.16 mmol) in CH2Cl2 (20 mL) was added N,N-dimethylcarbamyl chloride (0.18 mL, 1.96 mmol) at 0 °C. The mixture was warmed to rt and stirred overnight. The reaction mixture was diluted with CH2Cl2, washed with water and brine, dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the carbamoyl-protected phenol 50 (505 mg, 94%) as a white solid (mp 125-126 °C). IR (film) 1713, 1531, 1386, 1359, 1246, 1165 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.75 (dd, J = 7.9, 1.4 Hz, 1H), 7.60, (t, J = 8.0 Hz, 1H), 7.53 (dd, J = 8.1, 1.4 Hz, 1H), 3.35 (s, 3H), 3.19 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 155.1, 153.9, 153.4, 130.0, 127.3, 122.4, 86.1, 37.5, 37.3; HRMS-ES: [M+Na]+ calcd for C9H9IN2NaO4 358.9505; found, 358.9513.
Dimethylcarbamic Acid 3-Amino-2-iodophenyl Ester (51)
To a solution of the nitrobenzene 50 (446 mg, 1.33 mmol) in EtOH (10 mL) and glacial acetic acid (5 mL) was added iron powder (296 mg, 5.30 mmol). The mixture was heated at 60 °C for 4 h and then cooled to rt. The mixture was diluted with water and carefully neutralized with solid Na2CO3. The resulting solution was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the aniline 51 (375 mg, 93%) as a white solid (mp 113-114 °C). IR (film) 3333, 1714, 1617, 1466, 1386, 1170 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.33 (t, J = 7.9 Hz, 1H), 6.80 (dd, J = 8.0, 2.9 Hz, 2H), 4.50 (br s, 2H), 3.42 (s, 3H), 3.28 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 154.3, 152.7, 149.0, 129.8, 113.0, 112.2, 82.0, 37.3, 37.2; HRMS-ES: [M+H]+ calcd for C9H12IN2O2 306.9944; found, 306.9949.
Dimethylcarbamic Acid 3-{4-Hydroxy-2-[2-(4-methoxyphenoxymethyl)benzylidene]butyrylamino}-2-iodophenyl Ester (52)
To a stirred solution of aniline 51 (111 mg, 0.362 mmol) and lactone 4022 (107 mg, 0.345 mmol) in CH2Cl2 (10 mL) was added AlMe3 (0.19 mL, 2.0 M in hexane, 0.380 mmol) at 0 °C. The mixture was warmed to rt, stirred overnight and then carefully diluted with saturated aqueous NH4Cl at 0 °C, followed by CH2Cl2. The organic layer was washed with water, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (10:1 CH2Cl2:MeOH) to give the amide 52 (207 mg, 97%) as a white solid (mp 110-111 °C). IR (film) 3387, 2934, 1725, 1665, 1509, 1229 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.37 (s, 1H), 8.06 (dd, J = 8.2, 1.4 Hz, 1H), 7.64 (s, 1H), 7.53−7.51 (m, 1H), 7.40−7.31 (m, 4H), 6.97 (dd, J = 8.1, 1.4 Hz, 1H), 6.92−6.79 (m, 4H), 5.01 (s, 2H), 3.75 (t, J = 5.9 Hz, 2H), 3.74 (s, 3H), 3.18 (s, 3H), 3.03 (s, 3H), 2.74 (t, J = 5.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.7, 154.6, 154.0, 153.2, 152.4, 140.0, 138.5, 135.6, 135.2, 134.5, 129.9, 129.7, 129.6, 129.0, 128.8, 119.82, 119.76, 116.4, 115.2, 89.4, 69.6, 62.3, 56.2, 37.3, 37.2, 31.8; HRMS-ES: [M+H]+ calcd for C28H30IN2O6 617.1149; found, 617.1139.
Dimethylcarbamic Acid 2-Iodo-3-{4-methoxymethoxy-2-[2-(4-methoxyphenoxymethyl)benzylidene] butyrylamino}phenyl Ester (53)
To a mixture of amide 52 (961 mg, 1.56 mmol), (i-Pr)2NEt (0.54 mL, 3.10 mmol) and NaI (467 mg, 3.12 mmol) in CH2Cl2 (30 mL) was added MOMCl (0.18 mL, 2.37 mmol) at 0 °C. The mixture was warmed to rt, stirred overnight and then diluted with saturated aqueous NaHCO3 and CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the MOM ether 53 (914 mg, 89%) as a white solid (mp 107-108 °C). IR (film) 3389, 2937, 1729, 1673, 1509, 1385, 1232 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.32 (s, 1H), 8.17 (dd, J = 8.3, 1.3 Hz, 1H), 7.67 (s, 1H), 7.57−7.54 (m, 1H), 7.43−7.34 (m, 4H), 6.98 (dd, J = 8.3, 1.3 Hz, 1H), 6.93-6.80 (m, 4H), 5.02 (s, 2H), 4.58 (s, 2H), 3.75 (s, 3H), 3.70 (t, J = 6.3 Hz, 2H), 3.29 (s, 3H), 3.19 (s, 3H), 3.05 (s, 3H), 2.84 (t, J = 6.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 167.6, 154.6, 153.9, 153.2, 152.3, 140.2, 138.1, 135.8, 135.1, 134.4, 130.0, 129.5, 129.3, 128.9, 128.6, 119.5, 119.4, 116.4, 115.2, 96.7, 88.8, 69.4, 66.5, 56.2, 55.8, 37.3, 37.2, 29.0; HRMS-ES: [M+H]+ calcd for C30H34IN2O7 661.1411; found, 661.1397.
Dimethylcarbamic Acid 2-Iodo-3-({4-methoxymethoxy-2-[2-(4-methoxyphenoxymethyl)benzylidene]butyryl}methylamino)phenyl Ester (54)
To a stirred suspension of NaH (11.1 mg, 60% dispersion in mineral oil, 0.278 mmol) in THF (6 mL) was added a solution of lactam 53 (167 mg, 0.253 mmol) in THF (4 mL) at 0 °C. The mixture was stirred at rt for 1 h and recooled to 0 °C. To the solution was added MeI (0.019 mL, 0.305 mmol) and the reaction mixture was warmed to rt and stirred overnight. The reaction mixture was diluted with saturated aqueous NH4Cl and water and extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (10:10:1 CH2Cl2:EtOAc:MeOH) to give the N-methyl amide 54 (167 mg, 98%) as a colorless oil. IR (film) 2926, 1723, 1616, 1503, 1227, 1164 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.38−6.96 (m, 8H), 6.81 (br s, 4H), 4.68−4.48 (m, 4H), 3.77 (s, 3H), 3.67 (br s, 2H), 3.36 (s, 6H), 3.11 (s, 3H), 3.02 (s, 3H), 2.59 (br s, 1H), 2.43 (br s, 1H); 13C NMR (75 MHz, CDCl3) δ 171.6, 154.4, 153.8, 153.1, 148.8, 136.5, 135.9, 134.4, 133.2, 130.2, 129.1, 128.4, 127.8, 127.0, 122.8, 116.5, 114.9, 97.3, 96.6, 68.5, 66.2, 56.1, 55.7, 37.8, 37.3, 37.1, 30.3; HRMS-ES: [M+H]+ calcd for C31H36IN2O7 675.1567; found, 675.1562.
[4-Dimethylcarbamoyloxy-3-(2-methoxymethoxyethyl)-1-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-[2-(4-methoxyphenoxymethyl)phenyl]acetic Acid Methyl Ester (55)
To a solution of N-methyl amide 54 (280 mg, 0.415 mmol) in DMA (2.6 mL) and MeOH (1.3 mL) were added Pd(OAc)2 (18.6 mg, 0.083 mmol), P(o-Tol)3 (75.7 mg, 0.249 mmol) and NaOAc (68.0 mg, 0.829 mmol). The mixture was stirred at 90 °C under a CO atmosphere (1 atm) for 1 d. The reaction mixture was cooled to rt and diluted with EtOAc. The solution was washed with water and brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:2 EtOAc:hexanes) to give the ester 55 (199 mg, 79%) as a colorless oil. IR (film) 2938, 1729, 1616, 1509, 1226 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.53−7.50 (m, 1H), 7.32-7.16 (m, 4H), 6.97 (d, J = 8.4 Hz, 1H), 6.88−6.80 (m, 4H), 6.51 (d, J = 7.6 Hz, 1H), 5.08, 4.85 (ABq, J = 11.6 Hz, 2H), 4.68 (s, 1H), 4.32, 4.21 (ABq, J = 6.5 Hz, 2H), 3.78 (s, 3H), 3.61 (s, 3H), 3.19-3.03 (m, 2H), 3.12 (s, 3H), 3.09 (s, 3H), 2.88 (s, 3H), 2.64 (s, 3H), 2.63-2.55 (m, 1H), 2.47-2.40 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.9, 171.2, 154.5, 153.4, 153.1, 148.7, 146.3, 136.5, 133.5, 130.6, 129.9, 129.6, 128.3, 127.9, 118.6, 116.8, 116.3, 115.0, 104.9, 96.7, 69.2, 64.4, 56.2, 55.4, 54.5, 52.6, 37.2, 36.4, 34.2, 26.9; HRMS-ES: [M+H]+ calcd for C33H39N2O9 607.2656; found, 607.2645.
Synthesis of Spirolactone 57
To a stirred solution of ester 55 (61.1 mg, 0.101 mmol) in MeOH (5.0 mL) was added concentrated HCl (1 drop) at rt. The mixture was warmed to 60 °C and stirred for 4 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (3:1 EtOAc:hexanes) to give spiro lactone 57 (37.5 mg, 70%) as a white solid (mp 213-215 °C) and hydroxy ester 56 (9.5 mg, 17%) as a colorless oil. Spiro lactone 57: IR (film) 1729, 1712, 1622, 1509, 1226, 1158 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.47 (dd, J = 7.5, 1.1 Hz, 1H), 7.27-7.19 (m, 2H), 7.12 (td, J = 7.6, 1.3 Hz, 1H), 7.04 (dd, J = 7.6, 1.4 Hz, 1H), 6.82 (dd, J = 8.4, 0.6 Hz, 1H), 6.78-6.70 (m, 4H), 6.42 (d, J = 7.8 Hz, 1H), 5.01 (ddd, J = 11.4, 6.8, 4.7 Hz, 1H), 4.97 (s, 1H), 4.94 (d, J = 11.9 Hz, 1H), 4.56 (ddd, J = 11.7, 7.5, 4.2 Hz, 1H), 4.50 (d, J = 11.9 Hz, 1H), 3.75 (s, 3H), 3.00 (s, 3H), 2.95 (s, 3H), 2.94 (s, 3H), 2.55 (ddd, J = 14.8, 6.7, 4.4 Hz, 1H), 2.42 (ddd, J = 14.7, 7.7, 4.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 177.5, 170.8, 154.6, 154.3, 152.9, 148.2, 144.9, 136.5, 132.2, 131.4, 130.6, 129.5, 128.3, 128.0, 121.9, 118.0, 116.7, 115.0, 106.0, 69.8, 65.0, 56.1, 52.1, 46.6, 37.6, 36.8, 30.4, 26.7; HRMS-ES: [M+Na]+ calcd for C30H30N2NaO7 553.1951; found, 553.1963.
Hydroxy ester 56: IR (film) 3470, 2927, 1735, 1712, 1616, 1509, 1226, 1152 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.54−7.52 (m, 1H), 7.28−7.17 (m, 4H), 6.86−6.80 (m, 5H), 6.53 (d, J = 7.7 Hz, 1H), 5.07, 4.83 (ABq, J = 12.9 Hz, 2H), 4.70 (s, 1H), 3.78 (s, 3H), 3.64 (s, 3H), 3.34 (t, J = 6.3 Hz, 2H), 3.12 (s, 3H), 2.92 (s, 3H), 2.72 (s, 3H), 2.56 (dt, J = 13.8, 6.9 Hz, 1H), 2.34 (dt, J = 12.7, 6.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 177.9, 171.2, 154.5, 154.4, 153.0, 148.3, 146.2, 136.4, 133.4, 130.6, 130.0, 129.6, 128.2, 128.0, 120.3, 117.2, 116.2, 115.0, 105.5, 69.2, 59.6, 56.2, 54.6, 52.7, 37.4, 37.2, 36.6, 27.0; HRMS-ES: [M+H]+ calcd for C31H35N2O8 563.2393; found, 563.2392.
O-Allylation of Spirolactone 57
To a stirred suspension of spirolactone 57 (69.1 mg, 0.130 mmol) and NaH (6.2 mg, 60% dispersion in mineral oil, 0.155 mmol) in DMF (2 mL) was added allyl iodide (0.024 mL, 0.262 mmol) at rt. The mixture was stirred at rt overnight. The reaction mixture was diluted with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the O-allyl ketene acetal 58 (46.6 mg, 63%) as a colorless oil. IR (film) 2916, 1712, 1616, 1509, 1226 cm−1; 1H NMR (300 MHz, CDCl3, 2:1 atropisomeric mixture) δ 7.40 (d, J = 7.3 Hz, 1H),7.26−6.95 (m, 7.5H), 6.90−6.77 (m, 4.5 H), 6.73 (s, 2H), 6.46 (d, J = 7.7 Hz, 1H), 6.43 (d, J = 7.8 Hz, 0.5H), 5.80−5.64 (m, 1.5H), 5.24−4.86 (m, 7.5H), 4.38−4.27 (m, 4.5H), 3.78 (s, 3H), 3.76 (s, 1.5H), 3.18 (s, 3H), 3.76 (s, 1.5H), 3.18 (s, 3H), 3.073 (s, 3H), 3.067 (s, 3H), 2.99 (s, 1.5H), 2.95 (s, 3H), 2.49−2.39 (m, 1.5H), 2.08−1.99 (m, 1.5H); 13C NMR (75 MHz, CDCl3) δ 180.0, 179.0, 157.1, 156.2, 154.5, 154.3, 153.9, 153.8, 153.5, 148.2, 148.0, 145.1, 145.0, 138.6, 138.1, 134.0, 133.7, 132.9, 132.5, 131.5, 131.1, 129.6, 129.5, 127.5, 127.2, 126.7, 126.4, 126.2, 123.0, 122.5, 118.3, 118.2, 117.9, 117.7, 116.3, 116.2, 115.0, 114.8, 105.5, 105.3, 88.0, 84.6, 69.5, 69.4, 68.5, 68.3, 65.1, 65.0, 56.2, 50.20, 50.15, 37.5, 37.4, 36.9, 31.6, 29.8, 26.9, 26.8; HRMSES: [M+H]+ calcd for C33H35N2O7 571.2444; found, 571.2429.
Claisen Rearrangement of Ketene Acetal 58
A solution of ketene acetal 58 in DMF or toluene was heated at 110 °C and stirred overnight. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (1:1 EtOAc:hexanes) to give the allyl lacone diastereomers 59 and 60. More polar major diastereomer 59 (pale yellow oil): IR (film) 2927, 1729, 1712, 1616, 1502, 1226, 1158 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.46 (br s, 1H), 7.23 (t, J = 8.0 Hz, 2H), 7.17 (t, J = 7.0 Hz, 1H), 6.83 (d, J = 8.7 Hz, 2H), 6.77 (br s, 2H), 6.60−6.56 (m, 2H), 5.64 (m, 1H), 5.04−4.87 (m, 3H), 4.66 (br s, 1H), 4.47 (br s, 1H), 3.79 (s, 3H), 3.22 (br s, 1H), 3.10 (s, 3H), 2.92−2.87 (m, 4H), 2.78−2.70 (m, 4H), 2.29 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 175.8, 171.9, 154.0, 153.7, 153.0, 149.3, 145.0, 137.0, 134.9, 134.3, 131.7, 131.4, 130.1, 127.5, 126.8, 120.1, 118.3, 116.9, 115.0, 114.5, 105.2, 68.7, 65.4, 55.7, 55.4, 38.4, 36.9, 36.5, 29.7, 29.4, 24.6; HRMS-ES: [M+Na]+ calcd for C33H34N2NaO7 593.2264; found, 593.2263. Less polar minor diastereomer 60 (pale yellow oil): IR (film) 2927, 1729, 1712, 1616, 1509, 1226, 1164 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.71 (br s, 1H), 7.30 (t, J = 8.1 Hz, 2H), 7.17 (t, J = 7.5 Hz, 1H), 7.02-7.00 (m, 2H), 6.90- 6.83 (m, 4H), 6.54 (br s, 1H), 6.34 (d, J = 7.7 Hz, 1H), 5.63-5.56 (m, 2H), 5.06−4.96 (m, 3H), 4.79 (br s, 1H), 4.55 (dd, J = 11.3, 6.4 Hz, 1H), 3.81 (s, 3H), 3.61 (dd, J = 15.2, 5.9 Hz, 1H), 3.23 (s, 3H), 3.18−3.02 (m, 2H), 3.11 (s, 3H), 2.58 (s, 3H), 2.01 (dd, J =14.9, 3.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 176.8, 171.1, 154.6, 154.1, 154.0, 149.4, 146.4, 138.3, 133.8, 132.4, 130.8, 129.9, 127.8, 125.9, 119.4, 118.4, 118.2, 116.4, 115.0, 113.7, 105.7, 71.1, 65.5, 56.7, 56.2 41.9, 37.5, 37.1, 30.1, 26.1, 24.9; HRMS-ES: [M+Na]+ calcd for C33H34N2NaO7 593.2264; found, 593.2274.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (CA-034303) for financial support of this research. We also thank Dr. H. Yennawar (Penn State Small Molecule X-Ray Crystallographic Facility) for the X-ray structure determinations, and Dr. Alan Benesi for assistance with 2D NMR experiments.
Footnotes
Supporting Information Available
Copies of proton and carbon NMR spectra of new compounds along with X-ray data for compounds 30 and 32. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355. [Google Scholar]
- 2.Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J. Org. Chem. 2001;66:8717. doi: 10.1021/jo010335e. [DOI] [PubMed] [Google Scholar]; Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J. Org. Chem. 2003;68:1640. doi: 10.1021/jo010335e. For the retraction of the nomofungin structure see: [DOI] [PubMed] [Google Scholar]
- 3.(a) Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. J. Nat. Prod. 2004;67:78. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]; (b) Hayashi H, Matsumoto H, Akiyama K. Biosci. Biotechnol. Biochem. 2004;68:753. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]; (c) Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C. J. Nat. Prod. 2005;68:258. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]
- 4. Some structurally different compounds were initially designated as the same communesins due to simulaneous publication by two groups (see refs 3a and 3b). However, the current communesin nomenclature can be found in ref 3c.
- 5.Verbitski SM, Mayne CL, Davis RA, Concepcion GP, Ireland CM. J. Org. Chem. 2002;67:7124. doi: 10.1021/jo026012f. [DOI] [PubMed] [Google Scholar]; Verotta L, Pilati T, Tato M, Elisabetsky E, Amador TA, Nunes DS. J. Nat. Prod. 1998;61:392. doi: 10.1021/np9701642. For another related alkaloid see: [DOI] [PubMed] [Google Scholar]
- 6.Wigley LJ, Mantle PG, Perry DA. Phytochemistry. 2006;67:561. doi: 10.1016/j.phytochem.2005.10.011. For recent labelling studies on the biosynthesis of the communesins see: [DOI] [PubMed] [Google Scholar]
- 7.(a) May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203. [Google Scholar]; (b) May JA, Stoltz BM. Tetrahedron. 2006;62:5262. [Google Scholar]
- 8.Yang J, Song H, Xiao X, Wang J, Qin Y. Org. Lett. 2006;8:2187. doi: 10.1021/ol0607138. [DOI] [PubMed] [Google Scholar]
- 9.Crawley SL, Funk RL. Org. Lett. 2003;5:3169. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 11.Sabahi A, Novikov A, Rainier JD. Angew. Chem. Int. Ed. 2006;45:4317. doi: 10.1002/anie.200601278. [DOI] [PubMed] [Google Scholar]
- 12.(a) Artman GD, III, Weinreb SM. Org. Lett. 2003;5:1523. doi: 10.1021/ol034314d. [DOI] [PubMed] [Google Scholar]; (b) Artman GD., III . The Pennsylvania State University; University Park, PA: 2004. Ph.D. Thesis. [Google Scholar]
- 13.(a) Grigg R, Millington EL, Thornton-Pett M. Tetrahedron Lett. 2002;43:2605. [Google Scholar]; (b) Brown S, Clarkson S, Grigg R, Thomas WA, Sridharan V, Wilson DM. Tetrahedron. 2001;57:1347. [Google Scholar]; (c) Anwar U, Casaschi A, Grigg R, Sansano JM. Tetrahedron. 2001;57:1361. [Google Scholar]; (d) de Meijere A, Bräse S. J. Organomet. Chem. 1999;576:88. [Google Scholar]; (e) Poli G, Giambastiani G, Heumann A. Tetrahedron. 2000;56:5959. [Google Scholar]; (f) Negishi E, Ma S, Amanfu J, Copéret C, Miller JA, Tour JM. J. Am. Chem. Soc. 1996;118:5919. [Google Scholar]
- 14.(a) Vetelino MG, Coe JW. Tetrahedron Lett. 1994;35:219. [Google Scholar]; (b) Coe JW, Vetelino MG, Bradlee M. J. Tetrahedron Lett. 1996;37:6045. [Google Scholar]
- 15.Schumacher RW, Davidson BS. Tetrahedron. 1999;55:935. [Google Scholar]
- 16.(a) Baldwin JE, Moloney MG, Parsons AF. Tetrahedron. 1992;48:9373. [Google Scholar]; (b) McCort G, Hoornaert C, Aletru M, Denys C, Duclos O, Cadilhac C, Guilpain E, Dellac G, Janiak P, Galzin A-M, Delahaye M, Guilbert F, O'Connor S. Bioorg. Med. Chem. 2001;9:2129. doi: 10.1016/s0968-0896(01)00118-3. [DOI] [PubMed] [Google Scholar]
- 17.Lipton MF, Basha A, Weinreb SM. Org. Synth. 1979;59:49. [Google Scholar]
- 18.(a) Denmark SE, Schnute ME. J. Org. Chem. 1995;60:1013. For some examples of reductive Heck reactions see: [Google Scholar]; (b) Diaz P, Gendre F, Stella L, Charpentier B. Tetrahedron. 1998;54:4579. [Google Scholar]; (c) de Meijere A, Meyer FE. Angew. Chem. Int. Ed. Engl. 1994;33:2379. and references cited. [Google Scholar]; (d) Sajiki H, Ikawa T, Yamada H, Tsubouchi K, Hirota K. Tetrahedron Lett. 2003;44:171. For a recent example of MeOH acting as a hydride transfer agent, see: [Google Scholar]
- 19. For preparation of lactones 16 and 31 see Supporting Information in ref 12a.
- 20.Levin JI, Turos E, Weinreb SM. Synth. Commun. 1982;12:989. [Google Scholar]
- 21.(a) Seno K, Hagishita S, Sato T, Kuriyama KJ. Chem. Soc. Perkin Trans. 1984;1:2013. [Google Scholar]; (b) Ozlu Y, Cladingboel DE, Parsons P. J. Tetrahedron. 1994;50:2183. [Google Scholar]
-
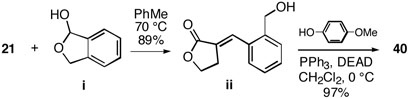
22.Prepared by reaction of Wittig reagent 2116 with known lactol i Mikami K, Ohmura H. Org. Lett. 2002;4:3355. doi: 10.1021/ol0265416. to afford benzylidene lactone ii, which was then protected as shown:
- 23.Dai W-M, Lai KW. Tetrahedron Lett. 2002;43:9377. [Google Scholar]
- 24.Tomioka K, Kawasaki H, Yasuda K, Koga K. J. Am. Chem. Soc. 1988;110:3597. Similar facial selectivity controlled by the conformation of an α-substituent has been proposed for the alkylation of 6-membered lactone enolates: [Google Scholar]
- 25.Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Pergamon; New York: 1983. pp. 274–284. [Google Scholar]
- 26.Imming P, Imhof I, Zentgraf M. Synth. Commun. 2001;31:3721. [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.