

Abstract
Cyclic GMP, guanosine 3′,5′-cyclic monophosphate, is a critical and multifunctional second messenger molecule that mediates diverse physiological and pathophysiological functions in cardiac and vascular tissues. Synthesized through nitric oxide, carbon monoxide, and/or natriuretic peptide-mediated guanylate cyclase stimulation and guanosine triphosphate dephosphorylation, cyclic GMP is capable of stimulating a cascade of serine/threonine kinase events including signaling through cyclic GMP- and/or cyclic AMP-dependent protein kinases, eliciting protein kinase-independent actions such as modulation of ion channels or transporters, or undergoing hydrolytic degradation through actions of cyclic GMP-regulated phosphodiesterases. Substrates, enzymes, co-factors and associated variables in this multifaceted system have historically been targets of vital pharmacotherapies with perhaps most common the use of vascular smooth muscle-targeting organonitrates in cardiac patients and phosphodiesterase inhibitors in individuals with erectile dysfunction. Accumulating basic science and clinical evidence, however, suggests that cyclic GMP signaling is compromised under conditions of disease or elevated physiological stresses. Moreover, nitric oxide can stimulate an array of cytotoxic effects and nitric oxide-based therapies can be limited by diminished bioactivity and the development of tachyphylaxis or tolerance after prolonged use. Consequently, an emerging area for clinical drug development and therapeutic drug evaluation for conditions of cardiovascular adversity has focused on identification of cyclic GMP signaling pathways that act under oxidized or nitric oxide-unresponsive conditions and/or that operate irrespective of nitric oxide-induced complications. The aim of this Therapeutic Review is to describe novel, NO-alternate avenues for cyclic GMP signaling in vascular smooth muscle growth with particular emphasis on pharmacotherapeutics of recently characterized cyclic GMP-specific approaches.
Keywords: BAY 41-2272, carbon monoxide, cyclic GMP, heme oxygenase, vascular smooth muscle, YC-1
Introduction & Background
Cardiovascular disease (CVD) comprises a large family of related disorders and includes angina pectoris, myocardial and systemic ischemia and/or infarct, coronary heart disease, heart failure, arteriosclerosis and atherosclerosis, hypertension, diabetes, metabolic syndrome, and stroke. An estimated 79,400,000 American adults currently have at least one of these forms of CVD.1 Mortality data show that CVD accounts for 36% of all deaths and that nearly 2400 Americans die from primary CVD or CVD-related secondary complications daily.1 Epidemiologic data designate CVD as the leading cause of death compared to any other single cause or group of causes including cancer, neurological and respiratory diseases, or accidental deaths and the total direct and indirect economic costs attributable to CVD diagnosis, treatment, and prevention is estimated at $432 billion for 2007.1 Clearly, identification of clinically feasible therapeutic approaches for the treatment of CVD and its complications is of utmost importance.
The expansive field of nitric oxide (NO) biology established its foundation through seminal studies on sympathetic cholinergic vasodilation in feline isolated perfused hindlimb vasculature.2,3,4 Serendipitously, these early efforts elucidated a critical link between vascular endothelium and vascular smooth muscle in terms of communicated signals responsible for induced relaxation. Continued studies identified an endothelium-derived relaxing factor, or EDRF, that upon acetylcholine stimulation diffuses from endothelial cells to adjacent vascular smooth muscle to initiate vasodilation in intact arteries.3,4,5 Mechanistic studies showed that EDRF stimulates this relaxation through activation of a soluble form of guanylate cyclase (sGC) and ensuing synthesis of the signaling molecule guanosine 3′,5′-cyclic monophosphate, or cyclic GMP, in vascular smooth muscle.6,7,8 In independent laboratories using bioassay and chemiluminescent approaches respectively, EDRF was unequivocally shown to be NO.9,10
Since this remarkable discovery of EDRF and NO homology, NO has been shown to be a ubiquitous signaling molecule that exists in diverse tissues and systems and across species. As mentioned, the major cardiovascular effects of NO are mediated through sGC activation and synthesis of cyclic GMP, and cyclic GMP can stimulate a myriad of downstream intracellular effectors such as serine/threonine cyclic GMP- and/or adenosine 3′,5′-cyclic monophosphate (cyclic AMP)-dependent protein kinases, protein kinase-independent pathways, or hydrolyzing cyclic GMP-specific phosphodiesterases (PDE). These cyclic GMP-mediated avenues play key roles in the regulation of a variety of biological and physiological processes as well as in the etiology, pathophysiology, and/or progression of assorted cardiac and vascular disorders.11, 12
Importantly, numerous substrates, enzymes, co-factors, and associated elements inherent in the NO signaling system have made it highly attractive for therapeutic targeting for a variety of complications, and cardiovascular pharmacotherapy based on NO biology has provided valuable remedy for patients for well over a century. Perhaps the most well-known NO-based pharmacotherapy is nitroglycerin, a compound generally considered to operate through metabolic liberation of NO in the vasculature.13 Nitroglycerin and associated “NO-donor” compounds are universal therapeutic agents for use in cardiovascular tissues and other organ systems and generally lead to beneficial outcomes based on enhanced NO-mediated cyclic GMP signaling. Interestingly however, drawbacks to the use of NO-based drugs for cardiovascular therapy were first reported in 1888 as the development of tolerance following repeated nitroglycerin treatment in hypertensive patients.14 Since this initial observation, emerging basic and clinical evidence convincingly demonstrate that NO-based interventions potentially suffer from the development of tolerance and tachyphylaxis, have capacity to exert significant cardiovascular toxicities, and often elicit broad deleterious consequences.15, 16, 17, 18 Moreover, clear experimental and clinical evidence showing that NO-based therapies reduce overall mortality is lacking. Indeed, there is great need to identify NO-alternate approaches that stimulate the salutary downstream actions of NO yet do not suffer from its limitations and/or harmful consequences.
A complementary pathway exists in mammalian cardiovascular tissues that parallels NO signaling and that is gaining support in terms of (patho)physiological significance and potential therapeutic utility. Heme oxygenase (HO) is the initial and rate-limiting enzyme for catabolism of mammalian heme and exists in both constitutive and inducible forms. Heme degradation by HO yields equimolar amounts of biliverdin IXa, ferrous iron, and carbon monoxide (CO). Biliverdin and sequential bilirubin along with ferrous iron serve as robust downstream anti-oxidants and, through a mechanism distinct from that for NO, CO stimulates sGC and produces biologically active cyclic GMP. Convincing scientific evidence is emerging in support of the HO/CO pathway as a NO-alternate system that has significant therapeutic promise yet does not suffer from many of the limitations associated with traditional NO signaling. One important caveat, however, regarding biological relevancy of CO is its relatively low potency for stimulating sGC, a valid concern for the notion of CO as a physiologically active signaling molecule in cardiovascular tissues.19
Based largely on limitations of (then) current NO and CO signaling, investigators spent much effort in trying to identify novel pharmacological approaches that could be used as alternative routes for control of sGC/cyclic GMP. Two distinct classes of compounds were discovered: heme-dependent sGC stimulators and heme-independent sGC activators.20, 21 Heme-dependent stimulators require an intact and functional prosthetic heme moiety in the cyclase and robustly synergize with NO- and/or CO-induced activation. Such compounds include YC-1, BAY 41-2272, and BAY 41-5843. Heme-independent activators such as BAY 58-2667 operate in heme-deficient or heme-oxidized tissues and can activate sGC even more potently under such conditions. This class of potential therapeutic agents is especially attractive considering the inimical oxidative setting of cardiovascular disease or injury.
This current article presents experimental and clinical evidence in support of NO-alternate, sGC/cyclic GMP-specific approaches that have significant pharmacotherapeutic potential in cardiac and vascular tissues. Based largely on published and preliminary results from the author's laboratory, particular emphasis is placed on HO- and CO-mediated cyclic GMP signaling and the newly described agents YC-1 and BAY 41-2272 and their positive influence on vascular smooth muscle growth. Description of these novel sGC/cyclic GMP-sensitizing approaches is included along with therapeutic rationale for their use, an area of study that is gaining critical importance in the basic and clinical sciences.21, 22, 23
Heme Oxygenase/Carbon Monoxide/Cyclic GMP Signaling
Brief background on the HO/CO system is essential for an understanding of its role as an NO-alternate, biologically active route for sGC stimulation and cyclic GMP production in vascular smooth muscle. Schmid and colleagues originally characterized HO as the rate-limiting microsomal enzyme that, in the presence of ferrous iron and stoichiometric amounts of NADPH and molecular oxygen, catalyzes oxidation of heme at the α-methene bridge to form biliverdin and its sequential reduced form bilirubin and liberates CO.24, 25 Assumingly based on caveats of their experimental setup and on several provocative observations, paradoxically these investigators stated that their findings “…argue strongly against a physiologic role for “heme α-methenyl oxygenase” in the degradation of heme compounds.”25 Since these original discoveries, the majority of studies have rightly focused on the biological anti-oxidant activities of biliverdin and bilirubin, the most potent anti-oxidant in mammalian tissues,26 as well as on ferritin-based apo-ferroxidase activity following conversion of ferrous iron.27 Interestingly, endogenous formation of HO-liberated CO was first described under both normal and pathophysiological conditions some 20 years earlier;28, 29 however, the notion of CO as a biologically relevant and functional signaling molecule in cardiovascular tissues has only recently been theorized.
Over the past decade notable experimental studies have provided much support for the biological relevance of HO and CO in multiple organ systems and under normal and pathophysiologic conditions. Moreover, a role for CO in mediating diverse cardiovascular functions is gaining popularity and has recently been the focus of several comprehensive review articles.30, 31, 32 As mentioned, CO stimulates sGC in vascular smooth muscle through a mechanism separate to that for NO. Specifically, activation of sGC by NO is biphasic, consisting of a transient inactive six-coordinate NO-heme iron of the β1 subunit of the cyclase,33 followed by a rate-limiting cleavage of the prosthetic iron-histidine105 bond to create a five-coordinate nitrosyl-heme complex and a catalytically active enzyme that achieves up to 400-fold induction.34, 35 It is proposed that CO binding to sGC also involves the primary formation of a six-coordinate ligand-enzyme complex; however, the iron-His105 bond remains intact following CO binding while a transient five-coordinate intermediate is formed only upon ligand-enzyme dissociation.35 36, 37, 38 This unique mechanism of enzyme activation by CO is thought to be the major limitation to the degree of CO-induced cyclase activation which has been reported to reach a relatively modest 4- to 5-fold.35 Following CO- (and/or NO-) mediated activation, sGC elicits cleavage of pyrophosphate from guanosine triphosphate (GTP) to yield the robust and multifunctional signaling messenger cyclic GMP. As with NO, the fate of CO-generated cyclic GMP is dependent upon intracellular localization in various cytosolic compartments or organelles, the presence and activities of cyclic GMP-degrading PDEs, direct and/or indirect communication with the parallel cyclic AMP system, and the existence of essential co-factors and target proteins and substrates.
Early findings from our laboratory and others in support of vascular smooth muscle HO as a biologically significant system were initially based on enhanced expression of its inducible form (HO-1) following experimental vascular injury.39, 40 These initial observations showed that arterial HO-1 transcript and protein levels are upregulated acutely following balloon distension injury, are subsequently reduced to lower than basal levels after 4 days coinciding with marked loss of medial smooth muscle cells, and then are stimulated again during the neointimal growth phase between 7 and 21 days post-injury. These findings provided basis for continued studies utilizing HO-1 over-expression approaches such as systemic pharmacologic dosing of the heme substrate and HO-1 inducer hemin41, 42, the phase II HO-1 inducer butylated hydroxyanisole,43 or localized luminal adenoviral-mediated HO-1 gene transfer following injury44. These results showed that elevated HO-1 signaling attenuates vascular wall remodeling and the neointimal growth response following distension injury and that this occurs through enhanced medial cell apoptosis concomitant with suppressed medial cell proliferation.44 These findings provide first evidence for a pathophysiologic role for the HO system in injured vasculature and strongly suggest HO as a protective signaling mechanism against aberrant vessel growth. Figure 1 shows cross-sectional photomicrographs of rat carotid arteries from uninjured animals (A) or from animals whose carotid arteries were balloon-injured and treated with vehicle (B), the heme substrate and HO-1 inducer hemin (C), or with hemin plus the selective HO-1 inhibitor tin protoporphyrin (D). One can clearly see a patent lumen with clearly defined medial elastic lamina in the uninjured artery. In the balloon-injured vessel (B), a marked matrix-rich neointima is apparent with significant luminal stenosis and moderate adventitial remodeling. In the HO-1-overexpressing artery (C), injury-induced vessel remodeling has been largely attenuated and luminal patency almost completely restored. Heme oxygenase-specificity was verified with concomitant hemin and HO-1 inhibition (D), which resembles an injured untreated vessel as seen in (B).
1.

Verhoff-van Gieson-stained photomicrographs of rat carotid artery 5 μm cross-sections from uninjured animals (A) or from animals whose arteries were balloon-injured and treated with DMSO vehicle (B), the heme substrate and HO-1 inducer hemin (C), or with hemin plus the selective HO-1 inhibitor tin protoporphyrin (D). A clear and patent lumen is observed with a thin smooth muscle-rich media in the uninjured arteries (A). A marked and concentric matrix-rich neointima with significant luminal stenosis is evident in vehicle-treated vessels 2 weeks following balloon injury (B), which is dramatically reduced in vessels exposed to HO-1 induction (C). This phenomenon is specific for HO-1, as concomitant treatment of vessels with hemin plus the HO-1 inhibitor tin protoporphyrin completely reverses the HO-1-mediated growth suppression (D).
Corresponding studies focused on the role of CO without the influence of upstream HO-stimulated anti-oxidant pathways in mediating vessel growth during disease or following injury. Endogenous HO-generated CO originating in vascular smooth muscle was shown to confer protection against neointimal development following balloon injury through anti-proliferative actions.45, 46 Subsequently and in support, exogenous CO administration via systemic dosing47, 48 or localized luminal incubation of a saturated CO solution49 reduced neointimal growth after injury and minimized atherosclerotic lesion formation. Proposed mechanisms for CO-mediated growth suppression include activation of p38 mitogen-activated protein kinases (MAPK), enhanced acute expression of the cyclin-dependent kinase (cdk) inhibitor p21Cip1,47 and CO-induced inhibition of the synthetic growth factor TGF-β1 and the G1 cyclins E and A.49 These findings directly implicate CO as a sGC/cyclic GMP-dependent salutary inhibitor of vascular smooth muscle growth and lend firm support for the biological relevance of this pathway.
sGC stimulators and sGC activators
Discovery of novel approaches for enhancing sGC-mediated cyclic GMP signaling in NO-independent fashion through heme-dependent sGC stimulators and/or heme-independent sGC activators has challenged the traditional view of NO biology and offers new insights into a promising pharmacological strategy with definitive therapeutic advantages. These novel approaches are even more appealing considering limitations and toxicities associated with NO-based therapies and the pro-oxidant local environment often present in diseased or abnormal states. Moreover, despite the profound findings that HO and the HO product CO have significant capacity to confer vascular protection against aberrant growth, skepticism still exists regarding biological relevancy of this pathway, a concern largely addressed through identification of cyclase stimulators and activators that act through CO-sensitive mechanisms. This section will include a summation of several of the latest sGC/cyclic GMP-specific approaches in light of several excellent and comprehensive reviews that have been recently published on this topic.20, 21, 22, 23 Published and preliminary data in support of the heme-dependent sGC-stimulating agents YC-1 and its analog BAY 41-2272 as biologically active therapeutic regulators of vascular smooth muscle growth will be presented.
1. YC-1
As mentioned, two classes of very promising compounds were recently discovered that have capacity to exert significant control over sGC and cyclic GMP signaling in the cardiovascular system: heme-dependent sGC stimulators that synergize with NO and/or CO and that require a functional cyclase heme, and heme-independent sGC activators that operate even more robustly under heme-jeopardized or heme-deficient conditions. The most characterized of these neoteric sGC-modifying agents is a benzyl indazole derivative named YC-1, first discovered by Teng and colleagues in 1994 as a NO-independent activator of platelet sGC and cyclic GMP synthesis in rabbits.50, 51 These influential early reports showed that YC-1 exerts potent anti-platelet effects under in vitro conditions such as inhibition of ATP release and platelet aggregation, reduction in intracellular calcium mobilization, and disaggregation of clumped platelets. Several of these findings were substantiated under in vivo conditions as prolonged tail bleeding times in YC-1-treated mice. Since these seminal discoveries, numerous experimental and clinical studies have investigated the actions of YC-1 in cardiac and vascular tissues under normal and pathologic conditions. Verifying the increasing popularity of this agent, in an invited review article on YC-1 prepared by this author in 200422 slightly over 120 publications were listed on the National Library of Medicine PubMed database related to YC-1. In a current PubMed search in preparation for this article 225 citations for YC-1 were listed, a dramatic increase in publications in only three years! Indeed, the broad utility of YC-1 in various cell types and organ systems makes it a highly attractive agent for use in both basic science and clinical studies. As mentioned, detailed discussions of the mechanisms of action of YC-1 and its manifold effects in the cardiovascular and hematological systems were recently published;22, 23, 52, 53 therefore, in this section only a brief summation of some of the crucial mechanisms of YC-1 and its targeted effects on vascular smooth muscle growth related to potential pharmacotherapeutic utility will be addressed.
YC-1 operates through complementary upstream and downstream mechanisms to robustly increase cyclic GMP signaling in vascular smooth muscle. At the upstream cyclase YC-1 stabilizes the active configuration of the enzyme,35,54 enhances the affinity of the activated enzyme for its substrate GTP,55 and reduces the dissociation of NO and/or CO from the enzyme thereby inhibiting ligand-enzyme de-activation.56 These processes strongly increase the production of sGC-mediated cyclic GMP. Downstream of cyclic GMP, YC-1 also serves to inhibit PDE isoenzymes responsible for cyclic GMP hydrolysis and degradation, thus enhancing its persistence and duration of action.57 YC-1 functions through these mechanisms in the absence of NO or under conditions of aberrant NO signaling, and the structure of YC-1 does not contain a group capable of NO donation (see Figure 8). YC-1 is therefore considered to be a NO-independent sGC stimulator; however, YC-1 does have ability to augment sGC activation and cyclic GMP synthesis elicited by both NO as well as CO. Notably, in the absence of NO YC-1 has capacity to stimulate CO-mediated sGC activation to a degree comparable to that of NO.35, 36 In fact, the notion of an endogenous “YC-1-like” compound that is up- or down-regulated in response to local stimuli in order to enhance CO-mediated signaling has been proposed by various investigators, and discovery of such a factor would lend credence to YC-1 as a therapeutic tool and substantiate HO/CO signaling as biologically important.
8.

Schematic of a novel pathway for heme oxygenase/carbon monoxide-dependent cyclic GMP signaling based on the newly described sGC/cyclic GMP-modulating agents YC-1 and BAY 41-2272 as shown. Also shown are characterized (*) and potential (#) sites of action of these agents that serve to enhance sGC function and stimulate cyclic GMP signaling. Through such pharmacologic modulation, downstream cyclic GMP-specific events are controlled including cellular proliferation and apoptosis and matrix balance via MMP biology. Through inhibition of cell proliferation and gelatinolytic MMP biology and exaggeration of cellular apoptosis, abnormal vessel growth is minimized and vascular protection conferred during states of injury or disease.
Regarding growth of vascular smooth muscle, YC-1 dose-dependently and significantly increases cyclic GMP content under in vitro58 and in vivo39 conditions. In rat primary (passage 3-7) vascular smooth muscle cells, YC-1 inhibited serum-stimulated proliferation in dose-dependent (10-50 μM) manner58. Verifying these data, previously unpublished findings show that YC-1 inhibits serum-induced proliferation of commercially-available rat vascular smooth muscle cells (A7R5, ATCC) in dose- and time-dependent fashion (see Figure 2) without evidence of cytotoxicity as estimated through trypan blue exclusion staining. In support of the anti-mitogenic effects of YC-1 observed in culture, in rat intact vasculature adventitially-applied YC-1 in pluronic gel attenuates vessel remodeling and neointimal growth 2 weeks after balloon distension injury.39, 58 Mechanistic analyses suggest that YC-1 reduces growth through inhibiting the proliferative factor TGF-β1 and via reducing focal adhesion kinase59 and through alteration of matrix balance by suppression of matrix metalloproteinase (MMP) biology.60 Corroborating unpublished preliminary findings show that YC-1 potently stimulates medial and neointimal cellular apoptosis 14 days post-balloon injury and significantly reduces medial and neointimal cellular proliferation at both acute (24, 48 hour) and prolonged (7, 14 days) time points after injury. Additional unpublished data show that selective inhibition of cyclic GMP-specific PDE by zaprinast in rat injured arteries mimics the growth-inhibitory effects of YC-1 while targeted sGC inhibition with ODQ fails to markedly alter the YC-1 effects. These novel findings suggest that YC-1 operates to reduce vascular smooth muscle growth after injury in cyclic GMP-specific manner. Moreover, the route of cyclic GMP action appears to be primarily due to enhanced cyclic GMP perseverance via inhibition of cyclic GMP hydrolysis by PDE and only minimally dependent upon cyclic GMP synthesis by sGC. Several representative photomicrographs for these YC-1 studies are shown in Figure 3 for balloon-injured untreated control (A) and balloon-injured YC-1-treated (B) arterial cross-sections 2 weeks post-injury. A marked diminution in neointimal growth and luminal stenosis is readily observed in vessels treated with YC-1 (B), thus firmly implicating YC-1 as a potent anti-growth agent and potential therapeutic tool against aberrant vascular growth and remodeling.
2.

Absolute rat aortic smooth muscle cell (RASMC) counts using commercially-available A7R5 cells (ATCC) grown in DMEM and treated with vehicle (DMSO) (A) or with varying concentrations of YC-1 (0.1-100 μM) and stimulated (10% FBS) for 24, 48, or 72 hours. Marked reduction in vascular smooth muscle cell proliferation by YC-1 is readily observed at all time points and in dose-dependent fashion. No evidence of cytotoxicity as estimated by trypan blue exclusion staining was evident in any cohort.
3.

Verhoff-van Gieson-stained photomicrographs of rat carotid artery 5 μm cross-sections from balloon-injured animals treated with DMSO vehicle (A) or with 1 mg YC-1 in adventitial pluronic gel (B) immediately after injury. Cross-sections were obtained 2 weeks post-injury. These data support an anti-proliferative action of YC-1 in the response to vessel injury. [Photo in (B) is modified from Ref. No. 39].
Indeed, YC-1 confers robust anti-mitogenic effects on vascular smooth muscle under culture conditions and in intact vasculature during the growth response to experimental injury. This anti-proliferative ability would be greatly warranted for pharmacologic treatment of a variety of abnormal growth conditions in cardiac and vascular tissues such as the de-differentiation that occurs in vascular smooth muscle cells during evolution of an atherosclerotic plaque or during angiogenesis and vasculogenesis that occur in vessel tumorigenesis. Of note, YC-1 has many other vasoactive properties that could provide added benefit under such conditions such as reduction of blood pressure and inhibition of platelet function. These multifaceted attributes of YC-1 make it very interesting and appealing in terms of pharmacotherpeutic utility in basic sciences and clinical medicine. Interestingly though, newly developed agents based on YC-1 have emerged that have more robust mechanisms of action and that might offer added promise for pharmacotherapeutic utility in cardiovascular disorders.
2. BAY 41-2272
Based in large part on the success of studies using YC-1 in cardiovascular tissues, utilizing YC-1 as the parent compound scientists discovered literally thousands of “YC-1 mimetics” that share similar characteristics to YC-1 and that exert significant and often more potent biological activities.61, 62 Based on structure-activity relationships and associated data, BAY 41-2272 (in addition to its analog BAY 41-8543) was selected as one of the two most promising heme-dependent sGC stimulators. Mechanistically, BAY 41-2272 and YC-1 operate in very similar fashion,63 and although an inhibitory effect for BAY 41-2272 on cyclic GMP-specific PDE has been suggested64 it has yet to be verified under physiological conditions.65 Like YC-1, BAY 41-2272 operates irrespective of NO yet synergizes with both NO and CO to potently stimulate sGC in heme-dependent fashion and to produce cyclic GMP. Notably, BAY 41-2272 is approximately 100-fold more potent than YC-1 in stimulating the enzyme, thus making it especially attractive for clinical use.
In vascular smooth muscle the growth-modulating effects of BAY 41-2272 have yet to be studied in detail although preliminary efforts in the author's laboratory are examining BAY 41-2272 under in vitro conditions and following experimental vascular injury in intact animal models. Unpublished results show that BAY 41-2272 significantly and dose-dependently reduces proliferation of rat A7R5 vascular smooth muscle cells at 24, 48, and 72 hours following serum (10% FBS) stimulation (see Figure 4). Interestingly, these anti-mitogenic properties of BAY 41-2272 are enhanced with concomitant HO-1 induction (by hemin dosing) primarily at lower BAY 41-2272 concentrations, and largely reversed with concomitant HO-1 inhibition (with tin protoporphyrin) primarily at higher BAY 41-2272 concentrations. These challenging findings imply that BAY 41-2272 operates to control proliferation of vascular smooth muscle through a “high/low gain switch” largely dependent on functional HO-1 signaling. Similar studies using BAY 41-2272 with/without concomitant induction (through SNP) or inhibition (with L-NAME) of NO signaling failed to demonstrate significant modulation of the BAY 41-2272 growth-modulating effects (data not shown). These promising early findings suggest that BAY 41-2272 operates in HO-dependent fashion in A7R5 vascular smooth muscle cells to reduce growth under culture conditions. Of note, however, recent experiments in the author's laboratory studying the effects of BAY 41-2272 on growth of rat primary vascular smooth muscle cells have yielded very interesting yet discordant findings. Rat primary vascular smooth muscle cells (at passage 2 and passage 7) treated with varying log concentrations of BAY 41-2272 and stimulated (10% FBS) for 48 hours showed dose-dependent increases in cell numbers compared to vehicle-treated cells (data not shown). The divergent results between commercial (and likely phenotypically-altered) and fresh (primary) cells in response to BAY 41-2272 may be partially dependent upon differential expressions of cyclic GMP-specific protein kinase and/or cyclic AMP-dependent kinase between phenotypically-altered cells versus primary cells, as has been shown previously by Lincoln and colleagues.66 Studies are currently underway in the author's laboratory to investigate these challenging findings.
4.
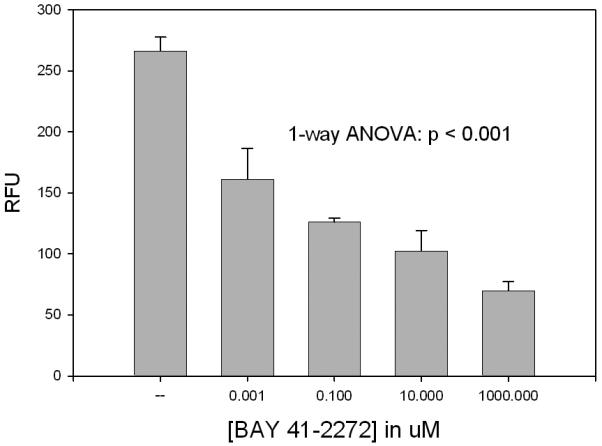
Cell proliferation assay on rat A7R5 vascular smooth muscle cells after 10% FBS stimulation and treatment with vehicle (DMSO) or log doses of BAY 41-2272 for 72 hours. One-way ANOVA indicates statistically significant reduction in growth in cells treated with BAY 41-2272. Interestingly, the anti-mitogenic effects of BAY 41-2272 were enhanced in cells simultaneously treated with hemin to induce HO-1 signaling (primarily at lower BAY 41-2272 concentrations) and reversed in cells simultaneously treated with tin protoporphyrin to inhibit HO-1 (primarily at higher BAY 41-2272 concentrations).
Related to growth, current studies are analyzing the effects of BAY 41-2272 on vascular smooth muscle matrix balance with particular emphasis on the gelatinolytic enzymes MMP-2 and MMP-9 and their roles in regulating the growth response to vascular trauma. These particular MMP enzymes are crucial for a variety of cellular functions in vascular smooth muscle including regulation of proliferation and migration that underlie growth in response to injury.67, 68 In initial efforts balloon injury was performed on rat carotid arteries and vessels were treated with perivascular BAY 41-2272 immediately after injury, and tissues were harvested after 48 hours for Western analysis. A Western blot on uninjured or balloon-injured carotid artery homogenates (to measure intracellular protein expression) with or without BAY 41-2272 treatment 48 hours post-intervention is shown in Figure 5A along with densitometric analyses for pro-MMP-9 (Fig. 5B) and active MMP-9 (Fig. 5C). BAY 41-2272 failed to exert significant effects on intracellular pro- or active MMP-9 in uninjured vessels. Balloon injury dramatically increased intracellular expression of both pro- and active forms of MMP-9, and these were completely reversed to levels equal to or below baseline with concomitant BAY 41-2272 treatment. This was the first indication that modulation of sGC/cyclic GMP signaling via BAY 41-2272 could impact MMP biology under in vivo conditions. Complementary experiments then addressed extracellular (secreted) MMP activity through use of in situ gel zymography. As shown in Figure 6, rat balloon-injured carotid arteries demonstrated marked elevations in extracellular gelatinase activity 24 hours post-injury (Lane 2 compared to Lane 1), as estimated by noticeable areas of “clearing” of the gelatin-containing gel. In balloon-injured arteries treated with perivascular BAY 41-2272 immediately after injury, significant reduction in extracellular gelatinase activity is certainly evident in Lane 3. These findings substantiate the initial Western blot data on MMP-9 expression and show that BAY 41-2272 confers similar inhibition on extracellular MMP-9 activity following injury. Of note, this technique estimates total extracellular gelatinase activity that includes both MMP-9 and MMP-2.
5.

A: Western blot analysis on rat uninjured or balloon-injured carotid artery homogenates with/without perivascular BAY 41-2272 treatment immediately after injury. Samples were obtained 48 hours post-injury. Lane 1: ladder; Lane 2: MMP-9 positive control; Lane 3: MMP-2 positive control; Lanes 4, 5: uninjured artery homogenates; Lanes 6, 7: balloon-injured artery homogenates. Both pro- and active forms of MMP-9 are represented. B, C: Densitometric analyses of Western blot data for pro-MMP-9 (Fig. 5B) and active MMP-9 (Fig. 5C). For both pro- and active forms, uninjured arteries show basal levels of MMP-9 expression which were not affected by BAY 41-2272 treatment. Balloon injury significantly elevates intracellular pro- and active MMP-9 which were both dramatically reduced with concomitant BAY 41-2272 treatment.
6.

In situ gel zymogram (using 10% Tris-glycine gels containing 0.1% gelatin as substrate, Invitrogen) showing rat uninjured (uninj.) or balloon-injured (BI) whole carotid artery sections with/without perivascular BAY 41-2272 (BAY) treatment given immediately after injury. Samples were obtained 24 hours post-intervention, and areas of “clearing” represent gelatinase activity. Lack of notable extracellular gelatinase activity is apparent in the uninjured artery sections (Lane 1) while the balloon-injured sections show robust activity in Lane 2 (n = 5 per group). Balloon-injured vessels treated with BAY 41-2272 show markedly reduced extracellular gelatinase activity as seen in Lane 3 (n = 6). Lines in Lane 3 represent experimental (technical) artifacts.
Next, in a series of detailed experiments the effects of BAY 41-2272 on independent intracellular and extracellular MMP-9 and MMP-2 activities following balloon injury were analyzed using column gel zymography, an approach that allows differentiation between these similar gelatinases based on molecular weight. Rat carotid arteries were uninjured or balloon-injured and treated with adventitial BAY 41-2272 as previously described. After 24 hours arteries were harvested and incubated in warm DMEM plus 10% FBS in culture for 24 hours. Conditioned media was obtained and analyzed for extracellular (secreted) MMP activity while arterial tissues were homogenized to measure intracellular MMP activity. Figure 7A shows column gel zymograms for extracellular and intracellular gelatinase activities, respectively, for data obtained from two rats in order to show reproducibility and consistency of results. Figures 7B-7E show densitometric results of extracellular and intracellular raw data from the zymograms shown in Figure 7A. In summary, these zymography data reveal considerable attenuation of injury-induced MMP activation by BAY 41-2272 which, combined with its ability to mitigate cell proliferation, makes BAY 41-2272 highly attractive for pharmacotherapeutic targeting of abnormal vascular smooth muscle growth. Specifically, in the column zymograms and corresponding densitometric data one can clearly see that BAY 41-2272 attenuates the injury-induced extracellular activities of the active forms of both MMP-9 (Fig. 7B) and MMP-2 (Fig. 7D). The influence of BAY 41-2272 on the inactive precursor (pro-) forms of the extracellular gelatinases is not as clear. For intracellular activities, BAY 41-2272 significantly minimizes injury-induced increases in both pro-MMP-9 and active MMP-9 (Fig. 7C). Correspondingly, BAY 41-2272 has comparable inhibitory effects on the intracellular activities of pro- and active MMP-2 (Fig. 7E).
7.

A: Column gel zymogram for extracellular and intracellular pro- and active forms of MMP-9 and MMP-2 from rat carotid arteries subjected to balloon injury and treated with/without BAY 41-2272. Arteries were uninjured or balloon-injured and treated with BAY 41-2272 immediately after injury. After 24 hours arteries were placed in DMEM plus 10% FBS and incubated at 37 °C for 24 hours. Conditioned media tissues and were then obtained for measures of extracellular (media) or intracellular (tissue homogenates) MMP activities. Shown are data from two rats to illustrate reproducibility of results. B-E: Densitometry results for column gel zymogram shown in Figure 7A for extracellular and intracellular pro- and active MMP-9 and MMP-2. Figures 7B and 7C show that BAY 41-2272 potently reverses injury-induced activation of MMP-9 towards control levels. Also, inhibitory effects of BAY 41-2272 are clearly seen for pro-MMP-9 in the intracellular compartment. Figures 7D and 7E similarly show that BAY 41-2272 reduces the active forms of extracellular and intracellular MMP-2. BAY 41-2272 also minimizes intracellular pro-MMP-2 as well.
In logical complement, under identical experimental conditions preliminary Western data show that BAY 41-2272 dramatically reduces injury-induced extracellular expression of the MMP-9-selective tissue inhibitor TIMP-1 (data not shown), which is particularly essential for cleaving pro-MMP-9 and forming active MMP-9. In the extracellular mileu with BAY 41-2272 reducing TIMP-1, the expected outcome would be enhanced pro-MMP-9 along with reduced active MMP-9. This exactly what is observed in these early findings (see Fig. 7). This novel early result suggests that BAY 41-2272 serves to reduce injury-mediated elevations in extracellular MMP-9 through reduction in extracellular TIMP-1, yet this line of investigation is currently underway to verify and elucidate these mechanisms. Clearly, based on these exciting new data BAY 41-2272 confers robust anti-growth properties on vascular smooth muscle under both eutrophic culture conditions as well as in the pathologic environment following vessel injury or disease. These results provide strong support for the therapeutic potential of this multifaceted sGC/cyclic GMP-regulating agent.
In summation, Figure 8 illustrates a novel pathway for cyclic GMP signaling based on published and preliminary data from studies examining HO and CO as well as YC-1 and its analog BAY 41-2272 and their effects on vascular smooth muscle growth. These NO-alternative approaches appear to mitigate growth through inhibition of cellular proliferation and MMP functions and stimulation of cellular apoptosis. Clearly, the therapeutic potential for these NO-independent sGC/cyclic GMP-modulating pharmacologic regimens for conditions of cardiovascular adversity is extremely high and most promising.
Conclusions
The critically important field of NO biology has provided foundation for countless basic science endeavors and the platform for broad clinical utility under conditions of cardiovascular adversity. Emerging evidence, however, mandates that alternate approaches are identified that elicit the beneficial aspects of NO-induced cyclic GMP signaling yet that do not suffer from inherent limitations associated with NO-based therapies. Abundant evidence supports HO and CO as a pivotal and (patho)physiologically relevant signaling pathway that has marked capacity to regulate growth of vascular smooth muscle. Moreover, the discovery of new cyclic GMP modulating agents such as YC-1 and BAY 41-2272 that operate in NO-independent manner but that augment NO and CO signals provides new routes for cyclic GMP control and sound basis for this novel pharmacotherapeutic regimen.
Acknowledgments
The author would like to apologize to investigators whose works were not included in this article due to space limitations and the concise nature of this report. Work in the preparation of this review was supported by National Heart, Lung, and Blood Institute grants HL-36045, HL-59976, HL-62467, HL-59868, and HL-81720; The Methodist Hospital Foundation; The Gillson-Longenbaugh Foundation, and The American Heart Association. The author would like to acknowledge Dr. William Durante of the University of Missouri, Drs. Elias Jackson, Jr. and Somnath Mukhopadhyay of North Carolina Central University, and Dr. Andrew Schafer at the University of Pennsylvania for their valuable contributions to this work. The author would also like to acknowledge Drs. A. Knorr and J.P. Stasch of BAYER Health Care AG, Germany for the generous contribution of BAY 41-2272 used in studies described herein.
Acknowledgment of Sources of Support:
National Heart, Lung, and Blood Institute grants HL-36045, HL-59976, HL-62467, HL-59868, and HL-81720; The Methodist Hospital Foundation; The Gillson-Longenbaugh Foundation, and The American Heart Association.
References
- 1.Rosamond W, Flegal K, Friday G, et al. Heart disease and stroke statistics – 2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Folkow B, Haeger K, Uvnas B. Cholinergic vasodilator nerves in the sympathetic outflow to the muscles of the hindlimb of the cat. Acta Physiol Scand. 1948;15:401–411. doi: 10.1111/j.1748-1716.1948.tb00515.x. [DOI] [PubMed] [Google Scholar]
- 3.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 4.Furchgott RF. Discovery of endothelium-derived relaxing factor and its identification as nitric oxide. In: Panza JA, Cannon RO III, editors. Endothelium, Nitric Oxide, and Atherosclerosis: From Basic Mechanisms to Clinical Implications. Futura Publishing Co., Inc.; Armonk, NY: 1999. pp. 3–11. [Google Scholar]
- 5.Furchgott RF. Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Biosci Rep. 1999;19:235–251. doi: 10.1023/a:1020537506008. [DOI] [PubMed] [Google Scholar]
- 6.Rapoport RM, Murad F. Endothelium-dependent and nitrovasodilator-induced relaxation of vascular smooth muscle: role of cyclic GMP. J Cyclic Nucleotide Protein Phosphor Res. 1983;9:281–296. [PubMed] [Google Scholar]
- 7.Martin W, Villani GM, Jothianandan D, et al. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J Pharmacol Exp Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- 8.Ignarro LJ, Harbison RG, Wood KS, et al. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther. 1986;237:893–900. [PubMed] [Google Scholar]
- 9.Ignarro LJ, Byrns RE, Buga GM, et al. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacological and chemical properties that are identical to those of nitric oxide radical. Circ Res. 1987;61:866–879. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 10.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 11.Loscalzo J, Vita JA, editors. Nitric oxide and the cardiovascular system. Humana Press; Totowa, NJ: 2000. [Google Scholar]
- 12.Panza JA, Cannon RO III, editors. Endothelium, Nitric Oxide, and Atherosclerosis: From Basic Mechanisms to Clinical Implications. Futura Publishing Co., Inc.; Armonk, NY: 1999. [Google Scholar]
- 13.Chung SJ, Fung HL. Identification of the subcellular site for nitroglycerin metabolism to nitric oxide in bovine coronary smooth muscle cells. J Pharmacol Exp Ther. 1990;253:614–619. [PubMed] [Google Scholar]
- 14.Stewart DD. Remarkable tolerance to nitroglycerin. Phila Polyclin. 1888;6:43. [Google Scholar]
- 15.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 16.Parker JD. Therapy with nitrates: increasing evidence of vascular toxicity? J Am Coll Cardiol. 2003;42:1835–1837. doi: 10.1016/j.jacc.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 17.Davis KL, Martin E, Turko IV, et al. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol. 2001;41:203–236. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- 18.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrate: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 19.Cary SP, Marletta MA. The case of CO signaling: why the jury is still out. J Clin Invest. 2001;107:1071–1073. doi: 10.1172/JCI12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hobbs AJ. Soluble guanylate cyclase: an old therapeutic target re-visited. Br J Pharmacol. 2002;136:637–640. doi: 10.1038/sj.bjp.0704779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evgenov OV, Pacher P, Schmidt PM, et al. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature Rev. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tulis DA. Salutary properties of YC-1 in the cardiovascular and hematological systems. Curr Med Chem. 2004;2:343–359. doi: 10.2174/1568016043356200. [DOI] [PubMed] [Google Scholar]
- 23.Jackson EB, Jr, Mukhopadhyay S, Tulis DA. Pharmacologic modulators of soluble guanylate cyclase/cyclic guanosine monophosphate in the vascular system – from bench top to bedside. Curr Vasc Pharmacol. 2007;5:1–14. [PMC free article] [PubMed] [Google Scholar]
- 24.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase: characterization of the enzyme. J Biol Chem. 1969;244:6388–6369. [PubMed] [Google Scholar]
- 26.Gopinathan V, Miller NJ, Milner AD, et al. Bilirubin and ascorbate antioxidant activity in neonatal plasma. FEBS Lett. 1994;349:197–200. doi: 10.1016/0014-5793(94)00666-0. [DOI] [PubMed] [Google Scholar]
- 27.Balla G, Jacob HS, Balla J, et al. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 28.Sjorstrand T. Endogenous formation of carbon monoxide in man under normal and pathophysiological conditions. Scand J Clin Lab Invest. 1949;1:201–214. [Google Scholar]
- 29.Sjostrand T. Endogenous formation of carbon monoxide; the CO concentration in the inspired and expired air of hospital patients. Acta Physiol Scand. 1951;22:137–141. doi: 10.1111/j.1748-1716.1951.tb00762.x. [DOI] [PubMed] [Google Scholar]
- 30.Durante W. Carbon monoxide and bile pigments: surprising mediators of vascular function. Vasc Med. 2002;7:195–202. doi: 10.1191/1358863x02vm424ra. [DOI] [PubMed] [Google Scholar]
- 31.Durante W, Johnson FK, Johnson RA. Role of carbon monoxide in cardiovascular function. J Cell Mol Med. 2006;10:672–686. doi: 10.1111/j.1582-4934.2006.tb00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182. doi: 10.1165/rcmb.2006-0333TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer B, Koesling D. cGMP signalling beyond nitric oxide. Trends Pharmacol Sci. 2001;22:546–548. doi: 10.1016/s0165-6147(00)01889-7. [DOI] [PubMed] [Google Scholar]
- 34.Wedel B, Humbert P, Harteneck C, et al. Mutation of His-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci USA. 1994;29:2592–2596. doi: 10.1073/pnas.91.7.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 36.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 37.Sharma VS, Magde D. Activation of soluble guanylate cyclase by carbon monoxide and nitric oxide: a mechanistic model. Methods. 1999;19:494–505. doi: 10.1006/meth.1999.0892. [DOI] [PubMed] [Google Scholar]
- 38.Deinum G, Stone JR, Babcock GT, et al. Binding of nitric oxide and carbon monoxide to soluble guanylate cyclase as observed with Resonance raman spectroscopy. Biochem. 1996;35:1540–1547. doi: 10.1021/bi952440m. [DOI] [PubMed] [Google Scholar]
- 39.Tulis DA, Durante W, Peyton KJ, et al. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem Biophys Res Commun. 2000;279:646–652. doi: 10.1006/bbrc.2000.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duckers HJ, Boehm M, True AL, et al. Heme oxygense-1 protects against vascular constriction and proliferation. Nature Med. 2001;7:693–698. doi: 10.1038/89068. [DOI] [PubMed] [Google Scholar]
- 41.Tulis DA, Durante W, Peyton KJ, et al. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis. 2001;155:113–122. doi: 10.1016/s0021-9150(00)00552-9. [DOI] [PubMed] [Google Scholar]
- 42.Aizawa T, Ishizaka N, Taguchi J, et al. Balloon injury does not induce heme oxygenase-1 expression, but administration of hemin inhibits neointimal formation in balloon-injured rat carotid artery. Biochem Biophys Res Commun. 1999;261:302–307. doi: 10.1006/bbrc.1999.1020. [DOI] [PubMed] [Google Scholar]
- 43.Liu XM, Azam MA, Peyton KJ, et al. Butylated hydroxyanisole stimulates heme oxygenase-1 gene expression and inhibits neointima formation in rat arteries. Cardiovasc Res. 2007;74:169–179. doi: 10.1016/j.cardiores.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tulis DA, Durante W, Liu XM, et al. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation. 2001;104:2710–2715. doi: 10.1161/hc4701.099585. [DOI] [PubMed] [Google Scholar]
- 45.Hesheng O, Jun Y, Ligia T, et al. Role of endogenous carbon monoxide in neointimal formation induced by balloon-injury in rat aorta. Chin Med Sci J. 1999;14:41–45. [PubMed] [Google Scholar]
- 46.Togane Y, Morita T, Suematsu M, et al. Protective roles of endogenous carbon monoxide in neointimal development elicited by arterial injury. Am J Physiol Heart Circ Physiol. 2000;278:H623–H632. doi: 10.1152/ajpheart.2000.278.2.H623. [DOI] [PubMed] [Google Scholar]
- 47.Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nature Med. 2003;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 48.Raman KG, Barbato JE, Ifedigbo E, et al. Inhaled carbon monoxide inhibits intimal hyperplasia and provides added benefit with nitric oxide. J Vasc Surg. 2006;44:151–158. doi: 10.1016/j.jvs.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Tulis DA, Keswani AN, Peyton KJ, et al. Local administration of carbon monoxide inhibits neointima formation in balloon injured rat carotid arteries. Cell Mol Biol. 2005;51:441–446. [PMC free article] [PubMed] [Google Scholar]
- 50.Ko FN, Wu CC, Kuo SC, et al. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- 51.Wu CC, Ko FN, Kuo SC, et al. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br J Pharmacol. 1995;116:1973–1978. doi: 10.1111/j.1476-5381.1995.tb16400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chun YS, Yeo EJ, Park JW. Versatile pharmacological actions of YC-1: anti-platelet to anticancer. Cancer Lett. 2004;207:1–7. doi: 10.1016/j.canlet.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Russwurm M, Koesling D. Guanylyl cyclase: NO hits its target. Biochem Soc Symp. 2004;71:51–63. doi: 10.1042/bss0710051. [DOI] [PubMed] [Google Scholar]
- 54.Friebe A, Koesling D. Mechanism of YC-1-induced activation of soluble guanylyl cyclase. Mol Pharmacol. 1998;53:123–127. doi: 10.1124/mol.53.1.123. [DOI] [PubMed] [Google Scholar]
- 55.Lee YC, Martin F, Murad F. Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci USA. 2000;97:10763–10768. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russwurm M, Mergia E, Mullershausen F, et al. Inhibition of deactivation of NO-sensitive guanylyl cyclase accounts for the sensitizing effect of YC-1. J Biol Chem. 2002;277:24883–24888. doi: 10.1074/jbc.M110570200. [DOI] [PubMed] [Google Scholar]
- 57.Galle J, Zabel U, Hubner U, et al. Effects of the soluble guanylate cyclase activator, YC-1, on vascular tone, cyclic GMP levels and phosphodiesterase activity. Br J Pharmacol. 1999;127:195–203. doi: 10.1038/sj.bjp.0702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tulis DA, Bohl Masters KS, Lipke EA, et al. YC-1-mediated vascular protection through inhibition of smooth muscle cell proliferation and platelet function. Biochem Biophys Res Commun. 2002;291:1014–1021. doi: 10.1006/bbrc.2002.6552. [DOI] [PubMed] [Google Scholar]
- 59.Wu CH, Chang WC, Chang GY, et al. The inhibitory mechanism of YC-1, a benzyl indazole, on smooth muscle cell proliferation: an in vitro and in vivo study. J Pharmacol Sci. 2004;94:252–260. doi: 10.1254/jphs.94.252. [DOI] [PubMed] [Google Scholar]
- 60.Liu YN, Pan SL, Peng CY, et al. YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] inhibits neointima formation in balloon-injured rat carotid through suppression of expressions and activities of matrix metalloproteinases 2 and 9. J Pharmacol Exp Ther. 2006;316:35–41. doi: 10.1124/jpet.105.090563. [DOI] [PubMed] [Google Scholar]
- 61.Straub A, Stasch JP, Alonso-Alija C, et al. NO-independent stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett. 2001;11:781–784. doi: 10.1016/s0960-894x(01)00073-7. [DOI] [PubMed] [Google Scholar]
- 62.Straub A, Benet-Buchholz J, Frode R, et al. Metabolites of orally active NO-independent pyrazolopyridine stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett. 2002;10:1711–1717. doi: 10.1016/s0968-0896(02)00034-2. [DOI] [PubMed] [Google Scholar]
- 63.Stasch JP, Becker EM, Alonso-Alija C, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 64.Mullershausen F, Russwurm M, Friebe A, et al. Inhibition of phosphodiesterase type 5 by the activator of nitric oxide-sensitive guanylyl cyclase BAY 41-2272. Circulation. 2004;109:1711–1713. doi: 10.1161/01.CIR.0000126286.47618.BD. [DOI] [PubMed] [Google Scholar]
- 65.Bischoff E, Stasch JP. Effects of the sGC stimulator BAY 41-2272 are not mediated by phosphodiesterase 5 inhibition. Circulation. 2004;110:e320–e321. doi: 10.1161/01.CIR.0000142209.28862.12. (Response) [DOI] [PubMed] [Google Scholar]
- 66.Cornwell TL, Soff GA, Traynor AE, et al. Regulation of the expression of cyclic GMP-dependent protein kinase by cell density in vascular smooth muscle cells. J Vasc Res. 1994;31:330–337. doi: 10.1159/000159061. [DOI] [PubMed] [Google Scholar]
- 67.Bendeck MP, Zempo N, Clowes AW, et al. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 68.Mason DP, Kenagy RD, Hasenstab D, et al. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ Res. 1999;85:179–185. doi: 10.1161/01.res.85.12.1179. [DOI] [PubMed] [Google Scholar]