

Abstract
Integrins, transmembrane glycoprotein receptors, play vital roles in pathological angiogenesis, but their precise regulatory functions are not completely understood and remain controversial. This study aims to assess the regulatory functions of individual beta subunits of endothelial integrins in angiogenic responses induced by vascular endothelial growth factor (VEGF). Inhibition of expression of β1, β3 or β5 integrins in endothelial cells resulted in down regulation of EC adhesion and migration on the primary ligand for the corresponding integrin receptor, while no effects on the recognition of other ligands were detected. Although inhibition of expression of each subunit substantially affected capillary growth stimulated by VEGF, the loss of β3 integrin was the most inhibitory. EC stimulation by VEGF induced formation of the high affinity (activated) state of αvβ3 in a monolayer and activated αvβ3 was co-localized with VEGF receptor-2 (VEGFR-2). Inhibition of expression of β1, β3 or β5 did not affect expression levels of VEGFR-2 in EC. However, inhibition of β3, but not β1 or β5, resulted in substantial inhibition of VEGFR-2 phosphorylation stimulated by VEGF. Exogenous stimulation of αvβ3 integrin with activating antibodies augmented VEGF-dependent phosphorylation of VEGFR-2, whereas integrin blockade suppressed this response. Most importantly, activated αvβ3 was detected on endothelial cells of tumor vasculature. Activation of αvβ3 was substantially increased in highly-vascularized tumors as compared to normal tissues. Moreover, activated αvβ3 was co-localized with VEGFR-2 on endothelial cells of proliferating blood vessels. Together, these results show the unique role of αvβ3 integrin in cross-talk with VEGFR-2 in the context of pathological angiogenesis.
Keywords: angiogenesis, endothelial cell, VEGF, VEGF receptor, integrin
Introduction
The integrins, a family of cell adhesion receptors, mediate multiple and diverse cellular responses, ranging from cell adhesion, migration and assembly of extracellular matrix to regulation of gene expression and cell proliferation. Integrins provide cells with means to sense and respond to their environment by transducing a signal across the cell membrane into the interior of the cell (“outside-in” signaling). Integrins recognize a wide spectrum of extracellular matrix proteins, including vitronectin, fibronectin and collagen.1 In many cases, an integrin preferentially binds to a primary ligand; however, some integrins can also recognize other extracellular matrix components.2,3,4 For example, αvβ3 integrin is primarily the vitronectin receptor, but it can also recognize fibrinogen and, to a lesser extent, fibronectin.5 At least seven members of the integrin family, αvβ3, αvβ5, αvβ1, α5β1, α3β1, α2β1 and α1β1, play important roles in endothelial cell biology.6 These adhesion receptors are classified into three subfamilies, fibronectin, laminin and collagen receptors, based on the ligand specificity of the integrin heterodimer. These receptors control cellular responses critical for the development of neovasculature in adult organisms, the process often referred to as angiogenesis.7 The process of angiogenesis involves coordinated endothelial cell proliferation, migration, invasion and tube formation, and is triggered by hypoxia-induced upregulation of VEGF, which in turn stimulates endothelial proliferation and migration.8 Importantly, VEGF induces activation of integrin receptors resulting in increased binding of soluble ligands as well as endothelial motility and adhesion.9,10
Integrin αvβ3 is known to be expressed on proliferating EC during the processes of angiogenesis and vascular remodeling. The disruption of αvβ3 integrin ligation by either blocking antibodies (LM609 or Vitaxin) or cyclic peptide antagonists (arginine-glycine-aspartic acid-containing peptides) prevents blood vessel formation in mouse retina, rabbit cornea and chick chorioallantoic membrane.11-14 An anti-β3 antibody that recognizes at least three integrins (αIIbβ3, αvβ3 and αMβ2) is beneficial in high risk angioplasty patients in part due to the blockade of αvβ315. Histological examination of human tumor tissue derived from mice treated with the αvβ3 integrin blockers revealed reduction not only in the tumor cell viability but also in the tumor’s vascular density.16 Indeed, angiogenesis induced by αvβ3-negative tumor cells can also be blocked with αvβ3 antagonists, indicating an obvious role for this integrin in the development of tumor vasculature. Results of positron emission tomography in patients have confirmed a strong correlation between expression of αvβ3 and the extent of tumor vasculature.17-19 αvβ3 antagonists disrupt vascular development in the embryo and block pathological angiogenesis in animal models with little negative impact on pre-existing blood vessels, making this integrin an attractive target for therapeutic intervention.20 Indeed, at least six integrin inhibitors are currently under evaluation as therapeutic agents for treatment of solid tumors and leukemias.21
From these pro-angiogenic activities of the vascular integrins, it would seem likely that genetic ablation of αvβ3 integrin would cause vascular defects; however, mice lacking β3 or both β3 and β5 integrins displayed enhanced tumor growth, as well as enhanced angiogenesis.22 Angiogenic responses to VEGF were augmented significantly in the absence of β3 integrins, most likely due to the elevated levels of VEGFR-2 observed in β3-null endothelial cells.23 Despite the clear pathological and clinical significance of integrins in angiogenesis and endothelial cell biology, numerous questions remain unanswered. Several crucial studies on integrin biology have been conducted using model cell systems and the results cannot be directly applied to endothelial cells. Analyses of integrin function using knockout mice are complicated by possible compensatory effects which usually occur during development and which cloud the interpretation of results.24 The aim of this study was to assess the role of each individual subfamily of integrin receptors in VEGF-induced angiogenic cellular responses using a siRNA-based short-term knockdown approach in primary endothelial cells.
Results
Silencing effects of siRNA on expression of integrins in endothelial cells
We utilized small interfering RNAs (siRNAs) to down regulate expression of the β subunits of various integrin heterodimers in primary EC. Upon transfection of EC with specific siRNAs, significant downregulation of β1, β3 and β5 subunit expression was achieved as evidenced by results of Western blotting analyses (Fig. 1A-C). Quantitative analysis revealed that the expression of β1, β3 and β5 integrins was downregulated by 75, 70 and 80%, respectively, whereas the control randomized non-specific siRNA oligos did not show any effect on expression of any of the integrins tested (Fig. 1A-C). Cell surface expression of β1, β3 and β5 integrins following siRNA transfection were confirmed by FACS analysis. Results revealed that transfection of HUVECs with specific siRNAs resulted in 74, 65 and 76% decreases in surface expression of β1, β3 and β5 integrins, respectively (Fig. 1D-F). Each siRNA was specific for its respective β subunit, since siRNAs did not affect expression levels of other subunits (not shown). These results conclusively show that siRNAs downregulated the total expression, as well as cell surface levels, of specific β integrins in endothelial cells.
Figure 1.

Knockdowns of integrin beta subunits on endothelial cells by specific siRNAs. HUVECs were transfected with control siRNA or integrin-specific siRNA and cell lysates were analyzed for expression of β1 (A), β3 (B) or β5 (C) integrin subunits using specific antibody. Densitometry analysis was performed and results are shown in bar graphs (lower). (D and E) Cell surface expression of β1 (D), β3 (E) or β5 (F) integrin subunits in endothelial cells was assessed by FACS analysis. Cells were fixed with 3% paraformaldehyde, stained with primary antibody against the corresponding integrin and with secondary antibody labeled with Alexa Fluor 488. The mean fluorescence intensity was determined; the value obtained using control cells was assigned 100%. Asterisks indicate significant difference over control (p < 0.0038).
Cell adhesion profiling of EC lacking β1, β3 and β5 integrin subunits
Adhesion of endothelial cells to extracellular matrix (ECM) components through cell surface integrins is known to required for endothelial cell growth, differentiation and survival.27 Endothelial cell adhesion to ECM induces cellular proliferation and programmed cell death of non-adherent cells in suspension.28 Ligation of Integrin ligands induces a wide variety of intracellular processes, including tyrosine phosphorylation of FAK, increased inositol lipid synthesis, cyclin synthesis and expression of several cell survival factors.29 Endothelial cell surface integrins mediate adhesion to ECM proteins, including vitronectin, laminin, collagen, von Willebrand factor and fibrinogen. To examine endothelial cell adhesion to various ECM proteins following inhibition of expression of specific integrin subunits, HUVECs transfected with integrin-specific siRNA were plated on various integrin ligand coated plates and the ability of EC to adhere to the distinct extracellular matrix components was tested. HUVECs transfected with randomized non-specific siRNA oligos were used as internal controls. Representative images and quantitative results are shown in Figure 2A and B, respectively. Inhibition of expression of the β1 subunit reduced EC adhesion to both collagen and laminin, but not to vitronectin. Silencing of the β3 subunit significantly inhibited EC adhesion to vitronectin, but not to collagen or laminin. Inhibition of expression of the β5 subunit only partially (∼50%) inhibited cell attachment to vitronectin and had no substantial effect on adhesion to other tested extracellular matrix components. These observations indicate that sister integrins did not functionally compensate for the suppressed expression of a specific integrin that was silenced in HUVECs.
Figure 2.

Specificity of integrins in reorganization on distinct ECM ligands. (A and B) HUVECs were transfected with control siRNA or siRNA specific for β1, β3 or β5 integrin. Wells of microtiter plates were coated with vitronectin, collagen or laminin-1 and were incubated overnight at 4°C. siRNA-transfected EC were harvested and resuspended in serum-free media at 5 × 105 cells/ml. The cell suspension (100 μL) was plated on the microtiter wells coated with integrin ligand. After incubation at 37°C for 45 min, wells were gently washed three times with DMEM and photographs were taken (A). The numbers of attached cells per field were counted and untransfected cells adherent to the individual ECM ligands were assigned a value of 100% (B). Asterisks indicate significant difference over control (p < 0.0046).
Vitronectin (αvβ3) and collagen (α5β1) receptors modulate endothelial cell migration
Extracellular matrix provides critical support for proliferating vascular endothelium through adhesive interactions with endothelial cell surface integrins.30 Extracellular matrix also provides the scaffold essential for maintaining the organization of vascular endothelial cells in blood vessels. Endothelial cell adhesion to extracellular matrix is required for endothelial cell proliferation, migration and morphogenesis.31,32 Integrin-mediated migration of endothelial cells also plays a crucial role in the vascular remodeling involved in angiogenesis, embryonic vasculogenesis and re-endothelialization in arteries following angioplasty. To examine which of the endothelial integrins trigger endothelial cell migration, HUVECs were transfected with various β-integrin specific siRNAs and these cells were plated on various extracellular matrix components. HUVECs transfected with randomized non-specific siRNA oligos were used as internal control. A wound was created across the cell monolayer by scraping away a swath of cells and the extent of wound healing due to the transfected EC was measured after 12 hours. Representative images are shown in Figure 3A. Percentages of wound recovery were quantified (Fig. 3B). Control EC were able to completely close the wound on vitronectin and were assigned a value of 100%. EC plated on collagen and laminin shown 85% and 30% recovery, respectively. EC transfected with siRNA targeting the β1 subunit were almost completely unable to heal wounds on collagen, but were not substantially different from control EC on vitronectin and laminin. Silencing of β3 and β5 subunits reduced wound recovery on vitronectin by 50 and 30%, respectively, without any effect on collagen and laminin. The results of these assays demonstrate that the critical integrin subunits for regulation of endothelial cell communication with extracellular matrix are β3 and β1. As above, no compensatory effects were observed.
Figure 3.
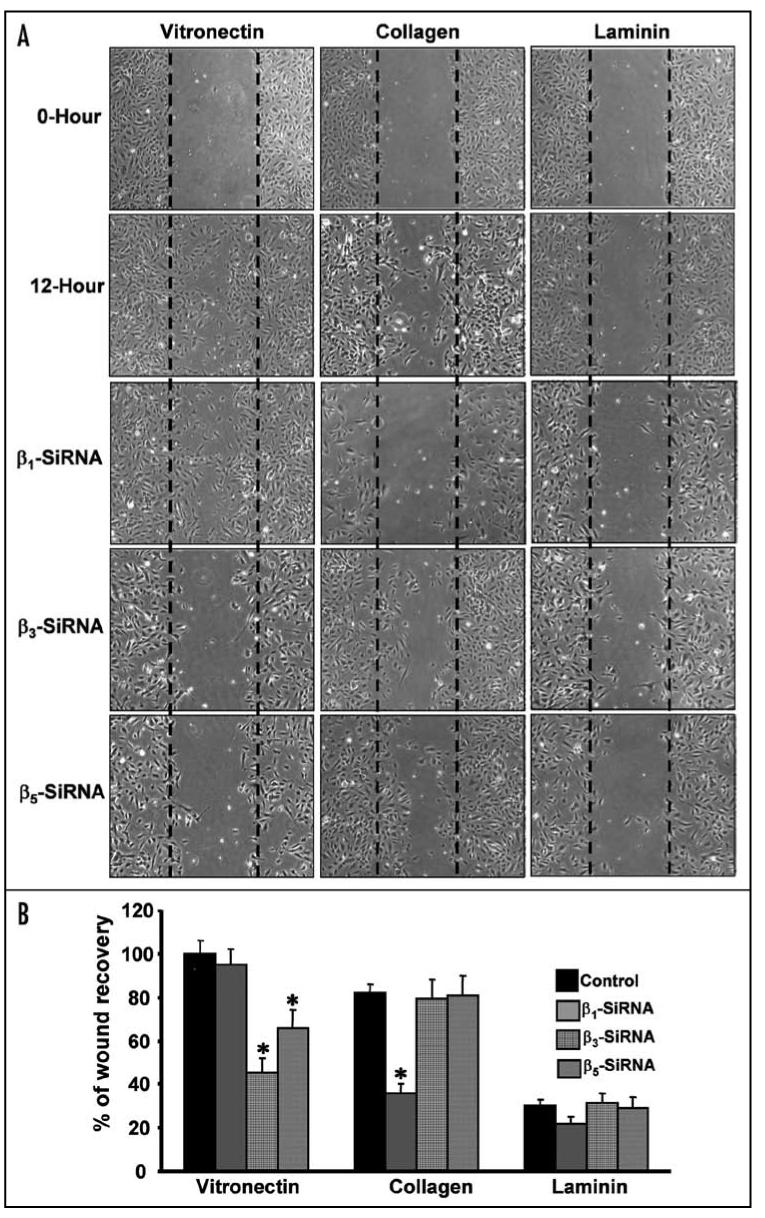
Vitronectin (αvβ3) and collagen (α5β1) receptors regulate endothelial cell migration. (A and B) HUVECs were transfected with control siRNA or siRNA specific for β1, β3 or β5 integrin. These cells were grown to confluence on 12-well plates precoated with individual integrin ligands. Cells were serum starved then wounded across the cell monolayer by scraping away a swath of cells. Wells were rinsed twice with sterile PBS and further cultured in DMEM medium containing 2% FBS. Sites were photographed immediately after wounding (zero hour) and 12 h later using a phase contrast microscope (A). Images were acquired using a Leica DMIRB phase contrast microscope, 5X objective, and a Micromax RTE/CCD-1300-V-HS camera. The mean wound area recovery by nontransfected endothelial cells on vitronectin for 12 hours was designated as 100% and the relative % of wound recovery for siRNA-transfected EC were determined (B). Asterisks indicate significant difference over control (p < 0.0058).
αvβ3 integrin is crucial for endothelial cell morphogenesis
During angiogenesis, proliferating and migrating endothelial cells organize to form three-dimensional capillary networks.33 This processes begins with the transition of endothelial cells into a spindle-shaped morphology. This is followed by endothelium alignment and connection into solid, multicellular, precapillary cord-like structures that form an integrated polygonal network.34 During this vascular morphogenesis, extracellular matrix serves as an adhesive support and, through interaction with integrins, provides crucial signaling to regulate endothelial cell shape and contractility. To examine which integrin is crucial during endothelial cell morphogenesis, HUVECs were transfected with various β-integrin specific siRNAs and the angiogenic properties of the EC were assessed using a capillary tube formation assay on Matrigel. HUVECs transfected with randomized non-specific siRNA oligos were used as internal control. Capillary tube forming ability of unstimulated as well as VEGF-stimulated cells was tested. Silencing of the β1 subunit affected capillary formation in the absence as well as in the presence of VEGF. As shown in Figure 4A, the regularity of the typical honeycomb-like pattern was disturbed resulting in incomplete connections between cellular cords. Among the three different siRNA transfections, knockdown of the β3 subunit produced the most severe inhibitory effect on capillary growth; silencing β3 significantly reduced the formation of cellular cords by these EC both in the absence and presence of VEGF (Fig. 4A). Down regulation of the β5 integrin subunit incompletely but substantially impaired capillary growth of unstimulated EC and, to a lesser extent, of VEGF-stimulated cells. The quantitative aspects of the capillary growth of all three EC lines are shown in Figure 4B. Thus, it appears that knockdown of the β3 subunit was the most effective in inhibiting an angiogenic response in vitro. Therefore, we further focused on β3 integrin, which forms a complex exclusively with the αv subunit on EC to produce the heterodimer αvβ3.
Figure 4.
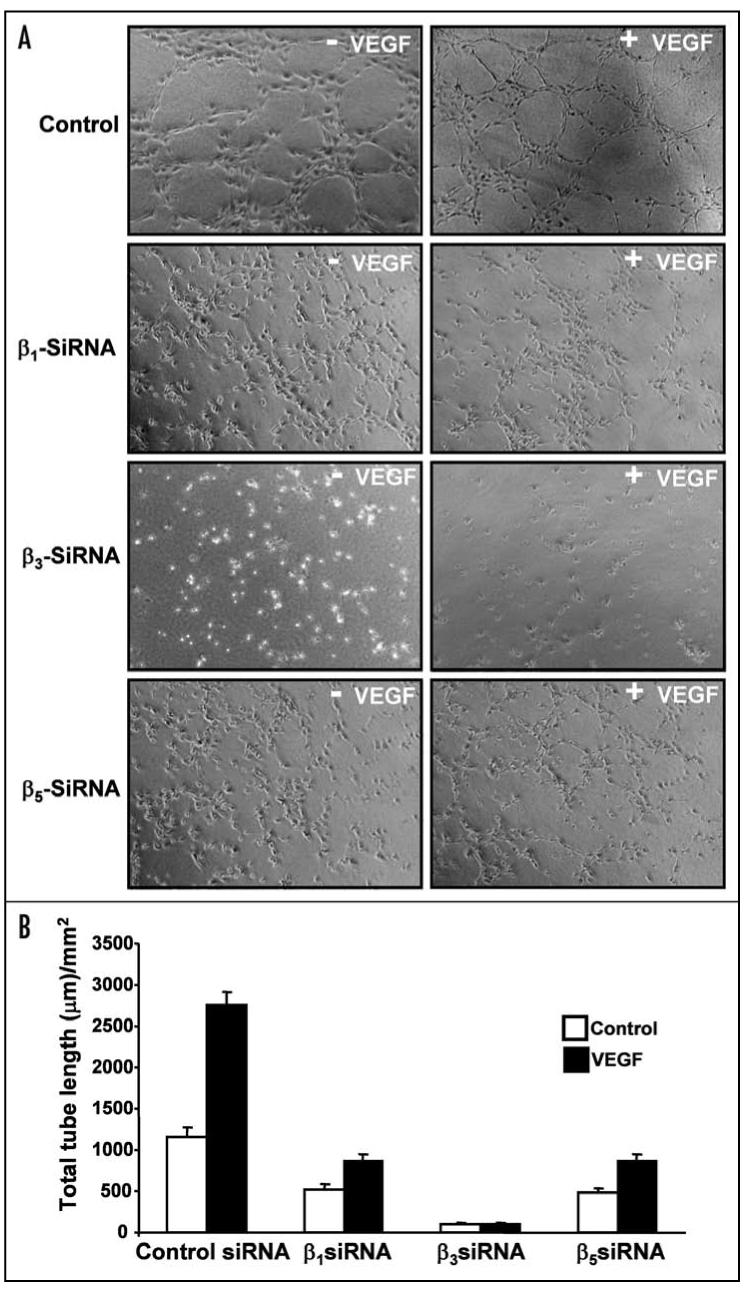
β3 integrin regulates endothelial cell morphogenesis in vitro. (A and B) HUVECs were transfected with control siRNA or siRNA specific for β1, β3 or β5 integrin. Cells were transferred to Matrigel coated plates and further incubated at 37°C for 8 h with or without 20 ng/mL VEGF. Endothelial capillary tubes formed in Matrigel were observed using an inverted phase contrast microscope and photographs were taken (A). Mean length of tubes from five random fields were measured using Image-Pro software (B).
αvβ3 integrin affinity modulation and association with VEGFR-2
Integrin affinity relates to conformational modifications in the integrin heterodimer and strength of ligand binding. Usually, non-integrin receptors cause alterations in the integrin cytoplasmic domain, ultimately modulating the integrin activation state.35 Several receptor tyrosine kinases (RTKs), G-protein coupled receptors and cytokines have been shown to modulate the integrin activation state.9,36 To study the synergism between αvβ3 integrin and RTKs such as VEGFR-2, FGF receptor and EGF receptor on cell surfaces, HUVECs, NIH-3T3 and PC-3 cells were induced with VEGF, FGF and EGF, respectively, and integrin affinity was estimated (Fig. 5A-C). The genetically engineered antibody WOW-1 was used as a probe to detect αvβ3 in its high affinity state on the cell surface. Stimulation with human VEGF-A165 increased WOW-1 binding to EC by 6-fold compared to resting HUVECs, as measured by FACS analysis (Fig. 5A). Importantly, the mature form of VEGF-D, VEGF-DΔNΔC, known to be specific for VEGFR-2 on EC of blood vessel origin, was also a potent inducer of WOW-1 Fab binding, indicating that VEGFR-2 but not VEGFR-1 is primarily the receptor mediating αvβ3 integrin activation. Similarly, bFGF induced a 3-fold increase in WOW-1 binding in NIH-3T3 cells (Fig. 5B) and EGF induced approximately a 4-fold increase in WOW-1 binding in PC-3 cells (Fig. 5C). These results indicated that growth factors other than VEGF also induce αvβ3 integrin activation in various cell types.
Figure 5.
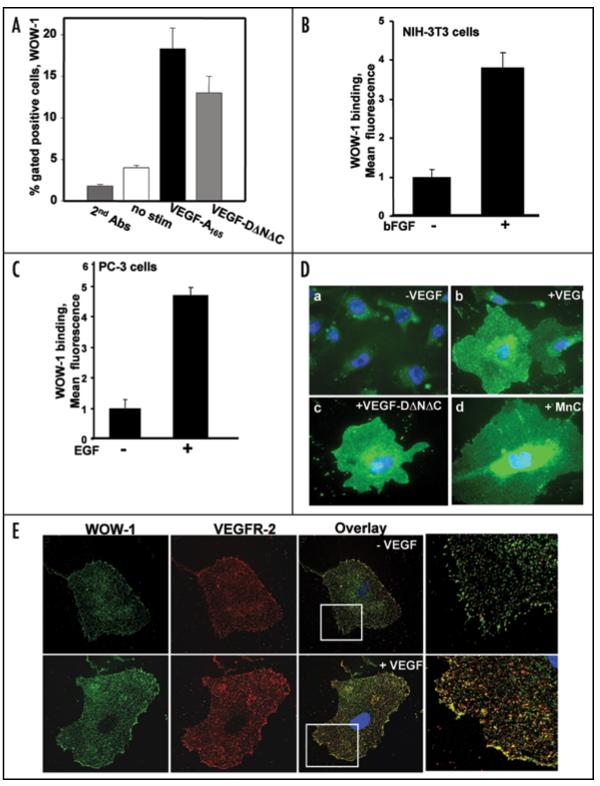
Activated αvβ3 integrin co-localizes with VEGFR-2 on endothelial cells. To evaluate growth factor-induced activation of αvβ3 integrin, semiconfluent, serum starved HUVECs were induced with VEGF-A165 (A), NIH-3T3 cells were induced with bFGF (Fig. 5B) and PC-3 cells were induced with EGF (Fig. 5C). These cells were further incubated with WOW-1 Fab and goat anti-mouse IgG labeled with Alexa Fluor 488. Fixed cells were then analyzed by flow cytometry. (D) HUVECs were grown on -gelatin-coated glass coverslips. These cells were serum starved and induced with VEGF-A165, VEGF-DΔNΔC or MnCl2 in the presence of WOW-1 Fab. Cells were washed and further incubated with goat anti-mouse IgG labeled with Alexa Fluor 488. Cells were fixed, observed via fluorescence microscopy and photographs were taken. (E) αvβ3 integrin and VEGFR-2 co-localize on endothelial cells. HUVECs were serum-starved overnight then stimulated with 20 ng/mL VEGF for 5 minutes. These cells were stained with WOW-1 and anti-VEGFR-2, followed by the incubation with goat anti-mouse IgG labeled with Alexa Fluor 488 and goat anti-rabbit IgG conjugated with Alexa Fluor 594. Without a stimulatory signal (upper), very little co-localization of β3 integrin and VEGFR-2 was observed. Upon VEGF stimulation (lower), the affinity of αvβ3 increases and co-localizes with VEGFR-2.
The results of WOW-1 binding to HUVECs in a pre-confluent monolayer in response to VEGF-A165, VEGF-DΔNΔC or Mn2+ are shown in Figure 5D. All treatments induced WOW-1 binding and binding was most evident at the cellular borders. To further investigate whether activated αvβ3 integrin forms a complex with VEGFR-2, HUVECs were induced with VEGF and stained with WOW-1 and VEGFR-2 antibody. Surprisingly, on VEGF-stimulated EC activated αvβ3 co-localized with VEGFR-2 at cell borders (Fig. 5E). Thus, our results establish that VEGFR-2-dependent activation of αvβ3 integrin leads to macromolecular interaction between αvβ3 integrin and VEGF receptor-2 in endothelial cells.
Activated αvβ3 integrin is an indicator of enhanced tumor vasculature
To further characterize αvβ3 integrin interaction with VEGFR-2 on endothelial cells of proliferating blood vessels, we utilized biopsy specimens of human prostate carcinomas that posses high expression of VEGF. Triple staining was performed on serial tissue sections of the prostate tumors with WOW-1 antibodies (to localize activated αvβ3), with antibodies against CD31 (to identify EC), and with antibodies to VEGFR-2 (to localize VEGFR-2) (Fig. 6A). As in the in vitro studies presented above, activated αvβ3 co-localized with VEGFR-2 in the vasculature of prostate carcinomas (Fig. 6A). The distribution of VEGFR-2 and WOW-1 staining was virtually identical in most carcinoma samples (Fig. 6B). This result demonstrates that activated αvβ3 integrin co-localizes with VEGFR-2 on endothelial cells of proliferating blood vessels. To further examine the VEGFR-2 association with the active form of αvβ3 integrin on endothelial cells of angiogenic blood vessels, robust angiogenesis was induced by left femoral artery ligation as described in Experimental Procedures. At the end of week 4 left leg muscle tissues were collected and immunohistochemical analyses were performed. Ischemia-induced angiogenic blood vessels showed intense staining for the activated form of integrin by WOW-1 Fab fragments and endothelial cells of the blood vessels were co-localized using anti-CD-31 antibody (Fig. 6C). These results clearly indicate that endothelial cells of angiogenic blood vessels in ischemic tissues were highly positive for WOW-1 staining, identifying the presence of activated αvβ3 integrin on these endothelial cells. To further investigate whether activated αvβ3 integrin is associated with VEGFR-2 on endothelial cells of angiogenic blood vessels in ischemic tissue, frozen tissue sections were stained for activated αvβ3 integrin using WOW-1 Fab fragments and rat anti-mouse VEGFR-2 antibody (Fig. 6D). The staining patterns demonstrate complete αvβ3 integrin co-localization with VEGFR-2 on endothelial cells of angiogenic blood vessels.
Figure 6.
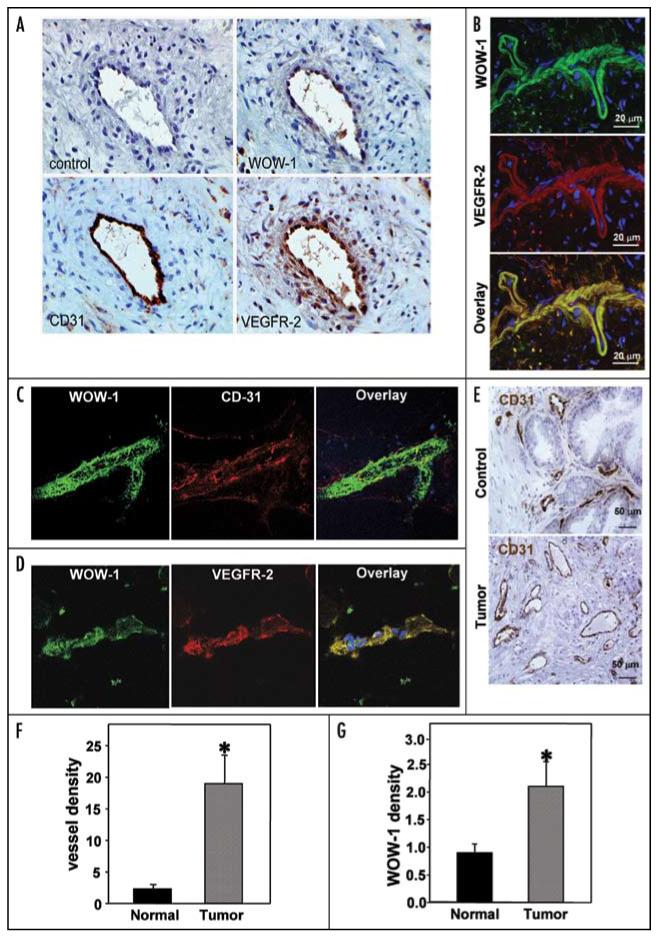
αvβ3 integrin activation level serve as a marker of tumor angiogenesis. (A) Activated αvβ3 integrin co-localizes with VEGFR-2 on endothelial cells of proliferating blood vessels. Parallel prostate tumor tissue sections were cut and stained for WOW-1 (activated αvβ3 integrin), CD31 (endothelial cell marker) and VEGFR-2. Blood vessels (revealed by CD31 staining) were positively stained for both WOW-1 and VEGFR-2, indicating the co-localization of activated αvβ3 with VEGFR-2 in tumor vasculature. (B) Frozen parallel prostate tumor sections were stained for activated αvβ3 integrin (WOW-1 Fab) and VEGFR-2. Tissue sections were analyzed with a confocal microscope and photographs were taken. (C and D) Association of VEGFR-2 with the active form of αvβ3 integrin on endothelial cells of angiogenic blood vessels. Robust angiogenesis was induced by left femoral artery ligation as described in Experimental Procedures and immunohistochemical analyses were performed. Ischemia-induced angiogenic blood vessels show intense staining for the activated form integrin as stained by WOW-1 Fab and endothelial cells on the blood vessels using anti-CD-31 and VEGFR-2 antibody (C and D). (E-G) Normal prostate tissue and prostate tumor sections were stained for CD-31 and WOW-1 (E). Vascular density was increased at least 6-fold in prostate tumors compared to normal prostate tissue (F). Vascular density was positively correlated with the density of WOW-1-positive vasculature in the two tissue samples (G). Asterisks indicate significant difference over normal tissue.
Next, we performed WOW-1 staining of normal prostate tissues for comparison with prostate carcinomas. Based on CD31 staining, the prostate carcinoma tissue is characterized by a 6-fold increase in vascular density compared to normal tissue (Fig. 6E and F). The vessels of normal prostate tissue were poorly stained with the WOW-1 antibody, whereas prostate tumor vessels were clearly WOW-1-positive, suggesting that activation of αvβ3 might serve as a marker of pathological angiogenesis. Quantification of WOW-1 density revealed a 2.7-fold increase in αvβ3 activation in prostate carcinomas compared to normal prostate tissues (Fig. 6G). Thus, quantification of αvβ3 integrin could be used as index for tumor angiogenesis. Thus, our in vivo analyses provide evidence that αvβ3 is activated on EC at sites of angiogenesis. Co-localization of activated αvβ3 with VEGFR-2 indicates possible cross-talk between these receptors not only in vitro but also in vivo.
VEGFR-2/β3 integrin cross-talk in EC
It was reported that knockouts of β3 and β5 subunits in mice resulted in upregulation of the expression level of VEGFR-2 on EC.37 However, as shown in Figure 7A-C, silencing the expression of β1, β3 or β5 integrin using siRNA had no effect on VEGFR-2 levels in EC. Thus, it is most likely that upregulation of VEGFR-2 in β3-null mice occurs during development as a compensatory mechanism due to the result of prolonged downregulation of αvβ3 integrin.38,39 We also assessed how inhibition of expression of β subunits affected VEGFR-2 activation in EC. In control EC, treatment with VEGF induced substantial phosphorylation of VEGFR-2 and this response was not affected by silencing of β1 integrin (Fig. 7D). In contrast, ablation of β3 expression by a specific siRNA resulted in ∼3-fold decrease of VEGFR-2 phosphorylation in response to VEGF. Silencing of the β5 subunit resulted in a modest decrease of phosphorylation of VEGFR-2 (Fig. 7D). Thus, it appears that β3 integrin, but not β1 or β5, influenced VEGFR-2 activation on EC. In order to confirm integrin-VEGFR interactions, detailed biochemical studies were undertaken. Accordingly, HUVECs were grown on vitronectin-coated plastic surface and stimulated with 20 ng/ml VEGF for 5 min. To study the interaction between β3 integrin and VEGF receptors, cell lysates were subjected to immunoprecipitation with anti-β3 integrin antibody and immunoblotted with anti-VEGFR-1, anti-VEGFR-2 and anti-VEGFR-3 antibodies separately (Fig. 7E). The blots indicate that VEGF induced interaction between VEGFR-2 and β3 integrin but not with VEGFR-1 or VEGFR-3.
Figure 7.

VEGF-induced VEGFR-2 phosphorylation is subordinate to αvβ3 integrin activation status. Effect of integrin knockdown on VEGFR-2 expression was evaluated by transfecting HUVECs with siRNA specific for (A) β1, (B) β3 or (C) β5 integrin. Cell lysates were analyzed for expression of VEGFR-2. Densitometry analysis was performed and results are shown as bar graphs (lower). (D) αvβ3 integrin activation dependent phosphorylation of VEGFR-2. HUVECs were transfected with β1, β3 or β5 integrin-specific siRNA and induced with 20 ng/mL VEGF. Cell lysates were analyzed for phosphorylation of VEGFR-2 using specific antibody. Densitometry analysis was performed and results are shown as bar graphs (lower D). (E-G) αvβ3 integrin and VEGF receptors interactions. HUVECs were stimulated with 20 ng/ml VEGF for 5 min. Cell lysate were subjected to immunoprecipitation with anti-β3 integrin antibody and immunoblotted with anti-VEGFR-1, anti-VEGFR-2 and anti-VEGFR-3 antibodies separately (E). HUVEC cell lysates were used as positive control (lane 1). Endothelial cells from wild type and β3 knockout mice were lysed and subjected to immunoprecipitation using anti-VEGFR-2 antibody and immunoblotted with anti-β1 integrin, anti-β3 integrin, anti-β5 integrin and anti-β6 integrin antibody (F). HUVECs were incubated with 1mM EDTA (lane 3) or 800 nM of SU1498 (lane 4), a potent VEGFR-2 specific inhibitor. These cells were stimulated with VEGF in presence of WOW-1 Fab fragments and lysed. Cell lysates subjected to immunoprecipitation with anti-VEGFR-2 antibody and immunoblotted with anti-β3 integrin or anti-His-Probe (recognizes WOW-1) antibody (G). (H) HUVECs were incubated with αvβ3 integrin-activating antibody (Libs-1, AP-7.3, CRC-54) or β3 integrin blocking antibody. These cells were induced with VEGF for 5 min and cell lysates were analyzed for phosphorylation of VEGFR-2 using specific antibody. Densitometry analysis was performed and results are shown as bar graphs (lower H).
To further examine whether VEGFR-2 is associated with other β integrin subunits in endothelial cells derived from β3 null mice the following experiment was performed. Endothelial cells from wild type and β3 knockout mice were from subcutaneous Matrigel implants as described in Experimental Procedures. Cell lysates were subjected to immunoprecipitation using anti-VEGFR-2 antibody and immunoblotted with anti-β1 integrin, anti-β3 integrin, anti-β5 integrin and anti-β6 integrin antibodies (Fig. 7F). The results indicate that β3 integrin is the major integrin associated with VEGFR-2 on actively proliferating endothelial cells of angiogenic blood vessels. A small fraction of β1 integrin was associated with VEGFR-2 in both wild type and β3 knockout endothelial cells. However, no traces of β5 or β6 integrins were associated with VEGFR-2 in wild type or β3 knockout endothelial cells. From this experiment it is clear that β3 is the major integrin associated with VEGFR-2 on proliferating endothelial cells. To further investigate whether VEGFR-2 binds to the activated form of β3 integrin (or even the inactive form), HUVECs were serum starved then incubated with 1 mM EDTA (lane 3), which is known to prevent integrins from achieving their active conformation, or 800 nM SU1498 (lane 4), a potent specific VEGFR-2 inhibitor. These cells were stimulated with 20 ng/ml VEGF in the presence of WOW-1 Fab fragments then lysed in ice-cold immunoprecipitation lysis buffer. Cell lysates were subjected to immunoprecipitation with anti-VEGFR-2 antibody and immunoblotted with anti-β3 integrin or anti-His-Probe (which recognizes WOW-1) antibody (Fig. 7G). Results indicate that no basal level of VEGFR-2 and β3 integrin interaction was detected (lane 1). VEGF stimulation induced VEGFR-2 and β3 integrin interaction as well as β3 integrin activation as evident by WOW-1 binding (lane 2). EDTA, known to prevent β3 integrin activation (lane 3), also prevented β3 integrin and VEGFR-2 interaction even in the presence of VEGF (lane 3). As expected, VEGFR-2 inhibitor SU1498 significantly reduced WOW-1 binding as well as VEGFR-2 β3 integrin interaction in HUVECs (Lane 4). These results clearly indicated that only the activated form of β3 integrin interacts with VEGFR-2, not the inactive forms.
Next, we assessed whether activation of αvβ3 by externally added activating antibodies influenced activation of VEGFR-2 in response to VEGF. As shown in Figure 7H, three different β3-specific activating antibodies, LIBS-1, AP-7.3 and CRC-54 augmented VEGFR-2 phosphorylation in VEGF-treated EC. In contrast, antibodies that blocked αvβ3 produced a 2-fold inhibition of VEGFR-2 phosphorylation. Thus, not only the expression but also activity of αvβ3 integrin appears to control VEGF-induced phosphorylation of VEGFR-2, demonstrating a truly functional cross-talk between these two receptors.
Discussion
Endothelial cells express a relatively wide range of integrin receptors, which permits interactions with numerous extracellular matrix ligands. Although the ligand recognition profile of each integrin is rather unique, there is a significant degree of redundancy and functional overlap. This is the case for αvβ3 and αvβ5, since both integrins are considered to be the primary receptors for vitronectin.40 However, our data indicate that of the several integrin receptors on EC, αvβ3 has a unique function in angiogenesis which cannot be duplicated by other integrins. First, the affinity of αvβ3 on EC can be rapidly modulated by treatment with VEGF, resulting in increased adhesive and migratory responses of EC. Importantly, we were able to document that αvβ3 is activated in the endothelium of highly vascularized tumors as compared to normal tissues. Second, the proangiogenic role of αvβ3 integrin is not limited to interactions with vitronectin and other substrates; it is tightly linked to its ability to augment activation of VEGFR-2. We demonstrated that activated αvβ3 is co-localized with VEGFR-2 not only in cultured EC, but also on endothelial cells of proliferating blood vessels at the sites of pathological angiogenesis. Several other studies have indicated the unique nature of association between these two receptors.41-46 The functional consequences of such association are of supreme importance, since VEGFR-2 mediates the majority of VEGF-induced responses, including EC proliferation, migration and permeability.47 The key role of the VEGF/VEGFR-2 system in angiogenesis is also underscored by the severe impairment of vasculature development leading to early embryonic lethality in mice lacking either of those genes.8,48,49 Thus, it appears that the ability of αvβ3 to regulate VEGFR-2 function makes this integrin a key player in VEGF-induced responses of endothelial cells.
Indeed, it has been previously demonstrated that the blockade of αvβ3 integrin with monoclonal antibodies or ligand antagonists leads to blunted blood vessel formation in the context of several models of pathological angiogenesis, including cancer, arthritis and ischemic retinopathy.21 However, vascular development appears to be normal in mice lacking β3 integrins and pathological angiogenesis is enhanced in β3, β5 or β3/β5 null mice.22 The major difference between the siRNA-based knockdown approach used in our study and genetic ablation in knockout animals is centered on the transient nature of siRNA-based silencing versus permanent and complete loss of protein expression in development. If the function of a targeted protein cannot be spared during development, such a loss will ultimately result in embryonic lethality. In contrast to its prominent role in development, VEGF does not seem to be absolutely required for the normal physiology of adult organisms, since neutralization of circulating VEGF has proven to be safe for patients.50 Likewise, based on results from β3 knockout, this integrin is not likely to be a crucial player during development, but it does mediate pathological neovascularization in adults.37 Thus, it is not surprising that its loss does not cause lethality but rather triggers compensatory upregulation of other pro-angiogenic molecules. Moreover, the lack of involvement of β3 in normal development and physiology makes it a very attractive therapeutic target for a number of diseases associated with pathological angiogenesis.
We demonstrated that αvβ3 integrin on EC exists and functions as part of a complex with VEGFR-2. This complex is formed not only upon cell adhesion to vitronectin, as was demonstrated previously,10 it can be triggered by VEGF activation of VEGFR-2 or, alternatively, by exogenous activation of αvβ3 even in the absence of adhesive ligand. Moreover, in vivo VEGFR2 seems to be associated with the activated form of αvβ3 integrin. Based on these results, one can predict that the physical loss of one part of this complex might provoke upregulation of its other part. It is possible that the physical presence of αvβ3 on the EC surface might also function to bind and sequester signaling molecules and otherwise enhance VEGFR-2-stimulated responses. Our current and previously published data10,51 in conjunction with the results of β3 knockout studies emphasize the intimate connection between αvβ3 and VEGFR-2. The differential consequences of short term vs. long term inhibition of αvβ3 may have relevance for pathological angiogenesis treatments with αvβ3 inhibitors, as prolonged therapy may trigger the same compensatory responses observed in β3 null mice, leading to treatment failure.
This study aimed to assess the regulatory functions of individual β subunits of the major endothelial cell surface integrins in VEGF-induced angiogenic responses. The major findings of this analysis are as follows: (1) Silencing of expression of individual β subunits resulted in downregulation of EC adhesion and migration on the corresponding primary ligand, while no effects on the recognition of other ligands were detected. (2) Among the three β subunits expressed on EC, loss of β3 had the most dramatic inhibitory effect on capillary growth of EC in response to VEGF. (3) αvβ3 activation triggered by VEGF/VEGFR-2 was detected in EC suspension as well as in a monolayer; on the monolayer activated αvβ3 was co-localized with VEGFR-2. (4) In vivo, activated αvβ3 co-localized with VEGFR-2 on endothelium. (5) Activation of αvβ3 in vivo was substantially increased in highly-vascularized tumors as compared to normal tissues. (6) Silencing of expression of β1, β3 or β5 in EC did not affect expression levels of VEGFR-2. However, inhibition of expression of β3, but not β1 or β5, resulted in inhibition of VEGFR-2 phosphorylation in response to VEGF. (7) Exogenous activation of αvβ3 integrin stimulated, and its blockade inhibited, VEGF-dependent phosphorylation of VEGFR-2. Together, these results show the prominent role of αvβ3 integrin in the functional cross-talk with VEGF/VEGFR-2 in a context of angiogenic endothelial functions.
Materials and Methods
Materials
Rabbit polyclonal anti-VEGFR-1, anti-VEGFR-2, anti-VEGFR-3, anti-β1-integrin, anti-β3-integrin, anti-β5-integrin, anti-β6-integrin, anti-His-Probe, mouse monoclonal anti-phospho tyrosine (PY20 and PY99) antibodies, and β1, β3 and β5 integrin-specific siRNAs were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-VEGFR-2 and anti-phospho VEGFR-2 were from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-β3-integrin blocking antibody was from Chemicon International, Inc. (Temecula, CA). Anti-CD31 antibody was obtained from DAKO (Kyoto, Japan). Mouse monoclonal anti-β3-integrin activating antibodies were generated in our laboratory. Purified collagen, laminin, vitronectin, bFGF, EGF and VEGF were purchased from R&D Systems (Minneapolis, MN). VEGFR-2 inhibitor SU1498 was obtained from Calbiochem (La Jolla, CA). Matrigel was obtained from BD Biosciences (San Jose, CA). The HUVEC nucleofector kit was obtained from Amaxa Biosystems (Gaithersburg, MD). Alexa Fluor 488-conjugated goat anti-rabbit, goat anti-mouse IgG and TRITC-conjugated goat anti-rabbit IgG were from Invitrogen (Carlsbad, CA). Purified WOW-1 Fab was provided by Dr. S.J. Shattil, The Scripps Research Institute, La Jolla, CA. All other chemicals were analytical grade.
Cell culture and transfection
The mouse embryonic fibroblast cell line NIH-3T3 and human prostate adenocarcinoma cell line PC-3 were obtained from ATCC and maintained according to their instructions. Human umbilical cord vein endothelial cells (HUVEC) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 90 μg/mL heparin sulfate, 90 μg/mL endothelial cell growth factor, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were used for experiments between the second and fifth passages. HUVECs were transiently transfected using the HUVEC nucleofector kit according to the manufacturer’s instructions. Forty-eight hr after transfection, cells were serum starved and used for experiments. Cell surface expressions of integrins were detected as described previously.25
Subcutaneous matrigel plug implantation and ischemic injury induced angiogenesis
β-3 integrin specific gene knockout mice were purchased from Jackson Laboratory and maintained on a C57/Bl6 background. Six- to eight-week old wild-type (WT) and β-3 gene knockout mice were used in this study. We performed all procedures according to protocols approved by the Cleveland Clinic Foundation Institutional Animal Care and Use Committee. Wild type and β-3 knockout mice were subcutaneously injected with Matrigel containing 40 ng/ml VEGF and 60 units/ml heparin. At the end of 14 days the subcutaneous implants of Matrigel were surgically removed from both wild type and β-3 knockout mice and preserved on ice before further processing. Matrigel plugs were enzymatically digested using collagenase/dispase solution at 4°C. The resultant clear solutions were centrifuged and cell pellets were used for experiments.
To initiate ischemic injury induced angiogenesis, C57BL/6 mice were dosed with an intraperitoneal injection of anesthesia (79.5 mg/kg ketamine, 9.1 mg/kg xylazine) and the proximal left femoral artery was ligated at two points 3mm apart. The artery between ligatures was excised. Blood flow was measured by the Moor Laser Doppler Imager Control system before and directly after surgery and then 3 days, 2 weeks and 4 weeks after surgery.
Cell adhesion assay
The cell adhesion assay was performed as described previously.9 HUVECs were detached from the tissue culture flasks using 20 mM EDTA. Cells were washed twice with sterile phosphate buffered saline (PBS) and re-suspended in serum-free DMEM. The cell suspensions were added to integrin ligand-coated wells and placed in a humidified incubator for 45 min. The wells were gently washed three times with DMEM and photographs were taken. The numbers of attached and spread cells per field were counted.
Endothelial wound healing assay
HUVECs were grown to confluence in 12 well plates precoated with various integrin ligands. Cells were serum starved for 4 h and then a wound was created by a pipette tip. Wells were rinsed twice with sterile PBS to remove wound-derived loose and dislodged cells and further cultured in DMEM medium containing 2% FBS. Images were recorded immediately after wounding (time zero) and 12 h later. Cell migration was quantified using image analysis of five randomly selected fields of denuded area. The mean wound area is expressed as percent of recovery (% R) from three identically treated plates using the equation % R = [1-(Tt/T0)] × 100, where T0 is the wounded area at 0 h and Tt is the wounded area after 12 h.
WOW-1 binding assay
WOW-1 Fab binding assay was performed as described previously.9 Semiconfluent HUVECs were serum starved for 4 h and further stimulated with 20 ng/mL VEGF-A165 or VEGFDΔNΔC. WOW-1 Fab was added to a final concentration of 30 μg/mL, followed by addition of FITC-conjugated goat antimouse IgG at 10 μg/mL. After 30 min cells were fixed with 3.7% formaldehyde in PBS for 15 min, washed twice with PBS, and fluorescence-activated cell sorting (FACS) was performed using a FACS Calibur (Becton Dickinson, San Jose, CA) and data were analyzed using CellQuest software.
Tube formation assay
The formation of vascular tube-like structures by HUVECs was assessed on a basement membrane matrix preparation. Twelve-well plates were coated with 0.5 mL of Matrigel according to the manufacturer’s instructions. HUVECs transfected with various β integrin-specific siRNAs while cells transfected with randomized oligos were used as internal controls. Cells were detached from tissue culture flasks using 20 mM EDTA in PBS, washed twice with sterile PBS and seeded on Matrigel-coated plates. Medium with or without 20 ng/mL VEGF was added and cells were further incubated at 37°C for 8 h. Tube formation was observed using an inverted phase contrast microscope (Leica, Wetzlar, Germany) and photographs were taken. Using Image-Pro software (Media Cybernetics, Silver Spring, MD), the degree of tube formation was quantified by measuring the length of tubes in three random fields.
Immunohistochemistry and immunocytochemistry analysis
Immunohistochemical analysis of prostate tumor tissues were performed as described previously.26 To examine the activation status of integrins on endothelial cells of angiogenic blood vessels, C57BL/6 mice were anesthetized with an intraperitoneal injection of anesthesia (79.5 mg/kg ketamine, 9.1 mg/kg xylazine). The proximal left femoral artery was ligated at two points 3 mm apart and the artery between ligatures was excised. Blood flow was measured by the Moor Laser Doppler Imager Control system before and directly after surgery and then at 3 days, 2 weeks and 4 weeks after surgery. At the end of 4 weeks the mice were anesthetized and muscle tissues were collected, embedded in O.C.T. medium and snap frozen in liquid nitrogen. Tissue sections were stained for activated integrin using WOW-1 Fab fragments and for endothelial cells on the blood vessels using anti-CD-31 antibody.
To study integrin activation status, HUVECs were grown to a monolayer on glass slides and then treated with 1 mM MnCl2 or 20 ng/mL VEGF for 10 min. These cells were further incubated with WOW-1 antibody for an additional 30 min, then were fixed with paraformaldehyde for 10 min, blocked with 5% bovine serum albumin for 30 min, and incubated with FITC-conjugated anti-mouse IgG. The cells were then washed, mounted with coverslips, and analyzed under a fluorescence microscope (Leica). Alternatively, cells were further incubated with anti-VEGFR-2 antibody and incubated with TRITC-conjugated anti-rabbit IgG. These cells were washed, mounted and analyzed by confocal microscopy (Leica).
Western blot analysis
HUVECs were lysed following the experiments using lysis buffer composed of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM iodoacetamide, 2 mM phenylmethylsulfonyl fluoride, 2 mM EDTA, 10 mM NaF, 10 mM Na2P2O7, 10 μg/mL leupeptin, 4 μg/mL pepstatin and 0.1 units/mL aprotinin. Cell lysates were centrifuged at 13,000 × g for 10 min at 4°C. Supernatants were collected and assayed for protein concentration using the Bio-Rad protein assay method (Hercules, CA). Cell lysates were denatured using Laemmli sample buffer and proteins were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis then probed with the indicated antibody. When appropriate, nitrocellulose membranes were stripped and blotted according to the manufacturer’s instructions.
Acknowledgements
We acknowledge financial support from the US National Institutes of Health (HL071625 and HL073311 to TVB) and American Heart Association (0625271B to GHM). We thank Judy Drazba and scientists at the Image Core Facility at the Cleveland Clinic for help in image analysis and Lori Mavrakis and Angela Money for providing HUVECs. The authors have no conflicting financial interests.
Abbreviations
- DMEM
dulbecco’s modified eagle’s medium
- EC
endothelial cells
- ECM
extracellular matrix
- FACS
fluorescence activated cell sorting
- GF
growth factor
- GFR
growth factor receptors
- HUVECs
human umbilical vein endothelial cells
- mAbs
monoclonal antibodies
- PBS
phosphate buffered saline
- RTKS
receptor tyrosine kinases
- siRNA
small interfering RNA
- VEGF
vascular endothelial growth factor
- VEGFR-2
vascular endothelial growth factor receptor-2
References
- 1.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–8. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 2.Arnaout MA, Goodman SL, Xiong JP. Coming to grips with integrin binding to ligands. Curr Opin Cell Biol. 2002;14:641–51. doi: 10.1016/s0955-0674(02)00371-x. [DOI] [PubMed] [Google Scholar]
- 3.Echarri A, Del Pozo MA. Caveolae internalization regulates integrin-dependent signaling pathways. Cell Cycle. 2006;5:2179–82. doi: 10.4161/cc.5.19.3264. [DOI] [PubMed] [Google Scholar]
- 4.Fang K, Fu W, Beardsley AR, Sun X, Lisanti MP, Liu J. Overexpression of caveolin-1 inhibits endothelial cell proliferation by arresting the cell cycle at G0/G1 phase. Cell Cycle. 2007;6:199–204. doi: 10.4161/cc.6.2.3740. [DOI] [PubMed] [Google Scholar]
- 5.Ruoslahti E. Integrin signaling and matrix assembly. Tumour Biol. 1996;17:117–24. doi: 10.1159/000217975. [DOI] [PubMed] [Google Scholar]
- 6.Alghisi GC, Ruegg C. Vascular integrins in tumor angiogenesis: mediators and therapeutic targets. Endothelium. 2006;13:113–35. doi: 10.1080/10623320600698037. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 9.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, Plow EF. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6:851–60. [PubMed] [Google Scholar]
- 10.Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. Embo J. 1999;18:882–92. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–2. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 12.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–71. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 13.Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest. 1995;96:1815–22. doi: 10.1172/JCI118227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake CJ, Cheresh DA, Little CD. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108:2655–61. doi: 10.1242/jcs.108.7.2655. [DOI] [PubMed] [Google Scholar]
- 15.Topol EJ, Califf RM, Weisman HF, Ellis SG, Tcheng JE, Worley S, Ivanhoe R, George BS, Fintel D, Weston M, et al. Randomised trial of coronary intervention with antibody against platelet IIb/IIIa integrin for reduction of clinical restenosis: results at six months. The EPIC Investigators. Lancet. 1994;343:881–6. doi: 10.1016/s0140-6736(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 16.Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–64. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 17.Beer AJ, Haubner R, Sarbia M, Goebel M, Luderschmidt S, Grosu AL, Schnell O, Niemeyer M, Kessler H, Wester HJ, Weber WA, Schwaiger M. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin alpha(v)beta3 expression in man. Clin Cancer Res. 2006;12:3942–9. doi: 10.1158/1078-0432.CCR-06-0266. [DOI] [PubMed] [Google Scholar]
- 18.Haubner R. alpha(v)beta (3)-integrin imaging: a new approach to characterise angiogenesis? Eur J Nucl Med Mol Imaging. 2006;33:54–63. doi: 10.1007/s00259-006-0136-0. [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Hynes RO. GPR56 and TG2: possible roles in suppression of tumor growth by the microenvironment. Cell Cycle. 2007;6:160–5. doi: 10.4161/cc.6.2.3760. [DOI] [PubMed] [Google Scholar]
- 20.Danen EH, Lafrenie RM, Miyamoto S, Yamada KM. Integrin signaling: cytoskeletal complexes, MAP kinase activation, and regulation of gene expression. Cell Adhes Commun. 1998;6:217–24. doi: 10.3109/15419069809004477. [DOI] [PubMed] [Google Scholar]
- 21.Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8:96–103. doi: 10.1007/s11912-006-0043-3. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, Huang X, Sheppard D, Hynes RO, Hodivala-Dilke KM. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- 23.Taverna D, Moher H, Crowley D, Borsig L, Varki A, Hynes RO. Increased primary tumor growth in mice null for beta3- or beta3/beta5-integrins or selectins. Proc Natl Acad Sci USA. 2004;101:763–8. doi: 10.1073/pnas.0307289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taverna D, Crowley D, Connolly M, Bronson RT, Hynes RO. A direct test of potential roles for beta3 and beta5 integrins in growth and metastasis of murine mammary carcinomas. Cancer Res. 2005;65:10324–9. doi: 10.1158/0008-5472.CAN-04-4098. [DOI] [PubMed] [Google Scholar]
- 25.Wee JL, Jackson DE. The Ig-ITIM superfamily member PECAM-1 regulates the “outside-in” signaling properties of integrin alpha(IIb)beta3 in platelets. Blood. 2005;106:3816–23. doi: 10.1182/blood-2005-03-0911. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–96. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 28.Meredith JE, Jr., Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–61. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–99. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 30.Iivanainen E, Kahari VM, Heino J, Elenius K. Endothelial cell-matrix interactions. Microsc Res Tech. 2003;60:13–22. doi: 10.1002/jemt.10238. [DOI] [PubMed] [Google Scholar]
- 31.Ruegg C, Mariotti A. Vascular integrins: pleiotropic adhesion and signaling molecules in vascular homeostasis and angiogenesis. Cell Mol Life Sci. 2003;60:1135–57. doi: 10.1007/s00018-003-2297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in endothelial cell and angiogenesis. Cell Cycle. 2006;5:512–8. doi: 10.4161/cc.5.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bautch VL, Ambler CA. Assembly and patterning of vertebrate blood vessels. Trends Cardiovasc Med. 2004;14:138–43. doi: 10.1016/j.tcm.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Bazzoni G, Dejana E, Lampugnani MG. Endothelial adhesion molecules in the development of the vascular tree: the garden of forking paths. Curr Opin Cell Biol. 1999;11:573–81. doi: 10.1016/s0955-0674(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 35.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 36.Byzova TV, Plow EF. Activation of alphaVbeta3 on vascular cells controls recognition of prothrombin. J Cell Biol. 1998;143:2081–92. doi: 10.1083/jcb.143.7.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds AR, Reynolds LE, Nagel TE, Lively JC, Robinson SD, Hicklin DJ, Bodary SC, Hodivala-Dilke KM. Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 2004;64:8643–50. doi: 10.1158/0008-5472.CAN-04-2760. [DOI] [PubMed] [Google Scholar]
- 38.Sheppard D. Endothelial integrins and angiogenesis: not so simple anymore. J Clin Invest. 2002;110:913–4. doi: 10.1172/JCI16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheresh DA, Stupack DG. Integrin-mediated death: an explanation of the integrin-knockout phenotype? Nat Med. 2002;8:193–4. doi: 10.1038/nm0302-193. [DOI] [PubMed] [Google Scholar]
- 40.Mousa SA. alphav Vitronectin receptors in vascular-mediated disorders. Med Res Rev. 2003;23:190–9. doi: 10.1002/med.10031. [DOI] [PubMed] [Google Scholar]
- 41.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–21. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- 42.Borges E, Jan Y, Ruoslahti E. Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J Biol Chem. 2000;275:39867–73. doi: 10.1074/jbc.M007040200. [DOI] [PubMed] [Google Scholar]
- 43.Vuori K, Ruoslahti E. Association of insulin receptor substrate-1 with integrins. Science. 1994;266:1576–8. doi: 10.1126/science.7527156. [DOI] [PubMed] [Google Scholar]
- 44.Schneller M, Vuori K, Ruoslahti E. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. Embo J. 1997;16:5600–7. doi: 10.1093/emboj/16.18.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woodard AS, Garcia-Cardena G, Leong M, Madri JA, Sessa WC, Languino LR. The synergistic activity of alphavbeta3 integrin and PDGF receptor increases cell migration. J Cell Sci. 1998;111:469–78. doi: 10.1242/jcs.111.4.469. [DOI] [PubMed] [Google Scholar]
- 46.Dardik R, Loscalzo J, Eskaraev R, Inbal A. Molecular mechanisms underlying the proangiogenic effect of factor XIII. Arterioscler Thromb Vasc Biol. 2005;25:526–32. doi: 10.1161/01.ATV.0000154137.21230.80. [DOI] [PubMed] [Google Scholar]
- 47.Eliceiri BP. Integrin and growth factor receptor crosstalk. Circ Res. 2001;89:1104–10. doi: 10.1161/hh2401.101084. [DOI] [PubMed] [Google Scholar]
- 48.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1- deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 49.Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999;13:1055–66. doi: 10.1101/gad.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 50.Schlaeppi JM, Wood JM. Targeting vascular endothelial growth factor (VEGF) for anti-tumor therapy, by anti-VEGF neutralizing monoclonal antibodies or by VEGF receptor tyrosine-kinase inhibitors. Cancer Metastasis Rev. 1999;18:473–81. doi: 10.1023/a:1006358220123. [DOI] [PubMed] [Google Scholar]
- 51.Mahabeleshwar GH, Feng W, Phillips DR, Byzova TV. Integrin signaling is critical for pathological angiogenesis. J Exp Med. 2006;203:2495–507. doi: 10.1084/jem.20060807. [DOI] [PMC free article] [PubMed] [Google Scholar]