

Abstract
Hg2+ is commonly used as an inhibitor of many aquaporins during measurements of transcellular water transport. To investigate whether it could also act on the paracellular water transport pathway, we asked whether addition of Hg2+ affected transport of radiolabeled probes through tight junctions of a salivary epithelial cell monolayer. Inclusion of 1 mM Hg2+ decreased transepithelial electrical resistance by 8-fold and augmented mannitol and raffinose flux by 13-fold, which translated into an estimated 44% increase in pore radius at the tight junction. These Hg2+-induced effects could be partially blocked by the protein kinase A (PKA) inhibitor N-[2-((p-bromocinnamyl) amino) ethyl]-5-isoquinolinesulfonamide, 2HCl (H89), suggesting that both-PKA dependent and PKA-independent mechanisms contribute to tight junction regulation. Western blot analyses showed a 2-fold decrease in tight junction-associated occludin after Hg2+ treatment and the presence of a novel hyperphosphorylated form of occludin in the cytoplasmic fraction. These findings were corroborated by confocal imaging. The results from this study reveal a novel contribution of the PKA pathway in Hg2+-induced regulation of tight junction permeability in the salivary epithelial barrier. Therapeutically, this could be explored for pharmacological intervention in the treatment of dry mouth, Sjögren’s syndrome, and possibly other disorders of fluid transport.
The cells forming epithelial and endothelial barriers play a vital role in separating the major fluid compartments in animals across the evolutionary spectrum. Efforts to quantitatively measure water transport in cells have relied substantially on the use of heavy metal ions such as Hg2+, which are known to inhibit many aquaporins at micromolar concentrations by cross-linking cysteine residues in the water-transporting pore of the channel (Macey, 1984; Preston et al., 1993; Savage and Stroud, 2007). Despite the fact that Hg2+ ions can possibly cross-link cysteine residues in a potentially larger number of cell proteins, inhibition of water transport by Hg2+ ions has evolved into a benchmark for transcellular water transport through epithelial and endothelial cells (Folkesson et al., 1994; Roberts et al., 1994; Schnitzer and Oh, 1996; Ko et al., 2002; Burghardt et al., 2006; Yang et al., 2006).
Much less is known about the effect of Hg2+ ions on other structures, such as the tight junction complex (TJC) that is the critical functional component in regulating water transport through the paracellular pathway (i.e., the spaces between the cells of epithelial or endothelial barriers). An early study by Böhme et al. (1992) described the disruption of the tight junction barrier by Hg2+ in rat colonic epithelium, but the molecular mechanisms underlying this phenomenon were not investigated. Therefore, we asked whether Hg2+ affects the permeability of the epithelial barrier by acting in a specific manner on the tight junction complex, and we sought to identify contributing molecular mechanisms.
Our results show that addition of Hg2+ activates the protein kinase A (PKA) pathway and causes a substantial increase in paracellular permeability that is associated with a reduction in occludin at the tight junction and appearance of novel, phosphorylated isoforms of occludin. In the salivary glands, this PKA pathway may therefore be a suitable pharmacological target for therapeutic interventions in the treatment of dry mouth, Sjögren’s syndrome, and other disorders of saliva formation.
Materials and Methods
Experimental Reagents
Mercuric chloride, myosin light chain kinase (MLCK) inhibitor ML-7 and β-mercaptoethanol were obtained from Sigma-Aldrich (St. Louis, MO). The PKA inhibitor H89 and the PKC inhibitor Ro-32-0432 were purchased from Calbiochem (San Diego, CA). [14C]Mannitol and [3H]raffinose were obtained from Moravek Biochemicals (Brea, CA). Mouse monoclonal antibody against occludin was obtained from Zymed Laboratories (South San Francisco, CA). Alexa-555-conjugated anti-mouse secondary antibody was purchased from Invitrogen (Carlsbad, CA). Horseradish peroxidase-conjugated secondary antibodies were from Vector Laboratories (Burlingame, CA). Protein concentration in each sample was measured using the bicinchoninic acid protein assay reagent kit (Pierce Chemical, Rockford, IL) according to manufacturer’s protocol. Nitrocellulose membrane and 4 to 15% linear gradient and 7.5% resolving Tris-hydrochloride ready gels were purchased from Bio-Rad (Hercules, CA). Immunoreactivity was detected by SuperSignal West Pico chemiluminescent reagents (Pierce Chemical). CellTiter-Glo Luminescent Cell Viability kit was purchased from Promega (Madison, WI).
Cell Culture
Rat submandibular gland epithelial cells-clone #6 (SMG-C6) cells were kindly provided by Dr. David Quissell (University of Colorado Health Sciences Center, Denver, CO) (Quissell et al., 1997). Cells were seeded at a density of 3 × 105 cells/cm2 on polyester Transwell filters (24 mm in diameter, 3.0-μm pore size; Costar, Lowell, MA). SMG-C6 cell monolayers formed stable tight junctions by day 8. Therefore, all the experiments were performed between day 9 and day 11.
Transepithelial Electrical Resistance
Cells were grown on polyester filters, and the resistance of the monolayers was determined with a Millicell ERS Volt-ohmeter (Millipore Corporation, Billerica, MA). Background resistance of a filter and media without cells was subtracted, and values were recorded as ohms × square centimeters.
Transport Studies
SMG-C6 cell monolayers cultured on permeable filter support for 9 to 11 days were used for transport studies with radioactive solutes following previously published protocols (Lindmark et al., 1995; Pauletti et al., 1996). In brief, cell monolayers were washed with prewarmed Hanks’ balanced salt solution (HBSS), pH 7.4, was added to receiver compartment (apical side, 1.5 ml) and [14C]mannitol or [3H]raffinose dissolved in HBSS was added to the donor compartment (basolateral side, 2.6 ml). Samples (120 μl, receiver side; 20 μl, donor side) were removed at 15-min intervals through 120 min from both sides. The volume removed from the receiver was always replaced with fresh, prewarmed HBSS. Each experiment was performed in triplicate.
Pore Radius at the Paracellular Junction
The aqueous pore radius at the paracellular junction of SMG-C6 cell monolayers was estimated in the presence and absence of 1 mM Hg2+ using the Renkin molecular sieving function (Renkin, 1954). This dimensionless parameter compares the molecular radius (r) of a solute with a cylindrical pore radius (R) and takes values of 0 < F(r/R) < 1:
The effective pore radius was calculated as described earlier by Adson et al. (1994) using the ratio of the paracellular permeabilities determined for two uncharged hydrophilic solutes of different molecular radius:
Papp,mannitol and Papp,raffinose are the apparent permeability coefficients of mannitol and raffinose, respectively, determined experimentally in the presence and absence of 1 mM Hg2+; r is the molecular radius of each solute [i.e., mannitol = 4.1 Å (Adson et al., 1994) and raffinose = 6.0 Å (Whittembury, 1962)]; and F(rsolute/R) is dimensionless Renkin molecular sieving function.
Preparation of Triton X-100-Soluble and -Insoluble Fractions
Cell monolayers cultured on permeable supports were exposed to various treatments and subsequently subjected to an extraction protocol adapted from Fey et al. (1984) and Stuart et al. (1994, 1996) to isolate Triton X-100-soluble and -insoluble fractions. In brief, cell monolayers were washed with phosphate-buffered saline (PBS) and incubated with 500 μl of cytoskeleton-1 buffer (0.5% Triton X-100, 100 mM NaCl, 10 mM Tris-HCl, pH 7.4, and 300 mM sucrose) in the presence of a protease inhibitor cocktail (Sigma-Aldrich). Extraction was performed for 20 min at 4°C on a gentle rocker. The Triton X-100-soluble fraction was completely removed and the remaining, insoluble residue was solubilized in sample buffer without dye (50 mM Tris, pH 6.8, 2% SDS, and 10% glycerol) containing protease inhibitor cocktail.
Cytotoxicity Assay
Potential cytotoxic effects of Hg2+ could include both apoptotic and necrotic cell death. To determine whether 1 mM Hg2+ is cytotoxic to SMG-C6 cells, viability was monitored using the CellTiter-Glo luminescent assay as described previously (Prichard et al., 2007). In brief, cells cultured on a 96-well plate were incubated in triplicate with HBSS (control), HBSS + 1 mM Hg2+, HBSS + 30 μM H89, or HBSS + 30 μM H89 + 1 mM Hg2+. After 0, 15, or 30 min, 50 μl of CellTiter-Glo reagent was added to each well, and the plates were mixed for 2 min on an orbital shaker to induce cell lysis. The plates were then incubated for an additional 10 min at room temperature, and the luminescence was quantified on a luminometer. Results were normalized to vehicle-treated controls and expressed as percentage of cell viability.
Western Blot Analysis
Western blots for Triton X-100-soluble and -insoluble fractions were performed as described previously (Kawedia et al., 2007). In brief, 10 μg of total protein was resolved using SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane. The membrane was probed with primary antibodies in 5% milk/Tris-buffered saline/Tween 20 overnight at 4°C. Secondary antibody conjugated to horseradish peroxidase enzyme (Vector Laboratories) was incubated in 5% milk/Tris-buffered saline/Tween 20 for 1 h. Immunoreactivity was detected by Super-Signal West Pico chemiluminescent reagents. Quantitation of the Western blots was done using ImageQuant software (GE Healthcare, Chalfont St. Giles, UK). β-Actin was used as loading control for total protein. For Triton X-100-insoluble fractions, where β-actin could not be used as a control, equal loading in individual lanes was confirmed by staining the membrane with Ponceau S after transfer to Immobilon (Millipore Corporation) membranes. Protein values are reported as a percentage of samples compared with control (100%).
Immunofluorescence Studies
Cell monolayers cultured for 9 to 11 days on permeable filter support were subjected to various treatments and fixed at room temperature for 30 min using 3.75% paraformaldehyde/PBS. Fixed cells were quenched using 0.1% sodium borohydride/PBS for 15 min at room temperature with moderate shaking, permeabilized with 0.1% Triton X-100/PBS for 4 min at room temperature. Permeabilized cells were blocked using 10% goat serum/PBS for 30 min at room temperature followed by incubation with primary antibodies in 10% goat serum/PBS for 30 min at room temperature followed by an incubation of secondary antibody. After secondary antibody incubation, cells were washed with PBS followed by two washes of 0.1% Triton X-100/PBS. Cells were postfixed for 5 min at room temperature using 3.75% paraformaldehyde/PBS solution. Cells were mounted using Vectashield (Vector Laboratories) hard set mounting media. Images were obtained in x-y-axis using a 510 upright confocal microscope (Carl Zeiss Inc., Thornwood, NY) at 20× using proprietary software (Carl Zeiss Inc.).
Statistics
Data are presented as mean ± S.E.M. or S.D., and differences within groups were tested with Student’s t test and analysis of variance. p < 0.05 was considered statistically significant.
Results
Addition of Hg2+ Increases Paracellular Permeability
To investigate the effect of Hg2+ on tight junction barrier, we measured transepithelial electric resistance (TEER) and transport of paracellular markers mannitol and raffinose. In all experiments, a baseline was recorded for 60 min before mercury was added to the basolateral chamber. As shown in Fig. 1, A and C, 1 mM mercury rapidly alters the tight junction barrier. Within 15 min, TEER values decreased by 75% to ~120 Ω · cm2 and gradually approached a steady-state value corresponding to 12.5% of the original cell monolayer resistance at the end of the experiment (n = 12). The cumulative flux of mannitol increased by 2-fold within 30 min and reached a 7-fold excess compared with control within 60 min (n = 12). This dramatic effect of mercury on mannitol flux was also demonstrated at a lower dose. As shown in Fig. 1A, 10 μM mercury enhanced cumulative mannitol flux by 3-fold at the end of the experiment, suggesting a dose-dependent effect of this heavy metal ion on paracellular permeability. Likewise, transepithelial transport of raffinose, a paracellular marker with a hydrodynamic radius of approximately twice the predicted size of mannitol, was dramatically increased in the presence of Hg2+ (Fig. 1B). Based on the apparent permeability coefficients calculated for these two uncharged, hydrophilic solutes (Fig. 1D), the pore size of the tight junction barrier before and after addition of mercury was estimated using the biophysical model established by Renkin (1954). The results from these predictions suggest that 1 mM mercury increases the average pore size at the tight junctions by 44% (58.0 ± 7.4 Å, n = 6 versus 83.6 ± 4.0 Å, n = 6; p = 0.0062).
Fig. 1.
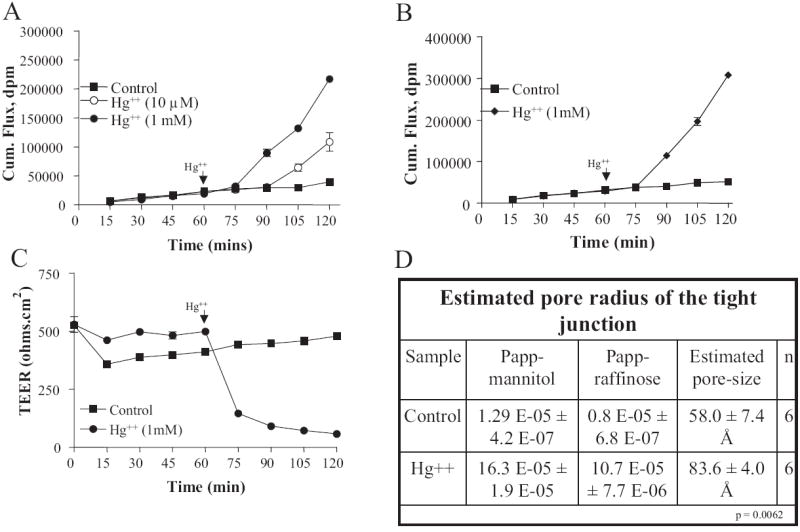
Addition of Hg2+ increases tight junction permeability of SMG-C6 cell monolayers. Cells were treated with 10 μM and 1 mM mercuric chloride, 60 min after the start of the experiment. A, cumulative mannitol flux as measured at 15-min intervals. B, cumulative raffinose flux as measured at 15-min intervals. C, transepithelial electric resistance as measured at 15-min intervals. D, estimated pore radius of tight junction before and after Hg2+ treatment.
Hg2+ Activates PKA Signal Transduction Pathway That Is Independent of MLCK
To determine the signaling mechanism through which Hg2+ modulated tight junction permeability, we tested the PKA inhibitor H89 and the myosin light chain kinase inhibitor ML-7. Inclusion of 30 μM H89 reduced the mercury-induced increase in paracellular permeability, but it could not completely block this effect, as shown in Fig. 2, A and B. At the end of 60 min, TEER only decreased by 50% in the presence of H89 compared with a 90% reduction in this value by mercury alone. Likewise, H89 limited mannitol flux increase to 4-fold instead of a 7-fold increase measured with mercury alone. In separate control experiments, H89 was shown not to affect TEER or mannitol flux. We also determined the effect of H89 inhibitor on Hg2+-induced increase in tight junction permeability at a much lower concentration (1 μM) to eliminate any nonspecific kinase inhibitory activity of H89. As shown in Fig. 2, C and D, addition of 1 μM H89 effectively reduced mercury-induced increases in raffinose and mannitol fluxes, further supporting earlier conclusions regarding the role of PKA in regulating tight junction permeability. In contrast, the MLCK inhibitor ML-7 did not block mercury-induced changes at the junctional complex, as shown in Fig. 2, E and F.
Fig. 2.
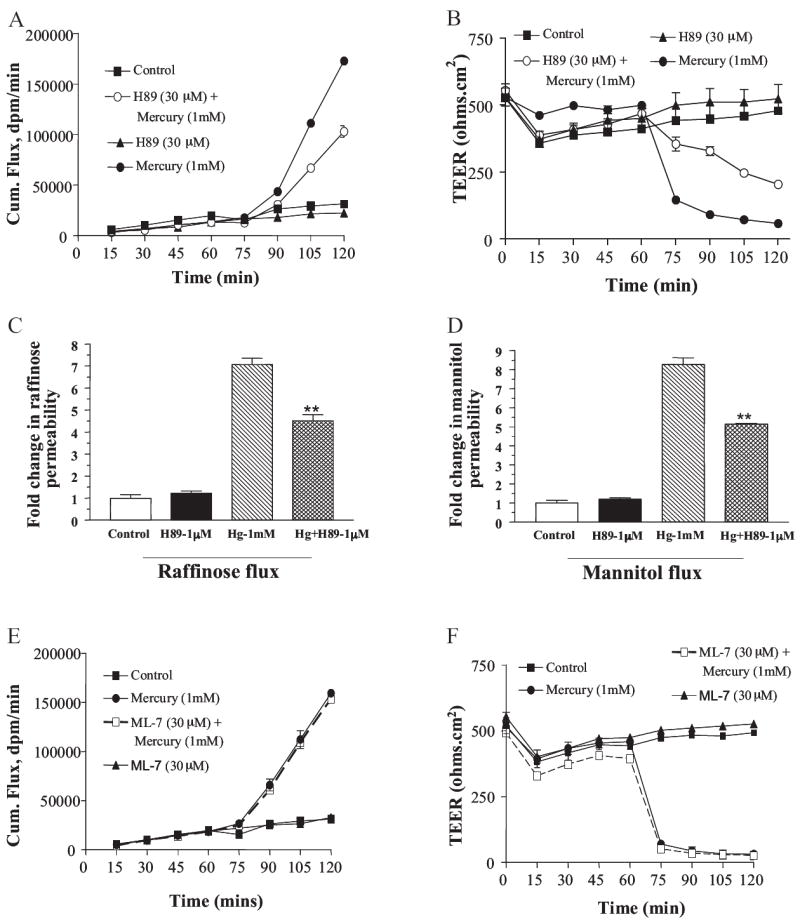
Hg2+ activates a signal transduction pathway that involves PKA and is independent of MLCK. Cells were treated with PKA inhibitor H89 and MLCK inhibitor ML-7 at time 0, and mercuric chloride was added after 60 min. A, cumulative mannitol flux as measured at 15-min intervals in presence of 30 μM H89 (n = 6). B, TEER as measured at 15-min intervals in presence of 30 μM H89 (n = 6). C, -fold change in raffinose permeability after addition of Hg2+ and its rescue by H89 at 1 μM concentration (n = 3). D, -fold change in mannitol permeability after addition of Hg2+ and its rescue by H89 at 1 μM concentration (n = 3). Cumulative mannitol flux as measured at 15-min intervals in presence of ML-7 (n = 6). D, TEER as measured at 15-min intervals in presence of ML-7 (n = 6).
Hg2+-Induced Increase in Paracellular Permeability Is Independent of Extensive Cross-Linking of Cysteine Residues
It is well known that addition of Hg2+ can cause cross-linking of sulfhydryl groups in the cysteine amino acid residues of proteins. We reasoned that if the effect of Hg2+ on paracellular permeability was due to cross-linking of proteins either in the tight junction itself or associated with the tight junctional complex, then reducing the cross-linked cysteine residues would result in altered transepithelial resistance. We therefore monitored TEER of cell monolayers treated with Hg2+ in the presence and absence of 1 mM β-mercaptoethanol. As shown in Fig. 3, addition of this reducing agent did not result in any significant changes in TEER across the monolayer, suggesting that the action of Hg2+ in altering TEER is unlikely to be caused by the cross-linking of cysteine residues in TJ proteins.
Fig. 3.
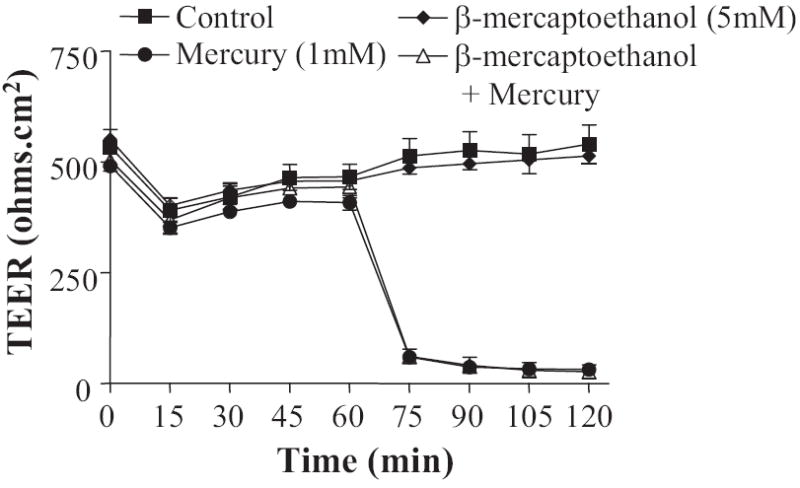
Hg2+-induced increase in the tight junction permeability is independent of its cysteine cross-linking property. Cells were treated with the reducing agent (cysteine crosslinking inhibitor) β-mercaptoethanol at time 0, and mercuric chloride was added after 60 min. TEER was measured at 15-min intervals in presence of β-mercaptoethanol (n = 6).
Hg2+ Induces Opening of Tight Junction Barrier before Activation of Cytotoxic Signals
To delineate whether the increase in paracellular permeability was the result of a specific action of Hg2+ on the tight junctions or whether it was due to its nonspecific cytotoxic effect on the cells. We determined total cell viability (apoptotic and necrotic cell death) as a function of time after addition of Hg2+ in presence and absence of PKA inhibitor H89. As shown in Fig. 4, A and B, at time 0 addition of Hg2+ had no effect on the TEER and the viability of the cells. Most importantly, 15 min after exposure to Hg2+ TEER dramatically decreased to 25% of the original value (Fig. 4B), whereas cell viability was not significantly changed (Fig. 4A). Mercury-induced cytotoxic effects were quantifiable only after 30 min were cell viability was reduced, on average, by 40% (Fig. 4A). However, this event did not correlate with a more dramatic reduction in TEER, as demonstrated in Fig. 4B. It is interesting to note that inclusion of the PKA inhibitor H89 that successfully rescued Hg2+-induced increase in tight junction permeability did not provide a significant protective effect on cell viability. At the concentration tested, H89 alone had no effect either on TEER or on cell viability (Fig. 4, A and B).
Fig. 4.
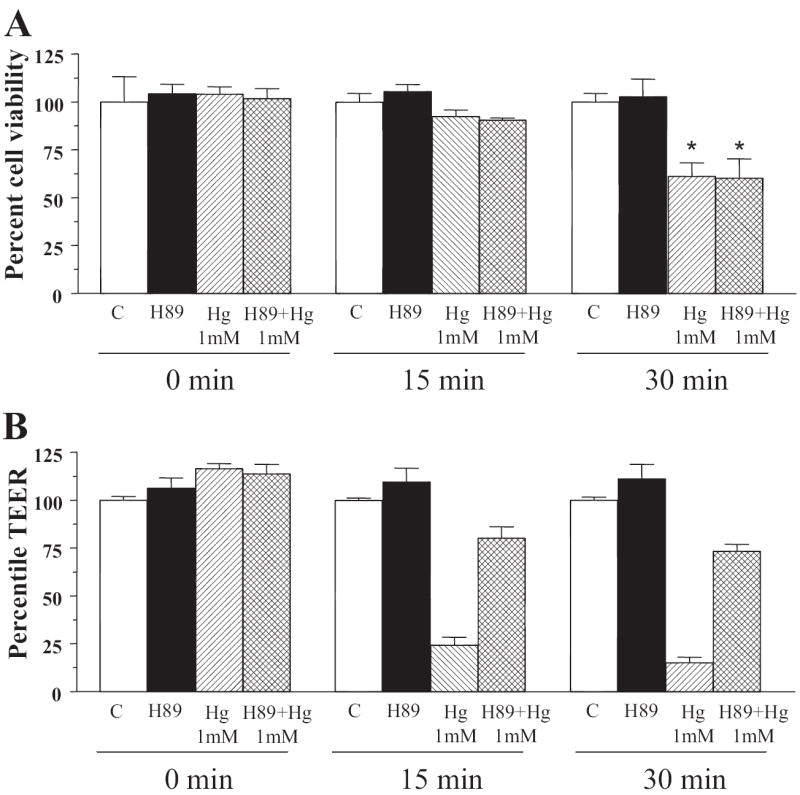
Hg2+ activates increase in tight junction permeability before the activation of cytotoxic signals in SMG-C6 cell monolayer. A, cells were treated with PKA inhibitor H89 at time 0 and 1 mM mercuric chloride was added after 60 min. Cell viability was determined using a commercially available kit (CellTiter-Glo luminescent cell viability assay; Promega) and is represented as percentage of cell viability compared with water-treated controls. Percentage of cell viability after the cell monolayers were treated with Hg2+ for 0, 15, and 30 min. B, transepithelial electric resistance of cell monolayers treated with PKA inhibitor and Hg2+ similar to in A for 0, 15, and 30 min (n = 4).
PKA Inhibitor Blocks Hg2+-Mediated Decrease in Occludin Expression at Tight Junctions
Confocal immunofluorescence microscopy revealed a significant reduction in occludin staining at the tight junctions after treatment with a 1 mM Hg2+ solution. Compared with vehicle-treated controls occludin in the TJs decreased 3-fold in response to Hg2+ (Fig. 5, A and B; n = 5; p = 0.005). Inclusion of H89 successfully blocked the action of Hg2+ (Fig. 5C; n = 5; p = 0.008). Fluorescence intensity was measured using ImageJ (National Institutes of Health, Bethesda, MD). The results are quantitated and compared in Fig. 5D.
Fig. 5.
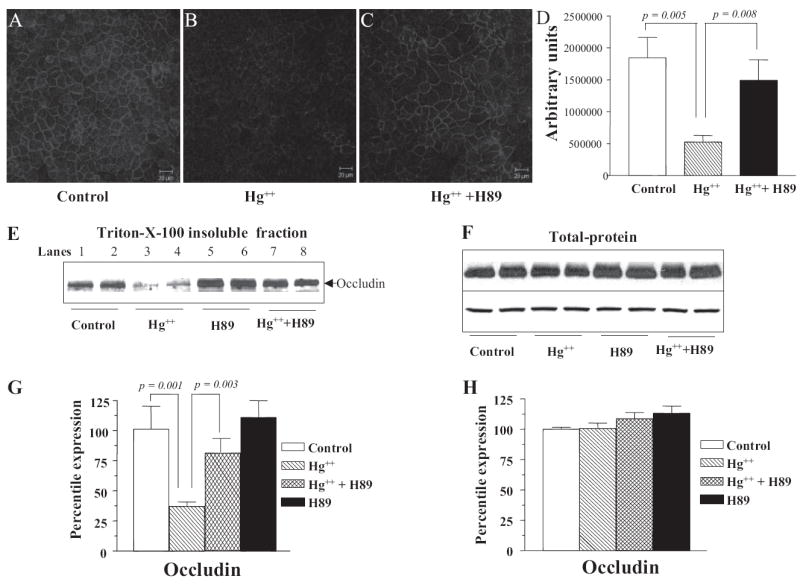
Addition of Hg2+ results in a decrease of occludin in the tight junctions via PKA-dependent mechanism. Confocal and Western blot analysis to determine occludin expression in the TJs of SMG-C6 cells: Cells were treated with PKA inhibitor H89 at time 0, and mercuric chloride was added after 60 min. Control (n = 3) (A), Hg2+ (n = 5) (B), and Hg2+ + H89 (n = 5) (C). D, fluorescence intensity was measured using ImageJ (National Institutes of Health). A minimum of seven fields were analyzed from each slide. E, Western blot of occludin in the tight junction fraction (Triton X-100-insoluble) of SMG-C6 cells. Equal loading in individual lanes was confirmed by staining the membrane with Ponceau S after transfer. F, Western blot of occludin in the total protein extracts of SMG-C6 cells. G, quantitation of the Western blots as shown in E from three individual experiments. H, quantitation of the Western blots as show in F from three individual experiments.
Western blot analysis of Triton X-100-insoluble cell fractions that contain TJ proteins demonstrated that addition of Hg2+ decreased the amount of occludin in these fractions by approximately 3-fold (Fig. 5, E and G; p = 0.001). This effect was prevented by addition of H89 (Fig. 5, E and G; p = 0.003), corroborating the results obtained by confocal imaging. H89 on its own was observed to cause a slight (but statistically insignificant) increase in occludin levels (Fig. 5, E and G). To investigate whether the decrease in occludin levels in Triton X-100-insoluble fractions was due to degradation or due to redistribution of occludin, the amount of total cellular occludin was determined in whole cell protein preparations. As shown in Fig. 5, F and H, Western blot analysis of total protein extracts showed no change in the expression of occludin in either controls or Hg2+-treated samples.
Hg2+ Triggers Phosphorylation of Occludin via a PKA-Dependent Mechanism
To investigate whether treatment with Hg2+ alters phosphorylation of occludin, we determined the shift in migration of occludin on SDS-polyacrylamide gel electrophoresis gels, after addition of Hg2+. We found that there are at least three distinct phosphorylated isoforms of occludin in SMG-C6 cells (Fig. 6). Form I (molecular mass, 60 kDa; Fig. 6A, lane 2) is solely present in the Triton X-100-soluble (cytosolic) fraction, whereas form II (molecular mass, 62 kDa; Fig. 6A, lane 1) is solely present in the Triton X-100-insoluble (membrane) fraction. Form III (molecular mass, 64 kDa; Fig. 6A, lane 3) is a novel, hyperphosphorylated form of occludin that is induced by adding Hg2+ and is also solely present in the Triton X-100-soluble fractions.
Fig. 6.
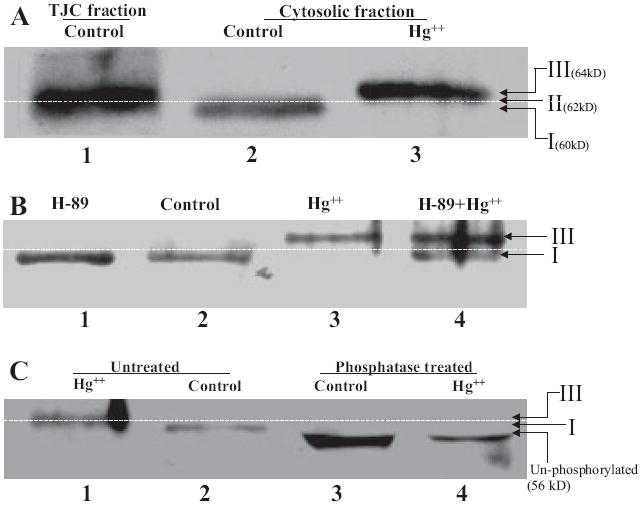
Hg2+ triggers phosphorylation and subcellular redistribution of occludin. Western blot analysis of the tight junction and cytosolic fraction of SMG-C6 cells. Cells were treated with PKA inhibitor H89 at time 0, and mercuric chloride was added after 60 min. A, Western blot of the three phosphorylated forms of occludin. Lane 1, form II (62 kDa), found in Triton X-100-insoluble (membrane) fractions of control and Hg2+-treated cells; lane 2, form I (60 kDa), found in Triton X-100-soluble (cytosolic) fraction of control cells; and lane 3, form III (64 kDa), found in Triton X-100-soluble (cytosolic) fractions of cells treated with Hg2+. B, Western blot of occludin in the soluble fraction (Triton X-100-soluble) of SMG-C6 cells. Lane 1, H89 alone; lane 2, control; lane 3, Hg2+; and lane 4, Hg2+ + H89. C, Western blot of occludin in the soluble fraction before phosphatase treatment (lane 1, Hg2+ and lane 2, control) and after phosphatase treatment (lane 3, control and lane 4, Hg2+).
Hg2+ induced a significant increase in molecular mass of occludin from 60 kDa (form I; Fig. 6B, lanes 1 and 2) to 64 kDa (form III; Fig. 6B, lane 3) in the Triton X-100-soluble fraction. This significant increase in molecular mass could be blocked by addition of PKA inhibitor H89 (Fig. 6B, lane, 4). To determine whether the increase in molecular mass was due to phosphorylation, the Triton X-100-soluble fractions were treated with alkaline phosphatase before gel electrophoresis. Treatment with alkaline phosphatase resulted in a reduction of molecular mass of occludin from 60 to 56 kDa in controls and from 64 to 56 kDa in Hg2+-treated samples (Fig. 6C), suggesting that the different molecular masses of occludin are due to differential levels of phosphorylation of this protein.
Discussion
The tight junctional complex is a critical cellular structure that regulates the passage of ions, water, nutrients, and drug molecules through the paracellular spaces in epithelial and endothelial barriers. Important epithelial barriers are found in the salivary glands, gastrointestinal tract, kidney, and lung, and strategies to effectively deliver pharmacological molecules through the pore of the tight junctional complex remain the subject of intense interest and investigations in the pharmaceutical industry. While studying the inhibitory properties of heavy metal ions on membrane water channels, we serendipitously discovered that Hg2+ enlarges the pore in the TJC and substantially increases paracellular permeability. Using the salivary epithelial SMG-C6 cell culture model, we show that Hg2+ decreases the physical barrier properties of tight junctions by activating a PKA pathway. However, it is apparent from the observation that addition of a PKA inhibitor only partially rescued the effect (approximately 50% rescue) that there is a contribution of other kinase-dependent and even nonkinase pathways that play a role in the Hg2+ action on tight junction permeability.
At the molecular level, this PKA pathway results in phosphorylation of occludin, a key TJ protein, and it causes removal of occludin from the tight junctions leading to an increase in paracellular permeability. Although we focused on occludin as a “proof of principle” due to its role in regulating TJ permeability, we cannot exclude the possibility that there may also be additional changes in other components of the TJC.
Addition of Hg2+ Increases the Permeability of the Epithelial Barrier
We measured the flux of radiolabeled mannitol and raffinose across polarized SMG-C6 cell monolayers in the presence and absence of Hg2+. Mannitol and raffinose are metabolically stable, uncharged hydrophilic space markers of different hydrodynamic radii that are not significantly taken up via endocytotic or transcytotic pathways and can thus be effectively used for estimating tight junction permeability (Hecht et al., 1988). In parallel, we measured TEER, which indicates the permeability of the monolayer to ions such as Na+, Cl−, and K+.
Addition of Hg2+ to the basolateral compartment of the Transwell significantly augmented transepithelial flux of mannitol and raffinose (Fig. 1, A and B). For mannitol, this effect was dose-dependent. The finding that TEER decreased by 75% within the first 15 min and subsequently stabilized at 60 min around 12.5% of the original value measured at t = 0 (Fig. 1C) indicates that the increased transport observed for hydrophilic solutes was paralleled by enhanced ion transport across the epithelial monolayer.
We analyzed the data obtained from experimental mannitol and raffinose transport studies using a biophysical model established by Renkin (1954) for molecular size-restricted diffusion, and we obtained a quantitative estimate of the effect of Hg2+ on the pore size at the TJs. The results from these predictions reveal that Hg2+ increases the average pore radius by almost 50% (Fig. 1D).
Hg2+ Activates a Signal Transduction Pathway That Involves PKA and Is Independent of MLCK, PKC, and Cysteine Cross-Linking Mechanisms
Previous studies have shown that MLCK and PKA pathways can regulate TJ permeability in monolayers of dog kidney collecting duct cells (Madin-Darby canine kidney) (Klingler et al., 2000), human colon adenocarcinoma cells (Caco-2) (Shen et al., 2006), and human colon carcinoma cells (T84) (Ma et al., 2000; Zolotarevsky et al., 2002). To delineate which of these pathways was required for the action of Hg2+ on the TJs of salivary epithelial cell monolayers, we added the MLCK inhibitor ML-7 before Hg2+ treatment. ML-7 did not block the effect of Hg2+ on mannitol flux (Fig. 2E) nor on TEER (Fig. 2F), suggesting that the effect of Hg2+ is independent of MLCK. In contrast, the PKA inhibitor H89 at concentrations as low as 1 μM (far below the Ki value for calmodulin kinase II, casein kinase I, MLCK, and PKC) significantly blocked the Hg2+-induced increase in permeability, as shown in Fig. 2, A to D, implicating for the first time that Hg2+ elicits its effect on TJs via a contribution of the PKA signaling pathway. In addition, PKC inhibitor Ro-32-0432 did not rescue Hg2+-induced increase in raffinose flux (8.5 ± 0.4-fold increase with Hg2+ alone versus 9.6 ± 0.9-fold increase with Hg2+ in presence of PKC inhibitor; n = 3; p = 0.11), suggesting that PKC does not seem to play a major role in the Hg2+ induced action on tight junctions.
Because Hg2+ has been demonstrated to cross-link cysteine residues of proteins, we asked whether the effect of Hg2+ was due to nonspecific cross-linked cysteine residues or due to more selective effects on TJs. Pretreatment of cell monolayers with the reducing agent β-mercaptoethanol did not rescue the effect of Hg2+ (Fig. 3), implying that the action of Hg2+ on tight junctions was unrelated to its cross-linking properties.
We also determined whether Hg2+-induced reduction in TJ barrier properties correlated with potential cytotoxic effects induced by this heavy metal. Using a cell viability assay (which measures both apoptosis and necrosis), we found that the temporal aspects of the observed increase in tight junction permeability did not support a significant role of Hg2+-induced cytotoxicity (Fig. 4, A and B). Furthermore, the PKA inhibitor that rescued cells from Hg2+ induced increase in tight junction permeability (Fig. 2) had no effect on cell viability. Combined, these results show that the effect of Hg2+ on tight junctions is not due to cytotoxicity. We therefore interpret these results to mean that Hg2+ disrupts the tight junction barrier either directly or indirectly through a PKA-dependent signaling mechanism.
Addition of Hg2+ Results in Less Occludin in the Tight Junctions
To investigate whether the increase in paracellular permeability was paralleled by molecular changes in the TJs, we initiated our analysis with the transmembrane protein occludin because 1) it interdigitates with similar occludins from adjacent cells to form the tight junction seal (Feldman et al., 2005); 2) other investigators had already established that occludins respond to heavy metal ions, such as Pb2+ (Wang et al., 2007), which we hypothesized might have similar actions as Hg2+; and 3) occludin is a benchmark indicator of TJ permeability (Kevil et al., 2000; Rao et al., 2002; Feldman et al., 2005).
Expression of occludin in the tight junctions was initially assessed by confocal immunofluorescence microscopy. Occludin staining in the tight junctions was significantly decreased after treatment with Hg2+ compared with untreated controls (Fig. 5, A and B). This effect was prevented by inclusion of H89 (Fig. 5C), corroborating the functional studies of TJ permeability described in this report.
To make a quantitative estimate of changes in the TJ, we used Western blot analysis of Triton X-100-insoluble fractions (enriched in TJs) prepared by subcellular fractionation (Fey et al., 1984; Stuart et al., 1994, 1996). Addition of Hg2+ significantly decreased the amount of occludin in TJ-enriched fractions (Fig. 5, E and G), and this decrease was blocked by the addition of H89 (Fig. 5, E and G). Addition of Hg2+ did not alter the levels of occludin in total protein extracts (Fig. 5, F and H), indicating that Hg2+ does not result in degradation of occludin but rather induces its sub-cellular redistribution into the TJ via a PKA-dependent mechanism.
Hg2+ Triggers Phosphorylation and Subcellular Redistribution of Occludin
Recruitment of occludin from the cytoplasm to the TJs, as well as shuttling of occludin from the TJs to the cytoplasmic compartment, is known to be regulated by phosphorylation (Cordenonsi et al., 1997; Sakakibara et al., 1997; Wong, 1997; Wong and Gumbiner, 1997). The identification of PKA as a downstream target of the action of Hg2+ prompted us to examine whether the degree of phosphorylation of occludin was altered in TJ-enriched and other subcellular fractions.
Our results show that there are at least three distinct phosphorylated isoforms of occludin in SMG-C6 cells (Fig. 6). Form I (molecular mass, 60 kDa; Fig. 6A, lane 2) is solely detectable in the Triton X-100-soluble fraction, whereas form II (molecular mass, 62 kDa; Fig. 6A, lane 1) was only detected in the Triton X-100-insoluble fraction. Form III (molecular mass, 64 kDa; Fig. 6A, lane 3) is a novel, hyperphosphorylated form of occludin that is induced by adding Hg2+ and was solely present in the Triton X-100-soluble fractions.
Hg2+ induced a significant increase in molecular mass of occludin from 60 kDa (form I) to 64 kDa (form III) in the Triton X-100-soluble fraction. This phosphorylation could be blocked by addition of H89 (Fig. 6B), suggesting that phosphorylation and subcellular relocation of occludin are mechanisms by which Hg2+ acts on TJ permeability. Treatment of the Triton X-100-soluble fraction with alkaline phosphatase before gel electrophoresis resulted in a 56-kDa occludin band in all samples, corroborating results from a previously proposed model in which the cytosolic form of occludin (Form I), is phosphorylated to Form II and recruited into the TJs (Cordenonsi et al., 1997; Sakakibara et al., 1997; Wong, 1997; Wong and Gumbiner, 1997). Because phosphorylation, especially at tyrosine residues, has been shown to increase TJ permeability by altered localization of occludin (Wachtel et al., 1999; Kevil et al., 2000; Hirase et al., 2001; Rao et al., 2002; Feldman et al., 2005; Harhaj et al., 2006; Rajasekaran et al., 2007), treatment with Hg2+ seems to activate a PKA pathway that either directly or indirectly converts occludin from Form II (which is in the tight junction) to a hitherto undescribed hyperphosphorylated form of occludin (Form III) that is solely detectable in the cytoplasm. Based on these observations, we propose that hyperphosphorylation of occludin from Form II to Form III is a novel mechanism by which occludin is removed from the TJs, and which results in increased permeability of the tight junction barrier as shown in the model (Fig. 7).
Fig. 7.
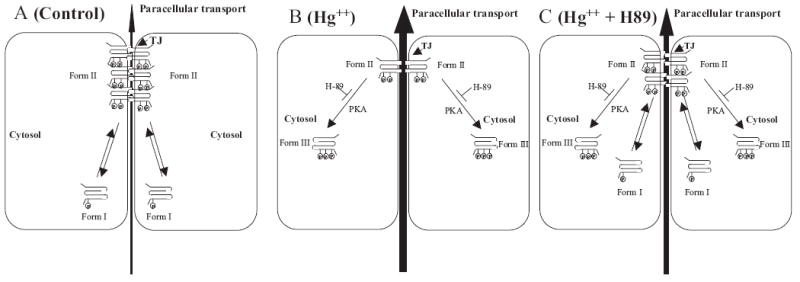
Model for Hg2+-triggered increase in tight junction permeability. A, in control cells, there are two phosphorylated forms of occludin: form I (molecular mass, 60 kDa) is detectable solely in the cytosol, and form II (62 kDa) is detected solely in the membrane fraction (mostly in the TJs). B, addition of Hg2+ results in a novel third form of occludin (form III; molecular mass, 64 kDa) that is seen solely in the cytosolic fraction similar to form I. C, PKA inhibitor H89 inhibits Hg2+-induced decrease in occludin at the tight junctions and formation of form III in the cytoplasm.
For more than two decades, Hg2+ inhibition of water transport has been assumed to operate by blocking transcellular movement of water through membrane water channels (aquaporins). Here, we show that treatment with Hg2+ activates a PKA-dependent pathway and also other pathways that have yet to be identified, the combined result of which alters the phosphorylation and subcellular distribution of occludin, and increases the permeability of TJs, thereby altering paracellular transport of hydrophilic solutes across the monolayer.
It is possible that PKA and other molecular components of this paracellular pathway in the salivary epithelium may provide novel targets for pharmacological modification to improve fluid secretion in conditions such as xerostomia (dry mouth), which occurs commonly in Sjögren’s syndrome, and in patients receiving radiation and chemotherapy for head and neck tumors (El-Sayed and Nelson, 1996; van der Reijden et al., 1999).
Acknowledgments
We are grateful to Drs. Wally Ip and Birgit Ehmer (University of Cincinnati Center for Biological Microscopy, Cincinnati, OH); David Quissel (University of Colorado Health Sciences Center, Denver, CO) for the SMG-C6 cell line; Santosh G. Dixit for help in mannitol and raffinose transport experiments; and our colleagues Drs. Roger Worrell and Paranthaman Senthamaraikannan for critical reading of and valuable suggestions on this manuscript.
This work was supported in part by Grant DE13823 from the National Institute of Dental and Craniofacial Research, National Institutes of Health.
ABBREVIATIONS
- TJC
tight junction complex
- PKA
protein kinase A
- MLCK
myosin light chain kinase
- ML-7
1-(5-iodonaphthalene-1-sulfonyl)homopiperazine, HCl
- H89
N-[2-((p-bromocinnamyl) amino) ethyl]-5-isoquinolinesulfonamide, 2HCl
- PKC
protein kinase C
- Ro-32-0432
2-{8-[(dimethylamino) methyl]-6,7,8,9-tetrahydropyrido[1,2-a]indol-3-yl}-3-(1-methylindol-3-yl) maleimide, hydrochloride
- SMG-C6
submandibular gland epithelial cells-clone #6
- HBSS
Hanks’ balanced salt solution
- PBS
phosphate-buffered saline
- TEER
transepithelial electric resistance
References
- Adson A, Raub TJ, Burton PS, Barsuhn CL, Hilgers AR, Audus KL, Ho NF. Quantitative approaches to delineate paracellular diffusion in cultured epithelial cell monolayers. J Pharm Sci. 1994;83:1529–1536. doi: 10.1002/jps.2600831103. [DOI] [PubMed] [Google Scholar]
- Böhme M, Diener M, Mestres P, Rummel W. Direct and indirect actions of HgCl2 and methyl mercury chloride on permeability and chloride secretion across the rat colonic mucosa. Toxicol Appl Pharmacol. 1992;114:285–294. doi: 10.1016/0041-008x(92)90079-8. [DOI] [PubMed] [Google Scholar]
- Burghardt B, Nielsen S, Steward MC. The role of aquaporin water channels in fluid secretion by the exocrine pancreas. J Membr Biol. 2006;210:143–153. doi: 10.1007/s00232-005-0852-6. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M, Mazzon E, De Rigo L, Baraldo S, Meggio F, Citi S. Occludin dephosphorylation in early development of Xenopus laevis. J Cell Sci. 1997;110:3131–3139. doi: 10.1242/jcs.110.24.3131. [DOI] [PubMed] [Google Scholar]
- El-Sayed S, Nelson N. Adjuvant and adjunctive chemotherapy in the management of squamous cell carcinoma of the head and neck region. A meta-analysis of prospective and randomized trials. J Clin Oncol. 1996;14:838–847. doi: 10.1200/JCO.1996.14.3.838. [DOI] [PubMed] [Google Scholar]
- Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57:883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Fey EG, Wan KM, Penman S. Epithelial cytoskeletal framework and nuclear matrix-intermediate filament scaffold: three-dimensional organization and protein composition. J Cell Biol. 1984;98:1973–1984. doi: 10.1083/jcb.98.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson HG, Matthay MA, Hasegawa H, Kheradmand F, Verkman AS. Transcellular water transport in lung alveolar epithelium through mercury-sensitive water channels. Proc Natl Acad Sci U S A. 1994;91:4970–4974. doi: 10.1073/pnas.91.11.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115. doi: 10.1167/iovs.06-0322. [DOI] [PubMed] [Google Scholar]
- Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest. 1988;82:1516–1524. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, Yokoyama M, Staddon JM. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem. 2001;276:10423–10431. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- Kawedia JD, Nieman ML, Boivin GP, Melvin JE, Kikuchi K, Hand AR, Lorenz JN, Menon AG. Interaction between transcellular and paracellular water transport pathways through Aquaporin 5 and the tight junction complex. Proc Natl Acad Sci U S A. 2007;104:3621–3626. doi: 10.1073/pnas.0608384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H(2)O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279:C21–C30. doi: 10.1152/ajpcell.2000.279.1.C21. [DOI] [PubMed] [Google Scholar]
- Klingler C, Kniesel U, Bamforth SD, Wolburg H, Engelhardt B, Risau W. Disruption of epithelial tight junctions is prevented by cyclic nucleotide-dependent protein kinase inhibitors. Histochem Cell Biol. 2000;113:349–361. doi: 10.1007/s004180000143. [DOI] [PubMed] [Google Scholar]
- Ko SB, Naruse S, Kitagawa M, Ishiguro H, Furuya S, Mizuno N, Wang Y, Yoshikawa T, Suzuki A, Shimano S, et al. Aquaporins in rat pancreatic interlobular ducts. Am J Physiol Gastrointest Liver Physiol. 2002;282:G324–G331. doi: 10.1152/ajpgi.00198.2001. [DOI] [PubMed] [Google Scholar]
- Lindmark T, Nikkila¨ T, Artursson P. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther. 1995;275:958–964. [PubMed] [Google Scholar]
- Ma TY, Hoa NT, Tran DD, Bui V, Pedram A, Mills S, Merryfield M. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol Gastrointest Liver Physiol. 2000;279:G875–G885. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- Macey RI. Transport of water and urea in red blood cells. Am J Physiol Cell Physiol. 1984;246:C195–C203. doi: 10.1152/ajpcell.1984.246.3.C195. [DOI] [PubMed] [Google Scholar]
- Pauletti GM, Gangwar S, Okumu FW, Siahaan TJ, Stella VJ, Borchardt RT. Esterase-sensitive cyclic prodrugs of peptides: evaluation of an acyloxy-alkoxy promoiety in a model hexapeptide. Pharm Res. 1996;13:1615–1623. doi: 10.1023/a:1016472119387. [DOI] [PubMed] [Google Scholar]
- Preston GM, Jung JS, Guggino WB, Agre P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J Biol Chem. 1993;268:17–20. [PubMed] [Google Scholar]
- Prichard MN, Daily SL, Jefferson GM, Perry AL, Kern ER. A rapid DNA hybridization assay for the evaluation of antiviral compounds against Epstein-Barr virus. J Virol Methods. 2007;144:86–90. doi: 10.1016/j.jviromet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quissell DO, Barzen KA, Gruenert DC, Redman RS, Camden JM, Turner JT. Development and characterization of SV40 immortalized rat submandibular acinar cell lines. In Vitro Cell Dev Biol Anim. 1997;33:164–173. doi: 10.1007/s11626-997-0137-8. [DOI] [PubMed] [Google Scholar]
- Rajasekaran SA, Barwe SP, Gopal J, Ryazantsev S, Schneeberger EE, Rajasekaran AK. Na-K-ATPase regulates tight junction permeability through occludin phosphorylation in pancreatic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G124–G133. doi: 10.1152/ajpgi.00297.2006. [DOI] [PubMed] [Google Scholar]
- Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368:471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkin EM. Filtration, diffusion, and molecular sieving through porous cellulose membranes. J Gen Physiol. 1954;38:225–243. [PMC free article] [PubMed] [Google Scholar]
- Roberts SK, Yano M, Ueno Y, Pham L, Alpini G, Agre P, LaRusso NF. Cholangiocytes express the aquaporin CHIP and transport water via a channel-mediated mechanism. Proc Natl Acad Sci U S A. 1994;91:13009–13013. doi: 10.1073/pnas.91.26.13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393–1401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DF, Stroud RM. Structural basis of aquaporin inhibition by mercury. J Mol Biol. 2007;368:607–617. doi: 10.1016/j.jmb.2007.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P. Aquaporin-1 in plasma membrane and caveolae provides mercury-sensitive water channels across lung endothelium. Am J Physiol Heart Circ Physiol. 1996;270:H416–H422. doi: 10.1152/ajpheart.1996.270.1.H416. [DOI] [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem. 1996;271:13636–13641. doi: 10.1074/jbc.271.23.13636. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Panichas M, Hebert SC, Brenner BM, Nigam SK. Critical role for intracellular calcium in tight junction biogenesis. J Cell Physiol. 1994;159:423–433. doi: 10.1002/jcp.1041590306. [DOI] [PubMed] [Google Scholar]
- van der Reijden WA, Vissink A, Veerman EC, Amerongen AV. Treatment of oral dryness related complaints (xerostomia) in Sjögren’s syndrome. Ann Rheum Dis. 1999;58:465–474. doi: 10.1136/ard.58.8.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtel M, Frei K, Ehler E, Fontana A, Winterhalter K, Gloor SM. Occludin proteolysis and increased permeability in endothelial cells through tyrosine phosphatase inhibition. J Cell Sci. 1999;112:4347–4356. doi: 10.1242/jcs.112.23.4347. [DOI] [PubMed] [Google Scholar]
- Wang Q, Luo W, Zheng W, Liu Y, Xu H, Zheng G, Dai Z, Zhang W, Chen Y, Chen J. Iron supplement prevents lead-induced disruption of the blood-brain barrier during rat development. Toxicol Appl Pharmacol. 2007;219:33–41. doi: 10.1016/j.taap.2006.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittembury G. Action of antidiuretic hormone on the equivalent pore radius at both surfaces of the epithelium of the isolated toad skin. J Gen Physiol. 1962;46:117–130. doi: 10.1085/jgp.46.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am J Physiol Cell Physiol. 1997;273:C1859–C1867. doi: 10.1152/ajpcell.1997.273.6.C1859. [DOI] [PubMed] [Google Scholar]
- Wong V, Gumbiner BM. A synthetic peptide corresponding to the extra-cellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Kim JK, Verkman AS. Comparative efficacy of HgCl2 with candidate aquaporin-1 inhibitors DMSO, gold, TEA+ and acetazolamide. FEBS Lett. 2006;580:6679–6684. doi: 10.1016/j.febslet.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]