

Abstract
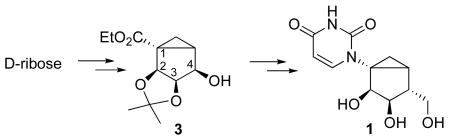
We have developed an approach toward enantiomerically pure (S)-methanocarba ribonucleosides based on several functional group transformations on a sensitive bicyclo[3.1.0]hexane system. D-Ribose was transformed into methanocarba alcohol 3 followed by conversion of the OH group to a nitrile with inversion of configuration at C4. The nitrile group was subsequently reduced in two stages to the 5′-hydroxymethyl group. An ester group appended to a tertiary carbon (C1) was transformed to an amino group as a nucleobase precursor.
Recent studies of the SAR of nucleosides and nucleotides as antiviral agents and receptor ligands have reported that conformational constraint of the ribose ring of such ligands allows the identification of preferred conformations of the normally freely-interconverting sugar ring. Use of the methanocarba, bicyclo[3.1.0]hexane ring system, which can be fixed in a North (N) or South (S) rigid envelope conformation,1 as a ribose substitute has greatly aided these studies. For example, (S)-methanocarba-thymidine specifically inhibits the growth of herpes simplex virus type 1 thymidine kinase-transduced osteosarcoma cells without the toxicity of its North counterpart.2 In the study of the GPCRs (G protein-coupled receptors) that respond to extracellular nucleosides (adenosine receptors) and nucleotides (P2Y receptors), the preference for a North (N) or South (S) ribose conformation depends on the receptor subtype.1,3,4,5 The P2Y6 receptor for UDP prefers the (S)-methanocarba analogue of dUDP, which is more potent than the corresponding 2′-deoxyriboside, dUDP. This knowledge is now being used to design additional nucleotide analogues having enhanced potency and selectivity for a given receptor subtype. (N)-methanocarba analogues of 2′-deoxyadenosine 3′,5′-bisphosphates constitute the most potent and selective antagonists of the P2Y1 receptor, which have potential as antithrombotic agents.5,6
Synthetic methods for methanocarba nucleosides have undergone refinement based on carbene cyclopropanation, metathesis, and other reactions.7,8,9,10 However, only the racemic form of (S)-methanocarba ribonucleosides has so far been reported.9 Our approach toward enantiomerically pure (S)-methanocarbanucleoside 1 (Scheme 1) was based on a previously published synthesis of (N)-methanocarba-based agonists of the A3 adenosine receptor.11 The synthesis of the North template started from L-ribose and involved as a key intermediate compound 2. A closer look at its enantiomer 3 and the target (S)-methanocarba derivative 1 revealed a similar carbocyclic skeleton possessing an identical configuration of the two OH groups at positions 2 and 3. Enantiomer 3 can be therefore converted into the target (S)-methanocarbanucleotide 1 through: (a) replacement of the C4 hydroxyl with a CH2OH group accompanied by inversion of configuration at C4; and (b) conversion of the ester group into a 2,4-pyrimidinedione ring.
Scheme 1.

Our initial synthetic plan (Scheme 1) involved conversion of alcohol 3 into alcohol 4 through substitution with a one-carbon nucleophile synthon. Subsequent transformations of alcohol 4 included converting the ester function into a primary amino group followed by final assembly of the uracil ring.
Preparation of alcohol 6 (Scheme 2) from D-ribose followed a published procedure for the synthesis of its enantiomer12 and was accomplished in 6% overall yield.13 Subsequent acid-catalyzed rearrangement of alcohol 6 provided a 60:40 equilibrium mixture of alcohols 3 and 6 that could not be separated chromatographically. The equilibrium mixture was enriched to a ratio of 80:20 by the selective crystallization of alcohol 6, leading to further enhancement to a ratio of 90:10 through recrystallization. The resultant mixture of alcohols 3 and 6 was converted into the corresponding triflates 7 and 8, which could be separated chromatographically. However, due to instability of triflates 7 and 8 all subsequent experiments were done using the mixture without purification.
Scheme 2.

A first attempt at substitution with a one-carbon nucleophilic synthon involved reaction of triflate 8 with 2-lithio 1,3-dithiane (Scheme 2). Although triflate 8 was found to be unstable under the reaction conditions the nucleophilic substitution provided the desired dithiane 9 in 35% yield. The reaction proceeded with inversion of configuration at C4, as evidenced by a change in the coupling constants J3,4 and J4,5 from 6 Hz to zero in the 1H NMR spectrum. Subsequent mercury perchlorate-assisted hydrolysis of dithiane 9 resulted in aldehyde 10 in low yield, most probably due to instability of the compound. Final reduction of aldehyde 10 with NaBH4 provided target alcohol 4.
Since the overall yield of alcohol 4 by this method was low, we tried an alternative one-carbon nucleophilic synthon. Nucleophilic substitution in triflate 8 with sodium cyanide (Scheme 2) provided the nitrile 11, although in only 40% yield; however, replacement of sodium cyanide with lithium cyanide resulted in improved yield. Furthermore, we found that separation of triflates 8 and 7 was not necessary, and a 90:10 mixture of these isomers could be subjected to nucleophilic substitution to provide nitrile 11 as the only isomer in 63% yield.14
Our original synthetic plan involved hydrolysis of the ester group in nitrile 11 followed by a Curtius rearrangement. However, basic hydrolysis of nitrile 11 with NaOH in MeOH-H2O (Scheme 3) resulted in formation of either ethyl 4-cyanobenzoate or 4-cyanobenzoic acid rather than the expected plain hydrolysis of the carboxylic group. The same outcome was observed with other reagents commonly used for basic hydrolysis, such as H2O-Et3N or K2CO3.
Scheme 3.

Since (N)-methanocarba derivatives are generally stable to bases, the observed transformation is undoubtedly related to the presence of an acidic proton at the α-position to the cyano group. The most probable mechanism of the rearrangement involves C4 deprotonation followed by ring-opening of the cyclopropane ring driven by the strain of the bicyclo[3.1.0]hexane system and aromatization of the resultant carbanion through sequential β-eliminations to give ethyl 4-cyanobenzoate 14.
We surmised that the undesired transformation could be avoided by decreasing the acidity of the α-proton at C4. This can be most easily accomplished through a reduction of the cyano group to obtain a CH2OH group. However, reduction of the cyano group of the nitrile was complicated since complex hydrides that are most commonly used for such transformations are incompatible with the ester functionality of nitrile 11. This problem was solved through the catalytic hydrogenation of nitrile 11 (Scheme 4). Although catalytic hydrogenation of nitriles is most commonly used for the preparation of amines,15 we found that the reaction could be stopped at the stage of aldehyde 10. Conducting the hydrogenation in a MeOH-H2O-AcOH solution resulted in rapid hydrolysis of the initially formed imine 15, thus preventing its further reduction. The resulting unstable aldehyde 10 was immediately reduced with NaBH4 to yield alcohol 4 in 55% overall yield.
Scheme 4.

TBDPS protection of alcohol 4 produced silyl ether 16, which was hydrolyzed into acid 17 (Scheme 4). Acid 17 was converted into the corresponding azide 18 through formation of a mixed anhydride followed by reaction with sodium azide. Thermal rearrangement of azide 18 was conducted in the presence of benzyl alcohol and resulted in the Cbz-protected amine 19, which was deprotected by hydrogenation to afford amine 20 (Scheme 5).
Scheme 5.

Building of the uracil ring from amine 20 followed a previously described procedure for the syntheses of chiral 2′-deoxyribo versions of (S)-methanocarba-nucleosides.16 Reaction of the amine 20 with an 3-ethoxyacryloyl isocyanate16 afforded the urea 21 in 70% yield. Cyclization of 21 with ethanolic HCl resulted in the concomitant removal of the acetal and TBDPS protection of the hydroxyl groups, to yield the target (S)-methanocarba nucleoside 1 in 21% yield from compound 21.
In conclusion, we have developed an approach toward enantiomerically pure (S)-methanocarba nucleosides based on functional group transformation on a sensitive bicyclo[3.1.0]hexane system. These derivatives are now suitable for detailed studies in biological systems.
Experimental Section
(1S,2S,5S,3R,4R)-1-Ethoxycarbonyl-4-cyano-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0] hexane (11)
A solution of alcohol 3 (0.97 g, 4 mmol) and pyridine (0.35 g, 4.4 mmol) in CH2Cl2 (8.8 mL) at 0 °C was treated dropwise over a period of 2 min with a solution of trifluoromethanesulfonic anhydride (1.24 g, 4.4 mmol) in CH2Cl2 (8.8 mL). The reaction mixture was stirred at 0 °C for 10 min more after completing the addition, and hexane (25 mL) was added. After 5 min the resultant suspension was filtered through pad of silica gel and the silica gel was washed with 30% EtOAc-hexane mixture (100 mL). The combined organic filtrates were evaporated at room temperature, the resultant residue of the triflate was dissolved in dry CH2Cl2 (2 mL) at 0 °C, and 0.5 M solution of lithium cyanide in DMF (7.8 mL) was added to the solution. The reaction mixture was stirred at 0 °C for 30 min, dissolved in a 50% EtOAc-hexane mixture (50 mL), washed with water (2 × 10 mL) and brine (1 × 10 mL), dried (Na2SO4), and evaporated. The residue was purified by flash chromatography (10% to 20% EtOAc-hexane) to afford 11 as colorless oil (0.63 g, 2.5 mmol, 63%); [α]D20 = −77.9° (c = 1.82, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.54 (d, 1H, J = 6.9 Hz), 4.90 (d, 1H, J =6.9 Hz), 4.20 (m, 2H), 3.18 (s, 1H), 2.40 (dd, 1H, J = 5.7, 9.6 Hz), 1.67 (dd, 1H, J = 5.4, 9.3 Hz), 1.50 (s, 3H), 1.42 (t, 1H, J =5.4 Hz), 1.30 (s, 3H), 1.29 (t, 3H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3): 170.9, 119.1, 112.9, 85.7, 80.7, 61.5, 35.6, 34.8, 26.0, 24.2, 19.3, 14.2.
(1S,2S,5S,3R,4R)-1-Ethoxycarbonyl-2,3-O-isopropylidene-2,3-dihydroxy-4-(hydroxymethyl)bicyclo[3.1.0]hexanecarboxylate (4)
Method A. A solution of nitrile 11 (0.25 g, 1 mmol) in MeOH (3 mL), H2O (1.5 mL), and HOAc (0.5 mL) was treated with 10% Pd/C (20 mg). The flask was filled with hydrogen and stirred for 2 h at room temperature. The mixture of unreacted nitrile 11 and aldehyde 10 was filtered, and the filtrate was evaporated at room temperature. The residue was dissolved in EtOAc (30 mL), washed with satd. NaHCO3-H2O solution until CO2 evolution ceased, dried (Na2SO4), and evaporated at room temperature. The residue was purified by flash chromatography (20% to 30% EtOAc-hexane) to afford the crude aldehyde, which was dissolved in MeOH (5 mL) and treated with NaBH4 (20 mg, 0.5 mmol), followed by acetone (1 mL) after 10 min.). The reaction mixture was evaporated and the residue was dissolved in EtOAc (20 mL), washed with brine (3 mL), dried (Na2SO4), and evaporated. The residue is purified by flash chromatography (20 to 30% EtOAc-hexane) to afford the title compound as colorless oil (0.14 g, 0.55 mmol, 55%). [α]D20 = −18.35°(c = 0.60, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.36 (d, 1H, J = 6.6 Hz), 4.59 (d, 1H, J = 6.9 Hz), 4.15 (m, 2H), 3.66 (br. m, 2H), 2.37 (t, 1H, J = 6.0 Hz), 1.99 (dd, 1H, J = 6.0, 9.3 Hz), 1.49 (s, 3H), 1.42 (t, 1H, J = 5.2 Hz), 1.29 (s, 3H), 1.25 (t, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 172.8, 110.0, 85.0, 80.5, 64.6, 60.9, 46.8, 37.6, 35,5, 26.2, 24.1, 19.6, 14.2.
(1S,2S,5S,3R,4R)-1-Ethoxycarbonyl-4-[(tert-butyldiphenylsilyloxy)methyl]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0]hexane (16)
To a stirred solution of alcohol 4 (0.80 g, 3.1 mmol) and imidazole (0.10 g, 1.5 mmol) in DMF (3 mL) was added neat tert-butyldiphenylchlorosilane (1.26 g, 4.5 mmol) followed by the dropwise addition of triethylamine (0.81 g, 8 mmol). The reaction mixture was stirred for 14 h at room temperature diluted with 20% EtOAc-hexane (50 mL), washed with water (2 × 20 mL), brine, dried (Na2SO4), and evaporated. The residue was purified by flash chromatography (0% to 20% EtOAc-hexane) to give silyl ether 16 as colorless oil (1.05 g, 2.1 mmol, 69%); [α]D20 = – 45.8° (c = 0.98, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.64 (m, 4H), 7.42 (m, 6H), 5.37 (d, 1H, J = 7.2 Hz), 4.57 (d, 1H, J = 7.2 Hz), 4.13 (m, 2H), 3.65 (br. m, 2H), 2.35 (t, 1H, J = 4.8 Hz), 2.07 (dd, 1H, J = 6.0, 9.6 Hz), 1.50 (s, 3H), 1.41 (t, 1H, J =5.1 Hz), 1.29 (s, 3H), 1.24 (t, 3H, J = 7.2 Hz), 1.05 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 172.9, 135.7, 133.1, 129.9, 127.8, 110.9, 85.5, 80.9, 65.9, 60.9, 46.7, 38.1, 36.2, 26.9, 26.3, 24.2, 19.9, 19.2, 14.3.
(1S,2S,5S,3R,4R)-4-[(tert-butyldiphenylsilyloxy)methyl]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylic acid (17)
A solution of silyl ether 16 (1.0 g, 2 mmol) in methanol (50 mL) was treated with 6 M NaOH (4 mL) and stirred under reflux for 2 h, evaporated, neutralized with conc. HCl, and extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were dried (Na2SO4), evaporated, and the residue was purified by flash chromatography (10% to 50% EtOAc-hexane) to afford 17 as colorless oil (0.71 g, 1.5 mmol, 76%); [α]D20 = −51.5° (c = 0.80, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.62 (m, 4H), 7.39 (m, 6H), 5.32 (d, 1H, J = 7.2 Hz), 4.53 (d, 1H, J = 7.2 Hz), 3.65 (br. m, 2H), 2.38 (t, 1H, J = 4.8 Hz), 2.15 (m, 1H), 1.60 (m, 1H), 1.50 (s, 3H), 1.29 (s, 3H), 1.27 (t, 3H, J = 7.2 Hz), 1.05 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 179.8, 135.7, 133.1, 129.9, 127.9, 111.1, 85.2, 80.4, 65.8, 46.8, 37.8, 36.9, 26.9, 26.3, 24.2, 20.7, 19.2.
(1S,2S,5S,3R,4R)-1-azidocarbonyl-4-[(tert-butyldiphenylsilyloxy)methyl]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0]hexane (18)
A solution of acid 17 (0.70 g, 1.5 mmol) and N,N-diisopropylethylamine (0.20 g, 1.6 mmol) in acetone (15 mL) at room temperature was treated with neat diphenylchlorophosphate (0.43 g, 1.6 mmol). The reaction mixture was stirred for 30 min followed by treatment with aqueous sodium azide (0.13 g in 2 mL, 2 mmol). The reaction mixture was stirred for 30 min, evaporated at room temperature, and the residue was partitioned between water (10 mL) and CH2Cl2 (50 mL). The organic layer was dried (Na2SO4), evaporated, and the residue was purified by flash chromatography (10 to 30% EtOAc-hexane) to afford the title compound as colorless oil (0.67 g, 0.13 mmol, 88%); [α]D20 = −63.0° (c = 0.48, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.63 (m, 4H), 7.35 (m, 6H), 5.21 (d, 1H, J = 6.3 Hz), 4.45 (d, 1H, J =7.2 Hz), 3.59 (d, 2H, J = 4.2 Hz), 2.29 (t, 1H, J =4.2 Hz), 2.10 (m, 1H), 1.56 (m, 1H), 1.50 (m, 1H), 1.41 (s, 3H), 1.20 (s, 3H), 0.99 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 179.4, 135.7, 133.1, 130.0, 127.9, 111.2, 85.4, 80.4, 65.8, 46.2, 41.1, 38.3, 26.9, 26.3, 24.2, 21.9, 19.3.
N-{(1S,2S,5S,3R,4R)-1-benzyloxycarbamino-4-[(tert-butyldiphenylsilyloxy)methyl]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0]hexane (19)
A solution of azidoketone 18 (0.67 g, 1.3 mmol) and benzyl alcohol (1 mL) in toluene (20 mL) was refluxed for 5 h. The reaction mixture was evaporated, the residue was twice purified by flash chromatography (5% methanol-chloroform, then 30% EtOAc-hexane) to afford title compound as colorless oil (0.54 g, 1.1 mmol, 69%); [α]D20 = −24.7° (c = 1.08, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.78 (br. d, 4H), 7.37 (m, 11H), 5.32 (br. s, 1H), 5.18 (br. s, 2H), 5.05 (br. d, 1H, J = 5 Hz), 4.57 (br. d, 1H, J = 6 Hz), 3.97 (br. s, 2H), 2.42 (br. t, 1H, J = 6.6 Hz), 1.72 (m, 1H), 1.60 (s, 3H), 1.29 (s, 3H), 1.43 (s, 3H), 1.19 (s, 9H), 1.08 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 159.1, 156.2, 136.6, 135.8, 135.7, 129.8, 127.9, 128.2, 128.6, 133.8, 111.0, 84.1, 83.4, 66.6, 65.3, 47.7, 46.0, 31.4, 27.0, 26.3, 24.2, 19.4, 17.7.
(1S,2S,5S,3R,4R)-1-Amino-4-[(t-butyldiphenylsilyloxy) methyl]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0] hexane (20)
A solution of benzyl carbamate 19 (0.63 g, 1.1 mmol) in methanol (5 mL) was stirred under hydrogen in the presence of 10% Pd on carbon (50 mg) for 2 h at atmospheric pressure. The reaction mixture was filtered and the residue was evaporated to afford title compound as colorless oil (0.42 g, 0.96 mmol, 87%); [α]D20 = −22.9° (c = 0.68, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.66 (m, 4H), 7.42 (m, 6H), 4.68 (d, 1H, J = 7.5 Hz), 4.48 (d, 1H, J = 7.5 Hz), 3.68 (m, 2H, J = 4.2 Hz), 2.17 (t, 1H, J = 4.2 Hz), 1.6 (m, 1H), 1.49 (s, 3H), 1.36 (m, 1H), 1.25 (s, 3H), 1.09 (s, 9H), 0.97 (t, 1H, J = 4.2 Hz); 13C NMR (75 MHz, CDCl3) δ 135.7, 133.4, 129.9, 127.8, 88.5, 84.7, 66.4, 48.5, 47.0, 32.0, 27.1, 26.9, 26.4, 24.4, 19.4, 18.1.
((1S,2S,3R,4R)-1-(3-Ethoxyacryloyl)aminocarbamoyl-4-[(tert-butyldiphenylsilyloxy]-2,3-O-isopropylidene-2,3-dihydroxybicyclo[3.1.0]hexane (21)
To a solution of 3-ethoxyacrylic acid (2 mmol) in CH2Cl2 (1 mL) was added neat oxalyl chloride (2 mmol) and DMF (0.02 g). The reaction mixture was stirred for 30 min, and was evaporated at room temperature. The residue was dissolved in toluene (5 mL) and stirred with silver cyanate (4 mmol) for 4 h. The resultant solution was filtered, and a 1.25 mL aliquot of the solution was evaporated for reaction with amine 20. The evaporated aliquot was dissolved in CH2Cl2 (2 mL) and added to a precooled (-78 °C) solution of amine 20 (0.20 g, 0.46 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 14 h. The reaction mixture was evaporated and the residue was purified by flash chromatography to afford the title urea as pale yellow oil (0.20 g, 0.34 mmol, 74%); [α]D20 = −32.7° (c = 0.44, CHCl3); 1H NMR (300 MHz, CDCl3) δ 9.09 (s, 1H, 8.92 (s, 1H), 7.66 (m, 4H), 7.57 (d, 1H, J = 12.0 Hz), 7.36 (m, 6H), 5.16 (d, 1H, J = 12 Hz), 5.05 (d, 1H, J = 7.2 Hz), 4.43 (d, 1H, J = 7.2 Hz), 3.82 (m, 4H), 2.34 (t, 1H, J = 7.8 Hz), 1.67 (dd, 1H, J = 4.8, 9.0 Hz), 1.50 (s, 3H), 1.23 (m, 7H), 1.05 (s, 9H); 13C (75 MHz, CDCl3) δ 167.4, 163.0, 155.1, 135.6, 133.8, 129.7, 127.7, 110.9, 97.8, 83.8, 83.5, 67.6, 65.3, 47.9, 45.2, 31.4, 27.0, 26.3, 24.1, 19.4, 17.4, 14.5.
1-[(1S,2S,5S,3R,4R)-2,3-Dihydroxy-4-(hydroxymethyl) bicyclo[3.1.0]hexyl]-1,3-dihydropyrimidine-2,4-dione (1)
A solution of urea 21 (0.20 g, 0.34 mmol) in ethanol (3 mL) was added conc. HCl (0.1 mL). The reaction mixture was refluxed for 5 h, and evaporated. The residue was purified by flash chromatography (0 to 20% MeOH-chloroform) to afford compound as amorphous solid (18 mg, 0.071 mmol, 21%); [α]D20 = -51.6 (c = 0.64, MeOH). 1H NMR (300 MHz, CD3OD) δ 7.50 (d, 1H, J = 7.5 Hz), 5.59 (d, 1H, J = 8.1 Hz), 4.43 (d, 1H, J = 6.6 Hz), 3.83 (d, 1H, J = 6.6 Hz), 3.60 (m, 2H), 2.07 (t, 1H, J = 4.8 Hz), 1.60-1.70-1 (m, 2H), 0.90 (m, 1H); 13C NMR (75 MHz, CD3OD) δ 184.9, 151.1, 105.3, 78.8, 76.0, 67.7, 56.2, 54.3, 29.4, 18.5, 13.5.
Supplementary Material
General experimental details, procedures for synthesis of compounds 9, 10, and 4 (method B); 1H and 13C NMR spectra and HRMS measurements of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.”
Acknowledgments
We thank Dr. Dina M. Sigano for optical rotation measurements and Drs. James A. Kelley, Christopher Lai, and Dilip Tosh for high resolution mass spectral measurements. This research was supported in part by the Intramural Research Programs of the National Institutes of Health, NIDDK and the Center for Cancer Research, National Cancer Institute at Frederick.
References
- 1.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 2.Schelling P, Claus MT, Johnert R, Marquez VE, Schulz GE, Scapozza L. J Biol Chem. 2004;279:32832–32838. doi: 10.1074/jbc.M313343200. [DOI] [PubMed] [Google Scholar]
- 3.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa M, Marquez VE, Harden TK, Jacobson KA. J Med Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA. J Med Chem. 2003;46:4974–4987. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, Jacobson KA, Gachet C. J Pharm Exp Therap. 2006;316:556–563. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee K, Cass C, Jacobson KA. Org Lett. 2001;3:597–599. doi: 10.1021/ol006999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallos JK, Koftis TV, Massen ZS, Dellios CC, Mourtzinos IT, Coutouli-Argyropoulou E, Koumbis AE. Tetrahedron. 2002;58:8043–8053. [Google Scholar]
- 9.Shin KJ, Moon HR, George C, Marquez VE. J Org Chem. 2000;65:2172–2178. doi: 10.1021/jo9917691. [DOI] [PubMed] [Google Scholar]
- 10.Lee JA, Kim HO, Tosh DK, Moon HR, Kim S, Jeong LS. Org Lett. 2006;8:5081–5083. doi: 10.1021/ol061959f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joshi BV, Moon HR, Fettinger JC, Marquez VE, Jacobson KA. J Org Chem. 2005;70:439–447. doi: 10.1021/jo0487606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi BV, Melman A, Mackman RL, Jacobson KA. Nucleosides, Nucleotides, and Nucleic Acids. 2007 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.All intermediate compounds possessed spectral data identical to the published ones in ref. 12.
- 14.Triflate 7 presumably does not produce the corresponding nitrile.
- 15.Hudlicky M. Reductions in Organic Chemistry. Washington, D.C.: American Chemical Society; 1984. pp. 239–240. [Google Scholar]
- 16.Ezzitouni A, Marquez VE. J Chem Soc, Perkin Trans. 1997;1:1073–1078. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental details, procedures for synthesis of compounds 9, 10, and 4 (method B); 1H and 13C NMR spectra and HRMS measurements of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.”