

Summary
The epithelial chloride channel CFTR is a glycoprotein that is modified by two N-linked oligosaccharides. The most common mutant CFTR protein in patients with cystic fibrosis, ΔF508, is misfolded and retained by ER quality control. As oligosaccharide moieties of glycoproteins are known to mediate interactions with ER lectin chaperones, we investigated the role of N-linked glycosylation in the processing of wild-type and ΔF508 CFTR. We found that N-glycosylation and ER lectin interactions are not major determinants of trafficking of wild-type and ΔF508 from the ER to the plasma membrane. Unglycosylated CFTR, generated by removal of glycosylation sites or treatment of cells with the N-glycosylation inhibitor tunicamycin, did not bind calnexin, but did traffic to the cell surface and exhibited chloride channel activity. Most importantly, unglycosylated Δ F508 CFTR still could not escape quality control in the early secretory pathway and remained associated with the ER. However, the absence of N-linked oligosaccharides did reduce the stability of wild-type CFTR, causing significantly more-rapid turnover in post-ER compartments. Surprisingly, the individual N-linked carbohydrates do not play equivalent roles and modulate the fate of the wild-type protein in different ways in its early biosynthetic pathway.
Keywords: CFTR, Glycosylation, Glycoprotein, Processing, Calnexin, EDEM
Introduction
The carbohydrate moieties of glycoproteins often do not play a role in their catalytic activities but contribute to stability, trafficking and localization within cells (Chow and Forte, 1995; de Souza and Simon, 2002; Hendriks et al., 2004; Li et al., 2007; Standley and Baudry, 2000).
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), including ΔF508, which is present in most cystic fibrosis patients, result in thermolability and mislocalization of the ion channel protein within epithelial cells (Cheng et al., 1990; Denning et al., 1992). As a result, the mutated nascent polypeptide cannot pass ER quality control. Although ΔF508 acquires core N-linked oligosaccharide chains in the ER, these are not extended because it cannot progress from the ER to the Golgi, where the glycosyltransferases required for chain extension are localized. Even the wild-type protein achieves a mature conformation state very inefficiently, so that only about a third of the core-glycosylated wild-type polypeptides are exported from the ER to the Golgi and, once glycosylation is complete there, progress to the plasma membrane (Lukacs et al., 1994; Ward and Kopito, 1994). Thus, the presence or absence of the complex oligosaccharide chains is diagnostic of the extent to which conformational maturation has enabled ER export to occur.
However, there have been proposals that CFTR oligosaccharide chains and their interactions with lectin chaperones in the ER are actively involved in the extent to which wild-type or mutant CFTR transits through steps in the early secretory pathway. It has been reported that ΔF508 CFTR stability and processing are modulated by the ER lectin calnexin (Okiyoneda et al., 2004; Okiyoneda et al., 2002), and inhibition of the association of CFTR with calnexin has been claimed to promote ΔF508 maturation (Egan et al., 2002; Egan et al., 2004; Harada et al., 2007; Norez et al., 2006a; Norez et al., 2006b); however, this effect could not be confirmed by others (Dragomir et al., 2004; Farinha and Amaral, 2005; Grubb et al., 2006; Loo et al., 2004; Song et al., 2004).
Here, we have addressed this issue in both non-polarized and polarized cells by assessing the effects of genetic and pharmacological prevention of the addition of one or both of the N-linked oligosaccharide chains on the trafficking and localization of wild-type and ΔF508 CFTR. Whereas these chains do not appear to be the primary determinants of ER retention or export, they do influence the stability and lifetime of wild-type CFTR, and the individual chains have different impacts on the fate of the protein in the ER.
Results
N-linked oligosaccharides of CFTR are required for calnexin binding, but not for channel function
The ER lectin chaperone calnexin has been shown to bind to monoglucosylated N-linked oligosaccharides of many glycoproteins until conformational maturation is complete (Helenius and Aebi, 2001; Parodi, 2000; Pind et al., 1994). CFTR binds to calnexin (Pind et al., 1994) and we mutated the two potential N-linked glycosylation sites of human CFTR (Fig. 1) to test whether this interaction is disrupted by removal of N-linked oligosaccharides. Mutation of native N-linked glycosylation sites (N894D/N900D) and treatment of cells with tunicamycin resulted in expression of unglycosylated CFTR (Fig. 2A). Calnexin co-immunoprecipitated with wild-type CFTR, whereas its soluble homolog calreticulin did not (supplementary material Fig. S2). The unglycosylated CFTR variant did not interact with calnexin (Fig. 2B) and, thus, the binding was solely mediated by the oligosaccharides. The unglycosylated CFTR, created either by mutation of the two glycosylation sites or by inhibition of N-linked glycosylation with tunicamycin, is functional and has similar channel properties to wild-type CFTR (Fig. 2C). Thus, N-linked oligosaccharides of wild-type CFTR are required for calnexin binding, but their absence does not prevent the protein from reaching a functional state.
Fig. 1.

Comparison of the sequences coding for the two potential N-glycosylation sites, 894 and 900, in extracellular loop 4 of CFTR. Sequences of mammalian CFTRs were aligned using FASTA. Amino acids fitting the consensus N-X-S/T for asparagine-linked glycosylation are highlighted. X can be any amino acid residue other than proline (Imperiali and Hendrickson, 1995).
Fig. 2.

Unglycosylated CFTR does not bind to calnexin but functions as a chloride channel. (A) Western blot of CFTR and unglycosylated CFTR created either by mutation of the glycosylation sites (N894D/N900D) or by expressing CFTR in tunicamycin-treated cells (+ Tunicamycin). CFTR and CFTR N894D/N900D variants were stably (left panel) or transiently (right panel) expressed in BHK-21 cells. Tunicamycin (5 μg/ml) was added directly after transient transfection to achieve complete inhibition of N-linked glycosylation. CFTR was detected in the microsomal membrane vesicle fraction by western blotting using mouse monoclonal anti-CFTR antibody 596. (B) CFTR N894D/N900D does not interact with calnexin. CFTR was immunoprecipitated by incubation with anti-CFTR antibody 596 crosslinked to Dynabeads (as indicated on the left). CFTR and calnexin (CNX) were detected by western blotting using mouse monoclonal antibody 596 for CFTR or rabbit polyclonal antibody SPA860 (Stressgen) for calnexin (as indicated on the right). Molecular weight marker positions (kDa) are indicated on the left. (C) The channel properties of unglycosylated CFTR are similar to wild-type CFTR. Single-channel measurements were performed using membrane vesicles prepared from stably transfected BHK-CFTR, BHK-N894D/N900D cells or from cells transiently transfected with CFTR that were treated with tunicamycin. Tunicamycin (5 μg/ml) was added directly after transient transfection to achieve complete inhibition of N-linked glycosylation.
Inhibition of interactions with ER lectin chaperones does not affect localization of wild-type or ΔF508 CFTR
To analyze whether the N-linked oligosaccharides are involved in retention of ΔF508 CFTR at the ER, we expressed ΔF508 with mutations at both N-glycosylation sites (ΔF508 N894D/N900D) (Fig. 3A). Similar to non-glycosylated wild-type CFTR, ΔF508 N894D/N900D did not interact with calnexin (Fig. 3B). To test the influence of the absence of N-linked oligosaccharides on the processing of ΔF508 CFTR, we analyzed localization and cAMP-stimulated channel activity at the cell surface. At physiological temperature (37°C), ΔF508 localized only to the ER regardless of whether or not it was glycosylated (ΔF508 or ΔF508 N894D/N900D), as observed by immunolocalization of permeabilized cells (Fig. 3C). Staining of the surface CFTR of intact cells with an antibody recognizing an external epitope confirmed that unglycosylated ΔF508 N894D/N900D was not present at the plasma membrane (Fig. 3D). ΔF508 CFTR is a temperature-sensitive mutation with the consequence that the protein cannot mature conformationally at 37°C, but is able to avoid ER quality control and proceed to the plasma membrane at 27°C (Denning et al., 1992). When cells were incubated at 27°C, both glycosylated ΔF508 and unglycosylated ΔF508 N894D/N900D could be detected at the cell surface (Fig. 3D). Thus, the absence of N-linked oligosaccharides did not seem to affect intracellular localization and processing of ΔF508 CFTR.
Fig. 3.

ΔF508 N894D/N900D cannot escape ER quality control. (A) Western blot of CFTR, CFTR N894D/N900D, ΔF508 and ΔF508 N894D/N900D expressed in BHK-21 cells. Lysates were separated by SDS-PAGE and CFTR was detected after transfer to nitrocellulose by mouse monoclonal anti-CFTR antibody 570. (B) ΔF508 N894D/N900D does not interact with calnexin. ΔF508 N894D/N900D was immunoprecipitated by incubation with anti-CFTR antibody 596 crosslinked to Dynabeads. CFTR and calnexin (CNX) were detected by western blotting using mouse monoclonal antibody 596 for CFTR or rabbit polyclonal antibody SPA860 for calnexin. Molecular weight marker positions (kDa) are indicated on the left. (C) Immunofluorescence microscopy of CFTR and ΔF508 glycosylation variants in permeabilized cells. Immunostaining was performed on permeabilized BHK-21 cells using anti-CFTR mouse monoclonal antibody 570, followed by goat anti-mouse IgG Alexa Fluor 488 conjugate. Calnexin was stained to visualize the ER compartment using rabbit anti-calnexin antibodies followed by goat anti-rabbit IgG Alexa Fluor 568 conjugate. (D) Visualization of cell-surface CFTR on non-permeabilized cells by applying antibody HA11 to detect the external HA epitope in EL2 of CFTR variants. Cells were grown at 37°C or incubated at 27°C for 48 hours in the presence of 2 mM sodium butyrate to promote cell-surface expression of Extope-ΔF508 CFTR. (E) cAMP-stimulated 36Cl- efflux measurements of stably expressing BHK-CFTR cells. Stimulation cocktail was added (+) at time 0. Each point represents the average of three independent samples and standard deviations are indicated. (F) Immunostaining of CFTR and ΔF508 glycosylation variants in virally transduced well-differentiated primary human airway epithelial cells. All pools of intracellular CFTRs were stained on frozen sections of cultures grown at 37°C with HA11 antibody followed by goat anti-mouse IgG Alexa 488 conjugate. (G) Labeling of apical CFTR in virally transduced well-differentiated primary human airway epithelial cells. Cultures were incubated at 27°C for 48 hours and the apical pool of CFTR variants was labeled with HA11 antibody; cultures were then frozen in OCT and frozen sections labeled with goat anti-mouse Alexa 488 conjugate. Scale bars: 10 μm.
Consistent with these findings, a functional assessment of the influence of the lack of glycosylation indicated that the wild-type protein was not severely impaired, nor the mutant facilitated, in reaching the cell surface (Fig. 3E). Cells expressing unglycosylated CFTR N894D/N900D showed a robust chloride efflux response to cAMP-elevating stimuli, whereas ΔF508 N894D/N900D did not display any activity, reflecting the absence of the channel protein at the plasma membrane.
To determine the influence of glycosylation on ER-to-cell-surface trafficking in a more native CFTR environment (Kreda et al., 2005), we transduced well-polarized human primary airway epithelial cells with viral vectors to express glycosylated and unglycosylated forms of wild-type and ΔF508 CFTR. At 37°C, the wild-type protein was observed primarily in its native location in the apical membrane regardless of its glycosylation state, whereas neither glycosylated ΔF508 nor unglycosylated ΔF508 N894D/N900D CFTR was able to reach the apical membrane and was instead localized intracellulary (Fig. 3F). However, when the cells were cultured at 27°C, the mutant protein became distinctly localized to the apical compartment, independently of its glycosylation state (Fig. 3G). Thus, the glycosylation state and the consequent interaction with calnexin did not appear to be primary determinants of the trafficking of either wild-type or ΔF508 CFTR, in either polarized or non-polarized cells.
In addition to calnexin, another ER lectin, EDEM (ER degradation-enhancing α-mannosidase-like protein; EDEM1), is able to bind CFTR (Fig. 4A) (Gnann et al., 2004). Overexpression of EDEM enhances the rate of ER-associated degradation (ERAD) of CFTR (Fig. 4B) (Farinha and Amaral, 2005; Gnann et al., 2004). We used glycosidase inhibitors to perturb the interaction of EDEM with nascent ΔF508 CFTR. Both 1-deoxymannojirimycin (DMM) and kifunensine inhibit the cleavage of a mannose residue required for interaction with EDEM (Elbein et al., 1990; Fuhrmann et al., 1984; Oda et al., 2003). Neither of these drugs influenced the residence of ΔF508 CFTR in the ER (Fig. 4C) or increased the amount of the mutant protein at the cell surface (Fig. 4D). A similar pharmacological experiment with castanospermine, which inhibits the glucosidase I activity (Sasak et al., 1985) necessary for calnexin binding, yielded the same result, consistent with our previous observations. Thus, neither of these ER lectins appears to be responsible for retention of the misassembled ΔF508 CFTR protein in the ER.
Fig. 4.
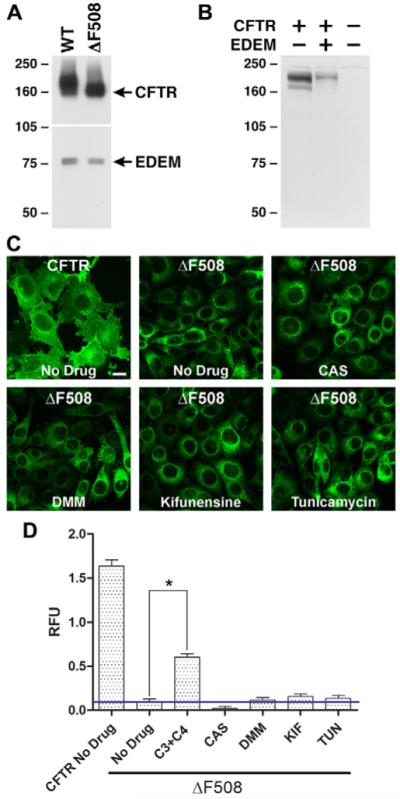
EDEM binds CFTR and accelerates its degradation, but neither the inhibition of EDEM interaction nor of calnexin interaction can rescue ΔF508 CFTR. (A) CFTR was immunoprecipitated from cells transiently expressing HA-tagged EDEM by anti-CFTR monoclonal antibody 596 crosslinked to Dynabeads. EDEM was detected by western blotting using monoclonal antibody HA11 and CFTR was detected by antibody 596. (B) CFTR was expressed transiently in BHK-21 cells alone or together with EDEM using pcDNA3 CFTR and pCMV-SPORT2 EDEM, respectively. In the case of CFTR expression alone, similar amounts of empty control vector were co-transfected with the CFTR plasmid. (C) Immunofluorescence microscopy of ΔF508 CFTR expressed in BHK-21 cells that were treated with various glycosidase inhibitors or tunicamycin. CAS (castanospermine, 0.2 mM) inhibits glucosidase I; DMM (1-deoxymannojirimycin, 0.5 mM) and kifunensine (0.2 mM) are inhibitors of mannosidase I; and tunicamycin (10 μg/ml) completely blocks N-glycosylation. The activity of these compounds was confirmed (supplementary material Fig. S3; Fig. 2A). Inhibitors were added to the growth media for 18 hours at the concentrations indicated and ΔF508 CFTR was visualized on permeabilized cells using anti-CFTR monoclonal antibody 596 followed by goat anti mouse IgG Alexa Fluor 488 conjugate. Scale bar: 10 μm. (D) Cell-surface ELISA showing that glycosidase inhibitors do not rescue ΔF508. BHK-21 cells expressing an externally tagged ΔF508 CFTR (Gentzsch et al., 2004) were seeded in a 96-well plate at 40,000 cells per well. Twenty-four hours after seeding, cells were treated for 20 hours with CAS (0.4 mM), DMM (0.5 mM), KIF (kifunensine, 0.2 mM) or tunicamycin (10 μg/ml). Corrector compounds C3 (VRT-325) and C4 (corr-4a), which have been reported to partially rescue ΔF508 CFTR (Loo et al., 2005; Pedemonte et al., 2005; Van Goor et al., 2006), were used as positive control at 20 μM each. Cells were fixed, labeled with HA11 antibody that recognizes the external tag, followed by goat anti-mouse IgG conjugated to X-Sight 761 and scanned with an infrared imaging system. A significant increase in the cell-surface pool of ΔF508 CFTR was observed on treatment with C3 and C4 (*P<0.0001). Neither treatment with glycosidase inhibitors nor complete inhibition of N-glycosylation by tunicamycin had an effect that was significantly different from the no-drug control. Data represent the average of eight wells; bars indicate s.e.m. Statistical significance was determined using an unpaired Student’s t-test. The blue line indicates the baseline level of the no-drug control.
Oligosaccharides impact CFTR maturation and turnover
Although the presence or absence of both oligosaccharide chains does not have a major effect on the steady-state localization of CFTR, the influence of the two together or individually on turnover is unknown. We performed pulse-chase labeling experiments with the variant in which both N-glycosylation sites were mutated, as in the localization experiments (Fig. 3), and also with variants with each individual site substituted. Prior to these kinetic experiments, the possibility that the two chains might have different effects on processing and turnover had not been explored. Western blots comparing the wild-type protein and the three different variants (Fig. 5A) detected only the unglycosylated form of the double mutant. The single-chain N894D species primarily formed a single band of mobility intermediate between the unglycosylated and wild-type forms, presumably with a complex N-linked chain at the N900 site. However, the other single-site variant, N900D, generated two strong bands, one with a similar mobility to that of N894D, probably representing the presence of the other single chain, and another more rapidly migrating band, most likely representative of a core rather than complex oligosaccharide chain at this site. The identity of the core-glycosylated forms of N894D and N900D was confirmed by digestion with endoglycosidase H, which selectively deglycosylates unprocessed core-glycosylated proteins and thus converted CFTR to a band with the same mobility as the N894D/N900D double mutant (Fig. 5B). Thus, the individual N-glycosylation sites are utilized when only one of them is present, and they undergo core and complex glycosylation. However, it appears that the nascent chain with only the N894 site available (N900D), progresses from the core to complex glycosylated state inefficiently.
Fig. 5.
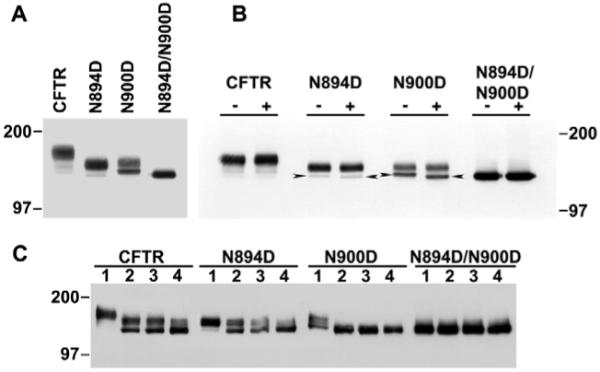
Mutagenesis and enzymatic deglycosylation of individual CFTR N-glycosylation sites. (A) Western blot of CFTR variants N-glycosylated at both, either, or neither of the two native sites. 1% SDS lysates of stably expressing CHO cells were resolved on 6% polyacrylamide gels, transferred to nitrocellulose and probed with mouse monoclonal antibody M3A7. (B) Endoglycosidase H treatment of CFTR individual glycosylation site mutants. Total cell lysates were incubated with (+) or without (-) endoglycosidase H at 37°C for 4 hours and analyzed by western blotting. The arrowheads indicate core-glycosylated protein or protein deglycosylated by endoglycosidase H. (C) Individual glycosylation site variants have different sensitivities towards deglycosylation by N-glycanase. Lysates of stably expressing CHO cells were incubated with 1 U N-glycosidase F at 37°C for 0 (lane 1), 10 minutes (lane 2), 20 minutes (lane 3) or 24 hours (lane 4) and subjected to SDS-PAGE and western blotting.
As a means of further probing the difference between the two single-oligosaccharide-chain species, we tested the susceptibility of each to enzymatic removal of the oligosaccharides with peptide:N-glycanase (PNGase). As shown in Fig. 5C, there was a striking difference in the ease of removal of the two chains. The oligosaccharide at the N900 site was efficiently attached (N894D in Fig. 5A) and was relatively refractory to removal by the PNGase (Fig. 5C, panel N894D), whereas that at the other site was rapidly removed to yield the unglycosylated species (Fig. 5C, compare panel N900D with N894D/N900D). Interestingly, the wild-type protein (Fig. 5C, panel CFTR) behaved similarly to N894D, strongly suggesting that its two chains were differentially sensitive in the same way as when present individually. The basis for this major difference is not yet clear, but emphasizes the non-identity of chains attached at the two sites.
To examine possible kinetic differences in the maturation and turnover of CFTR with either oligosaccharide chain, pulse-chase experiments were performed. Fig. 6A compares the conversion of the pulse-labeled core-glycosylated forms of each with the wild-type and unglycosylated forms and their turnover during long-term chases. Although unglycosylated N894D/N900D can mature conformationally and reach the cell surface where it functions (Figs 2 and 3), it clearly turns over much more rapidly than the wild-type protein (Fig. 6A). By contrast, the single-chain species N894D appeared to mature as effectively as the wild-type protein, with its two chains. As had been suggested by the western blots (Fig. 5A), the N900D species matured much less effectively than the wild-type or N894D proteins, and the mature product appeared to turn over faster than wild-type CFTR. To test the possibility that the differential impact of the two chains might be exerted at the early ER stage of processing, similar pulse-chase experiments were carried out in cells treated with brefeldin A (BFA) to prevent transport beyond the ER compartment. Several important results emerged from these experiments. First, turnover of the unglycosylated protein was similar to that of the wild-type protein (Fig. 6B,C), even though there was a large difference when the proteins were allowed to progress beyond the ER (Fig. 6A). Therefore, the complete absence of the oligosaccharide chains apparently has little influence on the turnover of the immature nascent chain in the ER, but strongly shortens its lifetime in more-distal compartments. The N894D species also turned over at a very similar rate to the wild-type protein in the BFA-exposed cells (Fig. 6B,C). Hence, this glycan at N900, which is relatively resistant to removal by N-glycanase, seems to be the dominant determinant of the ‘normal’ behavior of the wild-type protein in both the early and late stages of the secretory pathway. Again, in the BFA-treated cells, the N900D species was distinctly different from the others, turning over much more rapidly (Fig. 6B,C). Thus, N900D CFTR modified with an N-linked oligosaccharide chain only at the N894 site is clearly recognized differently in the ER, and enters primarily into a degradative pathway with a reduced proportion achieving maturation. This was further confirmed by the application of the proteasomal inhibitor ALLN, which significantly stabilized CFTR N900D, but neither CFTR N894D nor the double mutant (supplementary material Fig. S4).
Fig. 6.
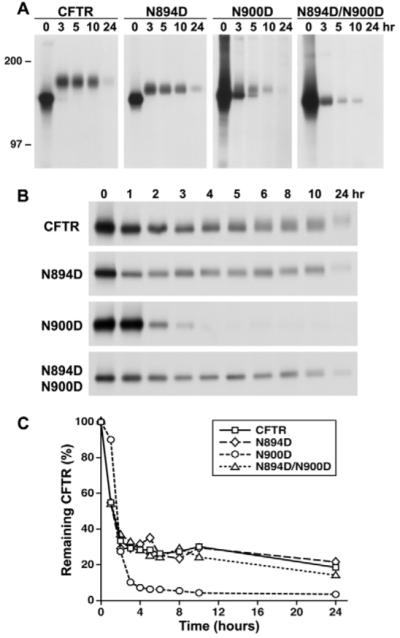
Turnover of CFTR glycosylation site variants with a single N-linked oligosaccharide. (A) Glycosylation site variant maturation and turnover as monitored by metabolic pulse-chase labeling. Cells were washed twice with methionine-free αMEM and starved for 30 minutes in the same medium. [35S]methionine was added to allow incorporation during a 20-minute pulse. Cells were then maintained in medium supplemented with 1 mM methionine, lysed with RIPA buffer at the times indicated, CFTR protein immunoprecipitated and the eluted proteins resolved on 7% polyacrylamide gels. (B) Effect of brefeldin A on turnover of glycosylation site variants. Wild-type and variant CFTR proteins were labeled in the presence of brefeldin A and pulse-chase experiments performed as described in A and Materials and Methods. (C) The amount of labeled protein remaining after the indicated chase periods was quantified by electronic autoradiography image analysis and is shown as a percentage of the initial label.
To investigate whether the single-glycosylation-site mutants have differential preference for ER lectins, we analyzed their interaction with calnexin and EDEM. We found that N894D interacted more strongly with calnexin than did N900D (Fig. 7A), but more of the latter was found in association with EDEM (Fig. 7B). The oligosaccharide at position 900 might mediate prolonged or preferential association with calnexin, as reflected in the decreased binding of the N894D mutant to EDEM but unaffected interaction with calnexin as compared with wild-type CFTR. By contrast, the oligosaccharide at position 894, which seems to direct the single mutant primarily into a degradative pathway at the ER, might support EDEM interaction in the N900D mutant, which is similar to that of the wild-type protein, whereas binding to calnexin is reduced.
Fig. 7.
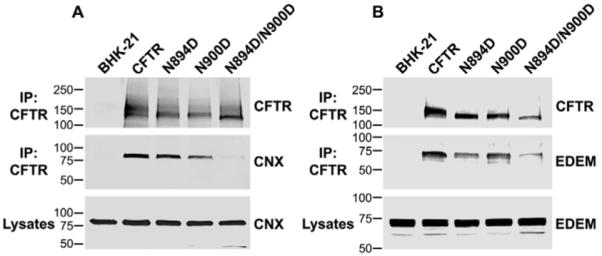
Interaction of N894D and N900D single-chain mutants with calnexin and EDEM. (A) Cell lysates from BHK-21 cells stably expressing CFTR, CFTR N894D, CFTR N900D or CFTR N894D/N900D were subjected to immunoprecipitation by incubation with anti-CFTR antibody 596 crosslinked to Dynabeads (IP: CFTR, as indicated on the left). CFTR and calnexin (CNX) were detected by western blotting using mouse monoclonal antibody 596 for CFTR or rabbit polyclonal antibody SPA860 for calnexin (as indicated on the right). Molecular weight marker positions (kDa) are indicated on the left. (B) CFTR was immunoprecipitated from cells that were stably expressing CFTR, CFTR N894D, CFTR N900D or CFTR N894D/N900D and that were also transiently expressing HA-tagged EDEM, by incubation with anti-CFTR monoclonal antibody 596 crosslinked to Dynabeads. EDEM was detected by western blotting using monoclonal antibody HA11 and CFTR using antibody 596.
Interestingly, a small amount of non-glycosylated CFTR was found to co-immunoprecipitate with EDEM. This could indicate that the CFTR protein itself, not just the oligosaccharides, is involved in this interaction. The ability of EDEM to interact with non-glycosylated substrate has been described previously. EDEM was found to bind to the α1-antitrypsin variant null Hong Kong (NHK), even when all the asparagines of the three potential N-glycosylation sites had been mutated (Oda et al., 2006), and the ER lectin also associated with ricin that did not contain any carbohydrates (Slominska-Wojewodzka et al., 2006). Another possible explanation for the association of non-glycosylated CFTR and EDEM is through an indirect interaction, mediated by another protein that is part of a chaperone complex. Possible candidates are, for example, members of the derlin family that have been shown to be involved in ER quality control and are known to interact with CFTR and EDEM (Oda et al., 2006; Younger et al., 2006).
Discussion
The mutant human CFTR protein ΔF508, which is present in 90% of cystic fibrosis patients, has some residual chloride channel activity (Dalemans et al., 1991; Drumm et al., 1991). However, it does not mature and traffic to the cell surface but is retained at the ER and eliminated by the proteasome (Jensen et al., 1995; Ward et al., 1995). Therefore, the mechanisms resulting in the different fates of the wild-type and mutant proteins are subject to intense investigation.
Although an increasing number of proteins, including components of the Hsp70 and Hsp90 chaperone complexes, have been identified that interact with ΔF508 CFTR at the ER and affect its processing to some extent, the detailed mechanism of its ER retention is not completely understood (Amaral and Kunzelmann, 2007; Riordan, 2008; Wang et al., 2006). It has been repeatedly proposed that calnexin might be a target for developing a therapeutic approach to rescue processing of ΔF508 CFTR and therefore it is important to clarify whether or not ΔF508 CFTR is retained by this lectin.
Numerous membrane glycoproteins have been shown to interact with calnexin, but in many cases it has not been clearly demonstrated that calnexin is a primary determinant of ER retention or export (Dickson et al., 2002; Halaban et al., 2000; Lu et al., 2003; Morello et al., 2001; Nagaya et al., 1999; Rigot et al., 1999). We show in this study that the failure to export the ΔF508 CFTR from the ER is independent of N-glycosylation and ER lectin interactions. The mutant protein was not able to reach the cell surface when interactions with ER-lectin chaperones were inhibited either by removal of N-linked glycosylation sites or by pharmacological inhibition (Figs 3 and 4). Another very recent study confirmed, using calnexin knockout mice, that calnexin is not necessary for ER retention of ΔF508 CFTR (Okiyoneda et al., 2008). This is in contrast to some other membrane glycoproteins, such as the major histocompatibility complex class I molecules and influenza hemagglutinin, which are retained by calnexin when not correctly assembled (Brothers et al., 2006; Jackson et al., 1994; Molinari et al., 2004; Rajagopalan and Brenner, 1994; Zhang et al., 1997).
A lack of N-linked oligosaccharides also did not affect sorting of wild-type CFTR to the plasma or apical membrane. Unlike CFTR, the N-glycans of some other membrane proteins, such as the β-subunits of the Na,K-ATPase or the gastric H,K-ATPase, carry apical sorting information (Vagin et al., 2007). Other examples of membrane proteins for which N-linked glycosylation is required for efficient trafficking to the cell surface are shaker K+ channels (de Souza and Simon, 2002), the glutamate receptor (Standley and Baudry, 2000), NKCC2 (SLC12A1) (Paredes et al., 2006) and aquaporin 2 (Hendriks et al., 2004).
We found that complete removal of N-linked glycosylation at both sites of CFTR did however influence turnover of the mature CFTR protein in post-ER compartments. The non-glycosylated double mutant turned over rapidly but behaved like wild-type CFTR in the presence of brefeldin A, which blocks transport from the ER to the Golgi apparatus (Fig. 6B,C). Farinha and Amaral (Farinha and Amaral, 2005) observed a shortened half-life of CFTR when both N-glycosylated asparagines were replaced by alanines or glutamines, but did not test whether degradation occurred at the ER or in a later compartment. Our results clearly show that unglycosylated CFTR is degraded after the protein has exited the ER.
Rapid turnover and decreased stability in the absence of N-linked glycosylation have also been observed in other membrane proteins. Mutation of N-linked glycosylation sites of the human κ opioid receptor led to a threefold faster turnover than that of the wild-type protein (Li et al., 2007). Similarly, another ABC transporter, P-glycoprotein (ABCB1), turns over three times faster when N-linked glycosylation is inhibited (Zhang et al., 2004). This degradation apparently occurs as a consequence of increased ubiquitylation. Ubiquitylation of mature CFTR has been shown to promote its targeting to lysosomal degradation (Sharma et al., 2004) and, therefore, a similar mechanism might contribute to the faster turnover of non-glycosylated CFTR.
As has been reported in previous studies, we did not find the chloride channel function of CFTR to be affected by lack of N-glycosylation (Chang et al., 1994; Gregory et al., 1991; Morris et al., 1993). Analogous to CFTR, the related ABC transporter P-glycoprotein functions without attached oligosaccharides (Gribar et al., 2000; Kramer et al., 1995; Schinkel et al., 1993; Urbatsch et al., 2001). Moreover, it has recently been shown that N-linked glycosylation is not essential for expression, transport activity, or trafficking of another human ABC protein, ABCG2 (Diop and Hrycyna, 2005; Mohrmann et al., 2005).
We found that the individual CFTR glycosylation mutants, with just one oligosaccharide chain attached, behaved differently to each other in several respects. The CFTR variant with a single oligosaccharide at position 894 (N900D) was much more susceptible to deglycosylation by PNGase, whereas the variant with glycosylation at position 900 (N894D) was surprisingly resistant to this treatment. PNGase is a cytoplasmic enzyme that catalyzes deglycosylation of proteins during ERAD, before they are degraded by the proteasome, and it has been shown that this N-glycanase acts specifically on denatured and misfolded proteins (Hirsch et al., 2003; Hirsch et al., 2004; Joshi et al., 2005; Suzuki and Lennarz, 2003). This suggests that N900D CFTR, with a glycan at position 894, might be a much better substrate for this pathway than the N894D variant.
Pulse-chase labeling experiments demonstrated that the individual oligosaccharides do affect the fate of the wild-type protein at the ER differently. With just the oligosaccharide chain at position 900 (N894D), turnover was similar to that of the wild-type protein; however, with an oligosaccharide attached only at position 894 (N900D), CFTR maturation was very inefficient. When transport from the ER was inhibited by brefeldin A, the CFTR mutant with an oligosaccharide attached to position 894 was very unstable and was rapidly removed by ERAD (Fig. 6B,C). The conclusion that N900D CFTR is a better substrate for ERAD than N894D CFTR was further strengthened by the preferential interaction of N900D CFTR with EDEM, which was similar to that of the wild-type protein, but which was reduced in the N894D mutant (Fig. 7), and by the observation that the proteasomal inhibitor ALNN stabilized N900D CFTR, but not N894D CFTR (supplementary material Fig. S4). It seems likely that the double mutant behaves in the ER like CFTR N894D rather than like CFTR N900D, because the presence of the oligosaccharide at N894 supports the ERAD pathway, whereas the absence of this oligosaccharide allows the double glycosylation mutant to exit the ER and proceed to the Golgi and plasma membrane.
The concept that each glycan of CFTR is capable of directing the processing of the protein at the ER in a different way fits extraordinarily well with the results obtained in this study. We suggest a model in which the oligosaccharide at position N894 accelerates ERAD, whereas the oligosaccharide attached to position 900 promotes maturation and allows the protein to progress to the Golgi (Fig. 8).
Fig. 8.
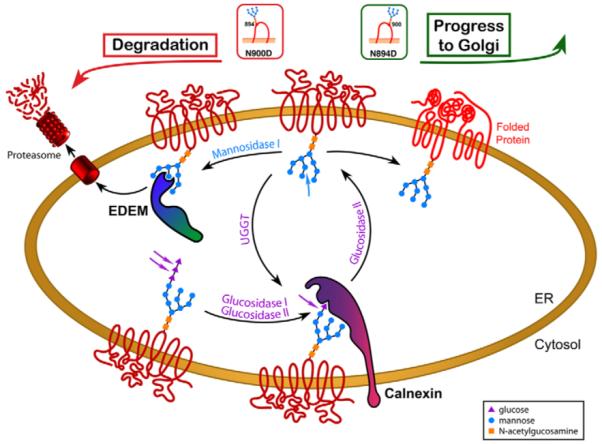
Individual N-linked oligosaccharides promote different fates in the processing of CFTR. Immediately after addition of the core glycan to the nascent polypeptide chain, the outermost of the three glucose residues is removed by glucosidase I. Subsequently, glucosidase II removes the next glucose residue. The resulting monoglucosylated core glycans bind to calnexin. When glucosidase II removes the remaining glucose residue, the glycoprotein dissociates from calnexin and, if properly folded, is free to leave the ER. If the protein is incompletely folded it is reglucosylated by the UDP-glucose:glycoprotein glucosyltransferase, creating again a monoglycosylated core oligosaccharide that can bind to calnexin. Proteins that have stayed in the ER for too long and are terminally misfolded become a substrate for α-mannosidase I. The trimmed oligosaccharide binds to EDEM (ER degradation-enhancing α-mannosidase-like protein) and the glycoprotein is consequently directed towards ERAD (ER-associated degradation). The carbohydrate at position 900 in CFTR N894D supports the route to maturation and progress to the Golgi, whereas the oligosaccharide at asparagine residue 894 in N900D promotes degradation.
We cannot rule out the possibility that the lack of glycans might affect the conformation of CFTR and that the N900D mutation is accompanied by a significant degree of misfolding that makes it a better ERAD substrate. However, this does not seem a very likely scenario because the double mutant, which lacks both oligosaccharide chains, is stable at the ER, similar to wild-type CFTR.
Interestingly, distinct functions of N-glycans have been observed in the processing of other glycoproteins. Hebert et al. (Hebert et al., 1997) reported that the number and location of glycans on influenza hemagglutinin determined its folding and association with calnexin. In another study, two out of eight N-glycosylation sites were shown to be the major determinants for efficient apical sorting of the transmembrane protein endolyn (CD164) (Potter et al., 2004). In case of the gastric H,K-ATPase β-subunit, just one out of four N-glycosylation sites affected ER-to-Golgi trafficking and enhanced endocytosis from the apical membrane (Vagin et al., 2004). In yeast, it has been observed that the N-linked oligosaccharides of CPY have different impacts on the processing of the protein and the most C-terminal of the four N-linked oligosaccharides is specifically required for ERAD (Kostova and Wolf, 2005; Spear and Ng, 2005).
The exact, molecular details of the means by which the individual glycans affect the processing of CFTR are not yet apparent, but CFTR-interacting lectins, including calnexin and EDEM, might act upon the variously glycosylated forms differentially (Fig. 8). The interactions of N-linked carbohydrates of CFTR at the ER might not be limited to these two chaperones, but could also involve lectin receptors such as ERGIC53 (LMAN1), VIP36 (LMAN2), VIPL (LMAN2L) or erlectin, which have all been shown to facilitate trafficking of certain glycoproteins in the early secretory pathway (Appenzeller-Herzog and Hauri, 2006; Appenzeller et al., 1999; Cruciat et al., 2006; Neve et al., 2003; Shimada et al., 2003).
In conclusion, the two N-linked oligosaccharides are not responsible for retention of ΔF508 CFTR at the ER, but they strongly influence turnover of the mature wild-type protein in post-ER compartments. In addition, it is notable that each individual oligosaccharide chain directs ER processing of CFTR differently. This might be due either to different oligosaccharide structures at the two sites, or to differences in the accessibility of the N-glycans at their separate locations in the polypeptide chain.
Materials and Methods
Mutagenesis and stable expression of CFTR
Replacement of asparagine residues N894 and N900 with aspartate residues was achieved by replacing a CFTR HpaI-DraIII cDNA fragment (bp 2463 to 3328) in pNUT CFTR with a counterpart generated by PCR coding for N894D, N900D or N894D/N900D, to produce CFTRs with only one or no glycosylation site (Chang et al., 1994). BHK-21 cells stably expressing CFTR containing an external HA epitope in EL2 (Extope-CFTR) have been described previously (Gentzsch et al., 2004). BHK-21 and CHO-K1 cells were transfected with Lipofectamine (Invitrogen) or using calcium phosphate and propagated in media with 500 or 50 μM methotrexate, respectively.
Human excess donor lungs and excised recipient lungs were obtained at the time of lung transplantation from portions of main stem or lumbar bronchi and cells were harvested by enzymatic digestion as previously described (Fulcher et al., 2005) under a protocol approved by the UNC Medical School institutional review board. To express Extope-CFTR in primary human airway epithelial cultures, cells were seeded on collagen-coated Millicell culture plate inserts and maintained at an air-liquid interface for 40-45 days. The well-differentiated cultures were then transduced with adenoviral vectors as described previously (Coyne et al., 2000) to express Extope-CFTR and Extope-ΔF508 with intact or mutated glycosylation sites (N894D/N900D).
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde for 10 minutes and immunostaining was either performed on non-permeabilized cells or on cells permeabilized with 0.1% saponin PBS. Cells were blocked with 1% BSA and 5% normal goat serum in PBS. Primary antibodies were mouse monoclonal anti-CFTR antibodies M3A7, 570 and 596 or anti-HA antibody HA11 (Covance). Detection of the external HA epitope in EL2 of CFTR (Extope-CFTR) on non-permeabilized BHK-21 cells was performed exactly as described previously (Gentzsch et al., 2004). Secondary antibodies were goat anti-mouse IgGs conjugated to Alexa Fluor 488 or 568 (Molecular Probes).
For detection of cell-surface Extope-CFTR and variants, primary human airway epithelial cultures were labeled with anti-HA antibody HA11 from the apical side for 30 minutes at room temperature, then frozen culture sections were prepared, fixed, permeabilized, blocked and treated with goat anti-mouse Alexa Fluor 488 conjugate.
Cell-surface ELISA
BHK-21 cells expressing externally tagged ΔF508 CFTR (Gentzsch et al., 2004) were seeded in a 96-well plate at 40,000 cells per well. Cells were fixed, labeled with the HA11 antibody that recognizes the external tag, followed by goat anti-mouse IgG conjugated to X-Sight 761 (Kodak). Plates were scanned with a LI-COR Odyssey infrared imaging system.
Immunoprecipitation and pulse-chase experiments
Immunoprecipitations were performed on NP40 cell lysates (1% Nonidet P40, 150 mM NaCl, 50 mM Tris-HCl pH 7.4, 10 mM NaMoO4) with protease inhibitors (1 μg/ml leupeptin, 2 μg/ml aprotinin, 50 μg/ml pefabloc, 121 μg/ml benzamidine, 3.5 μg/ml E64) using mouse monoclonal anti-CFTR antibody 596 crosslinked to Dynabeads Protein G (Invitrogen).
For metabolic labeling with [35S]methionine, cells were washed twice in methionine-free αMEM (Gibco) and starved in the same medium for 30 minutes. [35S]methionine (100 μCi/ml; Amersham) was then added and cells were labeled for 20 minutes. Cells were washed twice with regular growth medium supplemented with 1 mM methionine and chased for various times. In some experiments, 10 μg/ml brefeldin A was added to the growth medium 12 hours before labeling and was also present in all other incubations. Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% deoxycholic acid) with protease inhibitors and CFTR was immunoprecipitated with monoclonal antibody M3A7 followed by incubation with protein G-Sepharose beads (Sigma). Immunoprecipitated proteins were eluted with sample buffer and subjected to SDS-PAGE. The amount of labeled protein remaining after the indicated chase periods was quantified by electronic autoradiography image analysis using a Packard Instant Imager.
Deglycosylation and tunicamycin treatment
For N-glycosidase F digestion, 1% SDS cell lysates were diluted with 10 volumes of 100 mM sodium phosphate buffer (pH 7.5) supplemented with 25 mM EDTA, 0.5% NP40, 1% β-mercaptoethanol and protease inhibitors (1 μg/ml leupeptin, 2 μg/ml aprotinin, 50 μg/ml pefabloc, 121 μg/ml benzamidine, 3.5 μg/ml E64). One unit of N-glycosidase F (Boehringer Mannheim) was added and the samples incubated for various times at 37°C. To remove core-glycosylated oligosaccharides, the lysates were diluted with 10 volumes of buffer (50 mM sodium acetate pH 5.3, 0.5% NP40, protease inhibitors) to a final concentration of 0.1% SDS. The samples were than incubated for 4 hours at 37°C with 0.01 U endoglycosidase H (Boehringer Mannheim). Proteins were precipitated by addition of four volumes of cold ethanol, dissolved in sample buffer, and subjected to SDS-PAGE.
To completely inhibit N-linked glycosylation of CFTR, BHK-21 cells that had been transiently transfected with pcDNA3 CFTR were subsequently grown for 24 hours in medium containing 5 μg/ml tunicamycin.
Chloride efflux assay
Cells grown in 6-well culture dishes were washed twice with efflux buffer [136 mM NaNO3, 3 mM KNO3, 2 mM Ca(NO3)2, 2 mM Mg(NO3)2, 10 mM glucose, 20 mM HEPES pH 7.4] and loaded with 36Cl by a 1 hour incubation with 0.5 ml efflux buffer containing 1 μCi Na36Cl (Amersham). Chloride efflux was stimulated by addition of 1 mM IBMX, 10 μM forskolin and 100 μM dibutyryl cAMP at time 0. Samples were collected at 1-minute intervals into 24-well Top Count plates (Packard). MicroScint 40 (PerkinElmer) was added for scintillation counting.
Single-channel measurements
Microsomal membrane vesicles were isolated from BHK-21 cells stably expressing the different CFTR variants, phosphorylated with protein kinase A and single-channel kinetics were recorded as described previously (Aleksandrov and Riordan, 1998) (supplementary material Fig. S1).
Supplementary Material
Acknowledgments
We thank Nobuko Hosokawa and Kazuhiro Nagata for providing the plasmid for EDEM expression; the CFFT for supplying the ΔF508 corrector compounds C3 and C4; Scott H. Randell, as well as the affiliated Tissue Culture Core of the UNC Cystic Fibrosis Center, for providing primary human airway epithelial cells and we particularly appreciate Leslie Fulcher’s advice and guidance in the maintenance of these cultures; Kim Burns for preparation of frozen culture sections and Wendy Salmon and Michael Chua for outstanding assistance with confocal microscopy; Wanda K. O’Neal and the UNC Cystic Fibrosis Center Molecular Biology Core for construction of adenoviral vectors containing Extope-CFTR and glycosylation mutants and the UNC Vector Core Facility for production of adenoviral particles; and Tim Donovan and Casey Long for excellent assistance with tissue culture, plate assays and western blotting. This work was supported by the NIH (R01DK051870) and CFF.
References
- Aleksandrov AA, Riordan JR. Regulation of CFTR ion channel gating by MgATP. FEBS Lett. 1998;431:97–101. doi: 10.1016/s0014-5793(98)00713-3. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol. Sci. 2007;28:334–341. doi: 10.1016/j.tips.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Appenzeller C, Andersson H, Kappeler F, Hauri HP. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat. Cell Biol. 1999;1:330–334. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Janovick JA, Conn PM. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J. Mol. Endocrinol. 2006;37:479–488. doi: 10.1677/jme.1.02142. [DOI] [PubMed] [Google Scholar]
- Chang X-B, Hou Y-X, Jensen T, Riordan JR. Mapping of the Cystic Fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. J. Biol. Chem. 1994;269:18572–18575. [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chow DC, Forte JG. Functional significance of the beta-subunit for heterodimeric P-type ATPases. J. Exp. Biol. 1995;198:1–17. doi: 10.1242/jeb.198.1.1. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Kelly MM, Boucher RC, Johnson LG. Enhanced epithelial gene transfer by modulation of tight junctions with sodium caprate. Am. J. Respir. Cell Mol. Biol. 2000;23:602–609. doi: 10.1165/ajrcmb.23.5.4164. [DOI] [PubMed] [Google Scholar]
- Cruciat CM, Hassler C, Niehrs C. The MRH protein Erlectin is a member of the endoplasmic reticulum synexpression group and functions in N-glycan recognition. J. Biol. Chem. 2006;281:12986–12993. doi: 10.1074/jbc.M511872200. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavarani A, Lecocq J-P, Lazdunski M. Altered chloride ion channel kinetics associated with the DeltaF508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- de Souza NF, Simon SM. Glycosylation affects the rate of traffic of the Shaker potassium channel through the secretory pathway. Biochemistry. 2002;41:11351–11361. doi: 10.1021/bi0258041. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dickson KM, Bergeron JJ, Shames I, Colby J, Nguyen DT, Chevet E, Thomas DY, Snipes GJ. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc. Natl. Acad. Sci. USA. 2002;99:9852–9857. doi: 10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop NK, Hrycyna CA. N-Linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry. 2005;44:5420–5429. doi: 10.1021/bi0479858. [DOI] [PubMed] [Google Scholar]
- Dragomir A, Bjorstad J, Hjelte L, Roomans GM. Curcumin does not stimulate cAMP-mediated chloride transport in cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 2004;322:447–451. doi: 10.1016/j.bbrc.2004.07.146. [DOI] [PubMed] [Google Scholar]
- Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS. Chloride conductance expressed by Delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254:1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Egan ME, Glockner-Pagel J, Ambrose C, Cahill PA, Pappoe L, Balamuth N, Cho E, Canny S, Wagner CA, Geibel J, et al. Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 2002;8:485–492. doi: 10.1038/nm0502-485. [DOI] [PubMed] [Google Scholar]
- Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science. 2004;304:600–602. doi: 10.1126/science.1093941. [DOI] [PubMed] [Google Scholar]
- Elbein AD, Tropea JE, Mitchell M, Kaushal GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J. Biol. Chem. 1990;265:15599–15605. [PubMed] [Google Scholar]
- Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann U, Bause E, Legler G, Ploegh H. Novel mannosidase inhibitor blocking conversion of high mannose to complex oligosaccharides. Nature. 1984;307:755–758. doi: 10.1038/307755a0. [DOI] [PubMed] [Google Scholar]
- Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. In: Picot J, editor. Methods in Molecular Medicine: Human Cell Culture Protocols. Vol. 107. Humana Press; Totowa, NJ: 2005. pp. 183–206. [DOI] [PubMed] [Google Scholar]
- Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, Pagano RE, Riordan JR. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell. 2004;15:2684–2696. doi: 10.1091/mbc.E04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol. Biol. Cell. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, Anderson MP, Welsh MI, Smith AE. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell. Biol. 1991;11:3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Gabriel SE, Mengos A, Gentzsch M, Randell SH, Van Heeckeren AM, Knowles MR, Drumm ML, Riordan JR, Boucher RC. SERCA pump inhibitors do not correct biosynthetic arrest of deltaF508 CFTR in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2006;34:355–363. doi: 10.1165/rcmb.2005-0286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban R, Svedine S, Cheng E, Smicun Y, Aron R, Hebert DN. Endoplasmic reticulum retention is a common defect associated with tyrosinase-negative albinism. Proc. Natl. Acad. Sci. USA. 2000;97:5889–5894. doi: 10.1073/pnas.97.11.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Okiyoneda T, Hashimoto Y, Oyokawa K, Nakamura K, Suico MA, Shuto T, Kai H. Curcumin enhances cystic fibrosis transmembrane regulator expression by down-regulating calreticulin. Biochem. Biophys. Res. Commun. 2007;353:351–356. doi: 10.1016/j.bbrc.2006.12.036. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell Biol. 1997;139:613–623. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- Hendriks G, Koudijs M, van Balkom BW, Oorschot V, Klumperman J, Deen PM, van der Sluijs P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004;279:2975–2983. doi: 10.1074/jbc.M310767200. [DOI] [PubMed] [Google Scholar]
- Hirsch C, Blom D, Ploegh HL. A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 2003;22:1036–1046. doi: 10.1093/emboj/cdg107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Misaghi S, Blom D, Pacold ME, Ploegh HL. Yeast N-glycanase distinguishes between native and non-native glycoproteins. EMBO Rep. 2004;5:201–206. doi: 10.1038/sj.embor.7400066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiali B, Hendrickson TL. Asparagine-linked glycosylation: specificity and function of oligosaccharyl transferase. Bioorg. Med. Chem. 1995;3:1565–1578. doi: 10.1016/0968-0896(95)00142-5. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Cohen-Doyle MF, Peterson P, Williams DB. Regulation of MHC Class I transport by the molecular chaperone, calnexin (p88, IP90) Science. 1994;263:384–387. doi: 10.1126/science.8278813. [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Joshi S, Katiyar S, Lennarz WJ. Misfolding of glycoproteins is a prerequisite for peptide: N-glycanase mediated deglycosylation. FEBS Lett. 2005;579:823–826. doi: 10.1016/j.febslet.2004.12.060. [DOI] [PubMed] [Google Scholar]
- Kostova Z, Wolf DH. Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J. Cell Sci. 2005;118:1485–1492. doi: 10.1242/jcs.01740. [DOI] [PubMed] [Google Scholar]
- Kramer R, Weber TK, Arceci R, Ramchurren N, Kastrinakis WV, Steele G, Jr, Summerhayes IC. Inhibition of N-linked glycosylation of P-glycoprotein by tunicamycin results in a reduced multidrug resistance phenotype. Br. J. Cancer. 1995;71:670–675. doi: 10.1038/bjc.1995.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreda SM, Mall M, Mengos A, Rochelle L, Yankaskas J, Riordan JR, Boucher RC. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell. 2005;16:2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JG, Chen C, Liu-Chen LY. N-Glycosylation of the human kappa opioid receptor enhances its stability but slows its trafficking along the biosynthesis pathway. Biochemistry. 2007;46:10960–10970. doi: 10.1021/bi700443j. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Thapsigargin or curcumin does not promote maturation of processing mutants of the ABC transporters, CFTR, and P-glycoprotein. Biochem. Biophys. Res. Commun. 2004;325:580–585. doi: 10.1016/j.bbrc.2004.10.070. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Rescue of folding defects in ABC transporters using pharmacological chaperones. J. Bioenerg. Biomembr. 2005;37:501–507. doi: 10.1007/s10863-005-9499-3. [DOI] [PubMed] [Google Scholar]
- Lu M, Echeverri F, Moyer BD. Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic. 2003;4:416–433. doi: 10.1034/j.1600-0854.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Mohamed A, Kartner N, Chang X-B, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (DeltaF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann K, van Eijndhoven MA, Schinkel AH, Schellens JH. Absence of N-linked glycosylation does not affect plasma membrane localization of breast cancer resistance protein (BCRP/ABCG2) Cancer Chemother. Pharmacol. 2005;56:344–350. doi: 10.1007/s00280-005-1004-5. [DOI] [PubMed] [Google Scholar]
- Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol. Cell. 2004;13:125–135. doi: 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Petaja-Repo UE, Laperriere A, Lonergan M, Arthus MF, Nabi IR, Bichet DG, Bouvier M. Association of calnexin with wild type and mutant AVPR2 that causes nephrogenic diabetes insipidus. Biochemistry. 2001;40:6766–6775. doi: 10.1021/bi002699r. [DOI] [PubMed] [Google Scholar]
- Morris AP, Cunningham SA, Benos DJ, Frizzell RA. Glycosylation status of endogenous CFTR does not affect cAMP-stimulated Cl- secretion in epithelial cells. Am. J. Physiol. 1993;265:C688–C694. doi: 10.1152/ajpcell.1993.265.3.C688. [DOI] [PubMed] [Google Scholar]
- Nagaya N, Schulteis CT, Papazian DM. Calnexin associates with Shaker K+ channel protein but is not involved in quality control of subunit folding or assembly. Recept. Channels. 1999;6:229–239. [PubMed] [Google Scholar]
- Neve EP, Svensson K, Fuxe J, Pettersson RF. VIPL, a VIP36-like membrane protein with a putative function in the export of glycoproteins from the endoplasmic reticulum. Exp. Cell Res. 2003;288:70–83. doi: 10.1016/s0014-4827(03)00161-7. [DOI] [PubMed] [Google Scholar]
- Norez C, Antigny F, Becq F, Vandebrouck C. Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic. 2006a;7:562–573. doi: 10.1111/j.1600-0854.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- Norez C, Noel S, Wilke M, Bijvelds M, Jorna H, Melin P, DeJonge H, Becq F. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 2006b;580:2081–2086. doi: 10.1016/j.febslet.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006;172:383–393. doi: 10.1083/jcb.200507057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Wada I, Jono H, Shuto T, Yoshitake K, Nakano N, Nagayama S, Harada K, Isohama Y, Miyata T, et al. Calnexin Delta 185-520 partially reverses the misprocessing of the Delta F508 cystic fibrosis transmembrane conductance regulator. FEBS Lett. 2002;526:87–92. doi: 10.1016/s0014-5793(02)03134-4. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T, Harada K, Takeya M, Yamahira K, Wada I, Shuto T, Suico MA, Hashimoto Y, Kai H. Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell. 2004;15:563–574. doi: 10.1091/mbc.E03-06-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Niibori A, Harada K, Kohno T, Michalak M, Duszyk M, Wada I, Ikawa M, Shuto T, Suico MA, et al. Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and DeltaF508 CFTR. Biochim. Biophys. Acta. 2008;9:1585–1594. doi: 10.1016/j.bbamcr.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Paredes A, Plata C, Rivera M, Moreno E, Vazquez N, Munoz-Clares R, Hebert SC, Gamba G. Activity of the renal Na+-K+-2Cl- cotransporter is reduced by mutagenesis of N-glycosylation sites: role for protein surface charge in Cl- transport. Am. J. Physiol. Renal Physiol. 2006;290:F1094–F1102. doi: 10.1152/ajprenal.00071.2005. [DOI] [PubMed] [Google Scholar]
- Parodi AJ. Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994;269:12784–12788. [PubMed] [Google Scholar]
- Potter BA, Ihrke G, Bruns JR, Weixel KM, Weisz OA. Specific N-glycans direct apical delivery of transmembrane, but not soluble or glycosylphosphatidylinositol-anchored forms of endolyn in Madin-Darby canine kidney cells. Mol. Biol. Cell. 2004;15:1407–1416. doi: 10.1091/mbc.E03-08-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Brenner MB. Calnexin retains unassembled major histocompatibility complex class I free heavy chains in the endoplasmic reticulum. J. Exp. Med. 1994;180:407–412. doi: 10.1084/jem.180.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigot V, Andre F, Lehmann M, Lissitzky JC, Marvaldi J, Luis J. Biogenesis of alpha6beta4 integrin in a human colonic adenocarcinoma cell line involvement of calnexin. Eur. J. Biochem. 1999;261:659–666. doi: 10.1046/j.1432-1327.1999.00300.x. [DOI] [PubMed] [Google Scholar]
- Riordan JR. CFTR Function and Prospects for Therapy. Annu. Rev. Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Sasak VW, Ordovas JM, Elbein AD, Berninger RW. Castanospermine inhibits glucosidase I and glycoprotein secretion in human hepatoma cells. Biochem. J. 1985;232:759–766. doi: 10.1042/bj2320759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Kemp S, Dolle M, Rudenko G, Wagenaar E. N-glycosylation and deletion mutants of the human MDR1 P-glycoprotein. J. Biol. Chem. 1993;268:7474–7481. [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, Bache KG, Papsin B, Zerangue N, Stenmark H, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada O, Hara-Kuge S, Yamashita K, Tosaka-Shimada H, Yanchao L, Yongnan L, Atsumi S, Ishikawa H. Clusters of VIP-36-positive vesicles between endoplasmic reticulum and Golgi apparatus in GH3 cells. Cell Struct. Funct. 2003;28:155–163. doi: 10.1247/csf.28.155. [DOI] [PubMed] [Google Scholar]
- Slominska-Wojewodzka M, Gregers TF, Walchli S, Sandvig K. EDEM is involved in retrotranslocation of ricin from the endoplasmic reticulum to the cytosol. Mol. Biol. Cell. 2006;17:1664–1675. doi: 10.1091/mbc.E05-10-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Sonawane ND, Salinas D, Qian L, Pedemonte N, Galietta LJV, Verkman AS. Evidence against the rescue of defective deltaF508-CFTR cellular processing by curcumin in cell culture and mouse models. J. Biol. Chem. 2004;279:40629–40633. doi: 10.1074/jbc.M407308200. [DOI] [PubMed] [Google Scholar]
- Spear ED, Ng DT. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley S, Baudry M. The role of glycosylation in ionotropic glutamate receptor ligand binding, function, and trafficking. Cell. Mol. Life Sci. 2000;57:1508–1516. doi: 10.1007/PL00000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Lennarz WJ. Hypothesis: a glycoprotein-degradation complex formed by protein-protein interaction involves cytoplasmic peptide:N-glycanase. Biochem. Biophys. Res. Commun. 2003;302:1–5. doi: 10.1016/s0006-291x(03)00052-4. [DOI] [PubMed] [Google Scholar]
- Urbatsch IL, Wilke-Mounts S, Gimi K, Senior AE. Purification and characterization of N-glycosylation mutant mouse and human P-glycoproteins expressed in Pichia pastoris cells. Arch. Biochem. Biophys. 2001;388:171–177. doi: 10.1006/abbi.2001.2299. [DOI] [PubMed] [Google Scholar]
- Vagin O, Turdikulova S, Sachs G. The H,K-ATPase beta subunit as a model to study the role of N-glycosylation in membrane trafficking and apical sorting. J. Biol. Chem. 2004;279:39026–39034. doi: 10.1074/jbc.M405453200. [DOI] [PubMed] [Google Scholar]
- Vagin O, Turdikulova S, Tokhtaeva E. Polarized membrane distribution of potassium-dependent ion pumps in epithelial cells: different roles of the N-glycans of their beta subunits. Cell Biochem. Biophys. 2007;47:376–391. doi: 10.1007/s12013-007-0033-6. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Zhang JX, Braakman I, Matlack KE, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol. Biol. Cell. 1997;8:1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wu JY, Hait WN, Yang JM. Regulation of the stability of P-glycoprotein by ubiquitination. Mol. Pharmacol. 2004;66:395–403. doi: 10.1124/mol.104.001966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.