

Abstract
Objective:
To test the hypothesis that there is familial aggregation of dystonia and other movement disorders in relatives of patients with musician’s dystonia (MD) and to identify possible environmental triggers.
Methods:
The families of 28 index patients with MD (14 with a reported positive family history of focal task-specific dystonia [FTSD] and 14 with no known family history [FH−]) underwent a standardized telephone screening interview using a modified version of the Beth Israel Dystonia Screen. Videotaped neurologic examinations were performed on all participants who screened positive and consensus diagnoses established. All patients were investigated for DYT1 dystonia and suitable families were tested for linkage to DYT7. All family members were administered questionnaires covering potential triggers of FTSD.
Results:
A diagnosis of dystonia was established in all 28 index patients and in 19/97 examined relatives (MD: n = 8, other FTSD: n = 9, other dystonias: n = 2), 5 of whom were members of FH− families. In 27 of the 47 affected individuals, additional forms of dystonia were seen; other movement disorders were observed in 23 patients. In total, 18 families were multiplex families with two to four affected members. Autosomal dominant inheritance was compatible in at least 12 families. The GAG deletion in DYT1 was absent in all patients. Linkage to DYT7 could be excluded in 1 of the 11 informative families. With respect to potential environmental triggers, there was no significant difference between patients with MD/FTSD compared to unaffected family members.
Conclusion:
Our results suggest a genetic contribution to musician’s dystonia with phenotypic variability including focal task-specific dystonia.
GLOSSARY
- BIDS
= Beth Israel Dystonia Screen;
- FH+
= reported positive family history of focal task-specific dystonia;
- FH−
= no known family history of focal task-specific dystonia;
- FTSD
= focal task-specific dystonia;
- MD
= musician’s dystonia;
- WC
= writer’s cramp.
Musician’s dystonia (MD), a type of focal task-specific dystonia (FTSD), presents with painless muscular incoordination or loss of voluntary motor control of extensively trained movements when a musician is playing his or her instrument. It is usually confined to a limb or embouchure.1-4 The frequency of MD is estimated at about 1% among professional musicians; it is highly disabling and in many cases terminates performance careers.1
While the pathophysiology remains largely elusive, MD has been associated with intensive training regimes and thus been considered a form of occupational cramp. However, the epidemiology also suggests a possible hereditary component: 10% of patients with MD report a positive family history of dystonia.1 Clinical examination of relatives of patients with other forms of focal dystonia revealed even larger numbers of affected family members (23% to 27% of patients had relatives with dystonia5-7). In rare cases of focal dystonia, a hereditary component has been demonstrated, such as the GAG deletion in the DYT1 gene.8 However, this mutation was excluded in a small group of patients with MD.9 In two families, focal dystonia has been linked to a specific gene locus on chromosome 18 (DYT7 10,11). Most familial dystonias known today display an autosomal dominant pattern of inheritance with incomplete penetrance.
More recently, the report of three families with putative autosomal dominant inheritance of FTSD in relatives of patients with MD has lent support to the concept of a genetic contribution to MD.12 To test the hypothesis of a genetic etiology in at least a subset of MD, to explore a possible relationship between MD and other forms of FTSD, and to systematically study the role of potential environmental triggers, we initiated a large clinical genetic study of MD based on systematic examination of 28 families.
METHODS
Recruitment of study sample and onsite examination.
After approval of the study by the local ethics committee and obtaining informed consent, we included 28 professional musicians diagnosed with focal dystonia at the outpatient clinic of the Hanover Institute of Music Physiology and Musicians’ Medicine (index patients). The diagnostic procedure during their first visit included a detailed neurologic examination. Based on history and clinical features, all were classified as having likely primary dystonia. Fourteen of these index patients had a reported positive family history (FH+) of FTSD and were matched to 14 patients with no known family history (FH−) for age, sex, instrument group, and type of dystonia (limb vs embouchure). In a first telephone contact, all index patients were asked to report known cases of dystonia in their families (family history interview). All available first- and second-degree relatives with no known dystonia underwent a standardized telephone screening interview using a modified version of the Beth Israel Dystonia Screen containing additional questions screening for MD (BIDS; adapted from reference 13). Videotaped neurologic examinations were performed at a home visit by the same examiner (A.S.) in all 28 index patients, all 15 relatives with a known or reported form of FTSD, and in all 11 relatives who screened positive for dystonia in BIDS. Musicians were additionally examined while playing their instruments. Patients with focal symptoms were tested for a potential sensory trick using a latex glove. This may reduce dystonic contractions but the effect usually does not persist.1 Finally, pedigrees of all families were constructed using the Cyrillic 2.1 software (Wallingford, Oxfordshire, UK).
Questionnaires, risk factors, and statistical analyses.
Regardless of their clinical status, all available family members were administered previously published questionnaires covering demographic information, therapy, and medical and occupational history.12 Handedness was tested using the Edinburgh Handedness Inventory.14 Family members with dystonia were asked to estimate the course of their disease over time on a three-step scale (improvement, no change, deterioration). To identify potential triggers of dystonia, patients were asked whether they had had peripheral nerve trauma, pain, or overuse syndromes in the affected regions within 6 months before the onset of dystonia. Additionally, they reported whether they had considerably increased the time spent on any fine motor task within 6 months before dystonia onset. Likewise, unaffected relatives provided the same information. They were asked about peripheral trauma, pain, or overuse in the arms or mouth and about a considerable increase of time spent on any fine motor task for any time of their lives. χ2 Tests were applied to identify disproportionate frequencies in the occurrence of potential triggers in patients with dystonia and in healthy relatives. The level of significance was set at p < 0.05.
Diagnostic criteria, video rating, and consensus diagnosis.
A diagnosis of dystonia was made following previously published criteria1,15: 1) definite: muscle contractions producing characteristic twisting, flexion, or extension movements and postures consistently present; 2) probable: movements and postures of insufficient intensity or consistency to merit classification as definite; 3) possible: muscle contractions not considered abnormal but remotely suggestive of dystonia; 4) no dystonia. All examined family members were diagnosed in a three-step process: first, by onsite examination including information about pedigree structure and medical history (A.S.); second, by blinded independent video review by four movement disorders specialists (E.A., J.H., C.K., and A.M.), one of whom is an expert in MD (E.A.); third, by evaluation of questionable cases by two blinded external collaborators (S.B. and R.S.P.). Finally, a consensus diagnosis was established.
Molecular analysis.
Peripheral blood samples were collected from all probands, DNA extracted, and screened for the known three-nucleotide (GAG) deletion in the DYT1 gene. To test for a possible involvement of the DYT7 locus, suitable families were investigated for linkage using the following six DNA microsatellite markers: D18S481, D18S54, D18S976, D18S452, D18S843, and D18S1153. DYT7 haplotypes were constructed manually.
RESULTS
Patients and families.
The study procedure and main results are illustrated in figure 1. There were 28 index patients (14 FH+ of FTSD, 14 FH−), 20 men and 8 women, and their mean age was 43 ± 12 years (range: 27–74 years). The majority originated from Germany (25 patients); one patient was of French, one of Russian, and one of Korean background. Twenty-four patients (86%) had upper limb dystonias, 4 patients (14%) embouchure dystonias. The duration of dystonia was 12 ± 8 years (range: 1–33 years). Age at onset was 32 ± 11 years (range: 16–66 years).

Figure 1 Flow chart of the study displaying study procedure and results
FH = family history; FH+ = reported positive family history of focal task-specific dystonia; FH− = no known family history; pts = patients; MD = musician’s dystonia; FTSD = focal task-specific dystonia; BIDS = Beth Israel Dystonia Screen; def = definite; prob = probable; poss = possible; (+) = additional dystonias or additional other movement disorders in some individuals definitely, probably, or possibly present.
In total, 97 (56 FH+, 41 FH−) first- and second-degree relatives of these index patients, 54 men and 43 women, were examined at a mean age of 52 ± 22 years (range: 9–84 years).
Seventeen of the 56 FH+ relatives (32%) had a previously known FTSD (MD: n = 7, writer’s cramp [WC]: n = 10); two of these relatives were deceased (MD: n = 1, WC: n = 1).
Family history interview revealed another two deceased relatives with MD, one FH+ and one FH−.
Among a total of 78 (38 FH+, 40 FH−) putatively unaffected family members, 8 were professional musicians (10%), 30 amateur musicians (39%), and 40 were not playing an instrument (51%). Using the BIDS in these 78, an additional 11 individuals (14%) screened positive for dystonia, 7 of whom were members of FH− families.
Consensus diagnoses.
MD was established by onsite examination and videotape review in all 14 FH+ (definite: n = 13, probable: n = 1) and in all 14 FH− (definite: n = 14) index patients.
A type of dystonia was confirmed as definite or probable in 19 of 26 relatives (74%) who had a previously known form of FTSD or screened positive with the BIDS. Fourteen of these 19 probands were members of FH+ (MD: n = 7, other FTSD: n = 6, other dystonias: n = 1) and 5 of FH− (MD: n = 1, other FTSD: n = 3, other dystonias: n = 1) families.
In 27 of the 47 FH+ and FH− family members with definite or probable dystonia (62%), one (n = 18), two (n = 8), or three (n = 3) additional forms of dystonia were present at least possibly; i.e., these patients were not only affected with one dystonia type (e.g., MD) but also with another FTSD (e.g., WC) or additional other dystonias (e.g., cervical dystonia). In some of these patients, additional dystonias were the consequence of spread; conversely, some patients experienced dystonia outside the initially involved segment. In 23 of the aforementioned 47 family members (49%), one (n = 20), two (n = 2), or three (n = 1) additional other movement disorders or movement abnormalities were diagnosed as definitely or probably present, including tremor (n = 6), tics (n = 4), chorea (n = 2), involuntary perioral movements (n = 2), athetosis (n = 1), mirror movements (n = 8), and parkinsonian features (n = 4).
In total, 18 (13 FH+, 5 FH−) of the entire set of 28 families (64%) were multiplex families (based on definite, probable, and possible diagnoses of dystonia) with two to four affected family members in one (n = 6), two (n = 10), or three (n = 2) generations, compatible with autosomal dominant inheritance in at least 12 families. In 12 families, only one affected first-degree relative could be found. Ten of these 12 index patient-relative pairs (83%) were phenotypically concordant for distribution of symptoms, two discordant. In the six remaining families with three or more affected members, four were concordant for distribution of symptoms and two discordant. Pedigree information of all multiplex families is summarized in the table. Examples of representative pedigrees are shown in figure 2A. Detailed information on all 53 examined family members (one indi-vidual had declined examination) is given in table e-1 on the Neurology® Web site at www.neurology.org.
Table Characteristics of 18 multiplex families (summary of pedigree information)
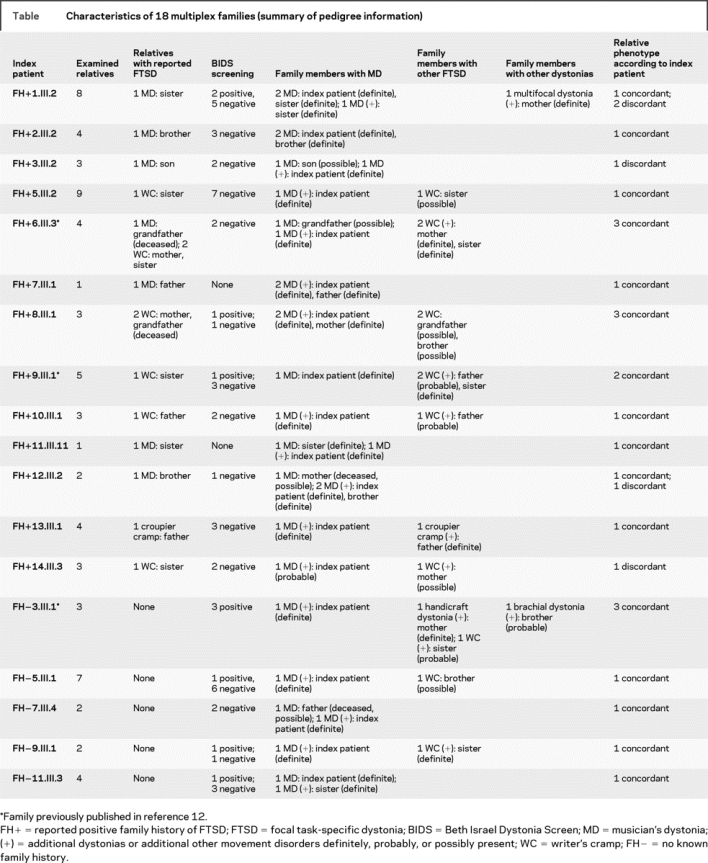

Figure 2 Examples of representative pedigrees of examined families
(A) Pedigrees of two families with a reported positive family history of focal task-specific dystonia (FTSD) (FH+ 1, FH+ 8). Upper right quadrant indicates family members with musician’s dystonia (MD), lower right quadrant members with other FTSD, lower left quadrant individuals with other dystonias, and upper left quadrant family members with other movement disorders. Symbols of definitely affected individuals are shaded in black, symbols of probably affected individuals are shaded in dark gray, and symbols of possibly affected individuals are shaded in light gray. Asterisks indicate personally examined and videotaped family members. Individuals screened positive with the BIDS are marked with a plus (+), family members who screened negative with a minus (−). The arrows indicate the index patients, deceased individuals are slashed. (B) Pedigree of one family with a reported positive family history of FTSD (FH+ 9) with DYT7 haplotypes. Linkage analysis at the DYT7 locus was performed in three family members with FTSD (II:2, III:1, and III:2) and in three unaffected individuals (II:1, II:3, and II:5). The affected offspring (III:1 and III:2) do not share a common haplotype from the affected father (II:2). Linkage in at least one offspring could be excluded. For pedigree symbol legend, see A.
Demographic and clinical data of patients identified.
All 47 family members definitely or probably affected with dystonia reported symptoms, but only 35 (75%) had previously been diagnosed with dystonia. Twelve family members (26%), all relatives of the index patients, did not carry an established diagnosis of any movement disorder. Nine individuals (19%) reported a partial remission of symptoms over the course of the disease, 30 individuals (64%) did not notice any changes, and 8 individuals (17%) reported progression. The mean age at onset of dystonic symptoms was 33 ± 13 years (range: 16–75 years). Patients with MD had a mean age at onset of 32 ± 11 years (range: 16–66 years), patients with WC of 35 ± 12 years (range: 20–55 years). Among patients with MD, the majority (23/36; 64%) were men.
Recent treatment.
In the 6 months prior to the examination, 12 individuals (26%), all with MD, received medical treatment. Botulinum toxin injections were given to 10/12 patients, 2/12 patients were taking phenytoin. Five patients with MD (11%) performed nonspecific exercises on the instrument.
Potential triggers.
As a potential trigger of dystonia, 5 individuals (11%), all relatives of the index patients, reported exposure to neuroleptic medication prior to the onset of dystonia. In 2 individuals (4%), this neuroleptic exposure occurred within 12 months before the onset. A considerable increase of time spent on the affected fine motor task within 6 months before the onset of the dystonia was reported by 9 of the 36 patients with MD (25%) and by 3 of the 7 patients with WC (43%). However, an increase of time spent on any fine motor task at any period during their lives was also reported by 8 of the 49 unaffected family members (16%). In this respect, there was no significant difference between patients with MD compared to unaffected family members (χ2 = 0.72; df = 1; p = 0.55), between patients with WC compared to unaffected family members (χ2 = 2.73; df = 1; p = 0.13), and between all patients with FTSD compared to unaffected family members (χ2 = 1.63; df = 1; p = 0.28). A history of pain or overuse syndromes within 6 months before the onset of dystonia was indicated by 3 of the 36 patients with MD (8%); none of the patients reported peripheral nerve entrapment. Two out of the 49 unaffected family members (4%) had a history of peripheral nerve trauma. A comparison between both groups revealed no difference regarding the occurrence of pain, overuse syndromes, or peripheral nerve traumas as potential triggers of dystonia (χ2 = 0.02; df = 1; p = 1.00).
Detailed demographic, clinical, and clinical genetic features of all 53 examined family members are summarized in table e-2.
Molecular findings.
The DYT1 GAG deletion was not present in any of the tested patients. Owing to the small number of affected family members, linkage to the DYT7 locus could not be excluded in 10 of the 11 informative families. In one family, affected offspring did not share a common DYT7 haplotype with their affected father. Therefore, linkage to the DYT7 locus could be excluded in this family as illustrated in figure 2B.
DISCUSSION
The present study expands previous findings of the presence of dystonia in a considerable number of relatives of index patients with MD with an autosomal dominant pattern of transmission.12 Affected relatives were identified both of index FH+ and FH− patients. Although none of the 14 FH− index patients reported any cases of dystonia in their families, 5 of them (36%) had affected relatives with dystonia on clinical examination. Several clinical studies on other forms of focal dystonia based on examination of family members revealed similar numbers of familial cases (23% to 27%).5-7 MD, however, has long served as a textbook example of a purely occupational dystonia, even more so than other forms of FTSD such as WC. Due to the large number of familial cases observed also in MD, the concept of MD as a sporadic and solely environmentally acquired type of dystonia needs to be reconsidered.
The present study focuses on clinical genetic data of MD based upon systematic family history taking and neurologic examination of both index patients and relatives. This approach resulted in a significant increase of familial cases compared to previous data based on family history interview,1 clearly indicating that the family history method is of limited use in genetic studies of primary adult-onset dystonia including MD.
The detection of a larger number of affected relatives in the present study compared to previous reports is largely the result of systematic history taking with the BIDS. Although superior to family history alone, we cannot fully exclude that a case with dystonia was not detected with the BIDS. While the utility of the BIDS as a screening tool was previously described to identify family members with cervical dystonia only,13 it is a computer-assisted telephone interview designed to detect many forms of dystonia. The present study demonstrated that the BIDS appears to be an excellent screening instrument also for genetic studies of FTSD but further validation is advisable.
An important goal for genetic studies of all types of focal dystonia is ascertainment and examination of as many affected cases as possible. The complex rating procedure of the videos was employed to ensure accurate diagnosis of dystonia. Establishing a diagnosis of MD requires specific expertise including experience in professional instrument playing. In some cases, a diagnosis is based only on hearing of subtle changes of complex sensorimotor skills, making it difficult to diagnose MD in some cases even for movement disorder specialists.
The majority of affected relatives had FTSD including MD and WC. Interestingly, most of them were not aware of the presence of dystonia, although all reported symptoms such as technical problems when playing an instrument or difficulties when writing. Only 7 of the 19 cases were previously diagnosed with dystonia. In addition, few relatives related their difficulties during motor performance to the dystonia of the index patients.
Surprisingly, a considerable number of identified patients with MD or WC displayed additional types of dystonia. In addition, an unexpected number of other hypokinetic and hyperkinetic movement disorders, some of them unusual, were present in a considerable number of participants. As a broad intrafamilial and interfamilial phenotypic spectrum is known for many genetic movement disorders, it is tempting to speculate that at least part of the observed additional movement disorders in our patients are due to a shared underlying genetic cause.
In a similar vein, the intrafamilial accumulation of MD and WC found in our families raises the question whether these disorders are indeed related or distinct entities. Recent studies comparing MD and WC patients using transcranial magnetic stimulation suggested pathophysiologic differences between the two conditions.16,17 Given a possible common genetic cause of MD and WC in at least a subset of patients as confirmed by the present study, however, these neurophysiologic differences could be interpreted as a secondary rather than a primary phenomenon.
Several environmental risk factors have been described as potential triggers for the development of MD, such as an increase of practice time spent on the instrument,1,3 or local pain or intensified sensory input due to various causes1,3,18 before the onset of dystonia. On the basis of a given, most probably genetic susceptibility, these factors were hypothesized to trigger the manifestation of MD.19 Triggers for the development of a genetic movement disorder have recently been identified in patients with rapid-onset dystonia-parkinsonism (DYT12).20 Although in our families some patients reported the aforementioned triggers before the onset of dystonia, no significant difference was observed compared to the unaffected family members, some of whom also reported potential triggers at different times of their lives. In view of these findings and the fact that triggers may be subject to substantial recall bias, the role of potential triggers in the pathophysiology of MD should be revisited.
Based on the results of the present study, our main hypothesis is that at least some cases of MD, other forms of FTSD, and possibly even other types of movement disorders may have a shared underlying genetic cause. A recent review article addressed the question whether the various types of focal dystonia may have a common etiologic background.21 Interestingly, available clinical genetic studies indicated that all types of focal dystonia are likely related disorders that share a common etiologic, most probably genetic background.21
Not surprisingly, none of our patients carried the GAG deletion in the DYT1 gene, which has only rarely been linked to focal dystonia.8 Owing to the relatively small number of affected family members, linkage to the DYT7 gene locus that has been described in two families with focal dystonia10,11 could not be definitively excluded in all but one family. To perform meaningful linkage analyses, the identification of large families with MD/FTSD suitable for a classic genome scan is needed. In addition, modern methods of genome-wide linkage in large numbers of MD/FTSD cases may lead to the identification of the genetic factors that cause or contribute to focal dystonia, the most common form of dystonia.
ACKNOWLEDGMENT
The authors thank the patients and family members for their participation in the study.
Supplementary Material
Address correspondence and reprint requests to Dr. Christine Klein, Dept. of Neurology, University of Lübeck, Ratzeburger Allee 160, 23538 Lübeck, Germany
christine.klein@neuro.uni-luebeck.de
Supplemental data at www.neurology.org
Supported by a grant from the Dystonia Medical Research Foundation, the Bachmann Strauss Foundation, and the Volkswagen Foundation. C.K. is a recipient of a Schilling Award from the Hermann and Lilly Schilling Foundation.
Disclosure: The authors report no disclosures.
Received September 23, 2008. Accepted in final form January 23, 2009.
REFERENCES
- 1.Altenmüller E. Focal dystonia: advances in brain imaging and understanding of fine motor control in musicians. Hand Clin 2003;19:523–538. [DOI] [PubMed] [Google Scholar]
- 2.Frucht SJ, Fahn S, Greene PE, et al. The natural history of embouchure dystonia. Mov Disord 2001;16:899–906. [DOI] [PubMed] [Google Scholar]
- 3.Lederman R. Focal dystonia in instrumentalists: clinical features. Med Prob Perform Art 1991;6:132–136. [Google Scholar]
- 4.Jankovic J, Ashoori A. Movement disorders in musicians. Mov Disord 2008;23:1957–1965. [DOI] [PubMed] [Google Scholar]
- 5.Waddy H, Fletcher N, Harding A, CD M. A genetic study of idiopathic focal dystonias. Ann Neurol 1991;29:320–324. [DOI] [PubMed] [Google Scholar]
- 6.Stojanovic M, Cvetkovic D, Kostic VS. A genetic study of idiopathic focal dystonias. J Neurol 1995;242:508–511. [DOI] [PubMed] [Google Scholar]
- 7.Defazio G, Martino D, Aniello MS, et al. A family study on primary blepharospasm. J Neurol Neurosurg Psychiatry 2006;77:252–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gasser T, Windgassen K, Bereznai B, Kabus C, Ludolph AC. Phenotypic expression of the DYT1 mutation: a family with writer’s cramp of juvenile onset. Ann Neurol 1998;44:126–128. [DOI] [PubMed] [Google Scholar]
- 9.Friedman JR, Klein C, Leung J, et al. The GAG deletion of the DYT1 gene is infrequent in musicians with focal dystonia. Neurology 2000;55:1417–1418. [DOI] [PubMed] [Google Scholar]
- 10.Leube B, Rudnicki D, Ratzlaff T, Kessler KR, Benecke R, Auburger G. Idiopathic torsion dystonia: assignment of a gene to chromosome 18p in a German family with adult onset, autosomal dominant inheritance and purely focal distribution. Hum Mol Genet 1996;5:1673–1677. [DOI] [PubMed] [Google Scholar]
- 11.Bhidayasiri R, Jen JC, Baloh RW. Three brothers with a very-late-onset writer’s cramp. Mov Disord 2005;20: 1375–1377. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt A, Jabusch HC, Altenmüller E, et al. Dominantly transmitted focal dystonia in families of patients with musician’s cramp. Neurology 2006;67:691–693. [DOI] [PubMed] [Google Scholar]
- 13.Saunders-Pullman R, Soto-Valencia J, Costan-Toth C, et al. A new screening tool for cervical dystonia. Neurology 2005;64:2046–2049. [DOI] [PubMed] [Google Scholar]
- 14.Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 1971;9:97–113. [DOI] [PubMed] [Google Scholar]
- 15.Bressman SB, Raymond D, Wendt K, et al. Diagnostic criteria for dystonia in DYT1 families. Neurology 2002;59:1780–1782. [DOI] [PubMed] [Google Scholar]
- 16.Rosenkranz K, Williamon A, Butler K, Cordivari C, Lees AJ, Rothwell JC. Pathophysiological differences between musician’s dystonia and writer’s cramp. Brain 2005 128(Pt 4):918–931. [DOI] [PubMed] [Google Scholar]
- 17.Rosenkranz K, Butler K, Williamon A, Cordivari C, Lees AJ, Rothwell JC. Sensorimotor reorganization by proprioceptive training in musician’s dystonia and writer’s cramp. Neurology 2008;70:304–315. [DOI] [PubMed] [Google Scholar]
- 18.Charness ME, Ross MH, Shefner JM. Ulnar neuropathy and dystonic flexion of the fourth and fifth digits: clinical correlation in musicians. Muscle Nerve 1996;19:431–437. [DOI] [PubMed] [Google Scholar]
- 19.Jabusch HC, Altenmüller E. Focal dystonia in musicians: from phenomenology to therapy. Adv Cogn Psychol 2006;2:207–220. [Google Scholar]
- 20.Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007 130(Pt 3):828–835. [DOI] [PubMed] [Google Scholar]
- 21.Defazio G, Berardelli A, Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain 2007;130(Pt 5):1183–1193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.