

Abstract
Background:
Rett syndrome (RTT) is caused by mutations in the transcriptional repressor methyl CpG-binding protein 2 (MECP2). Brain-derived neurotrophic factor (BDNF) is a neurotrophic factor playing a major role in neuronal survival, neurogenesis, and plasticity, and it has been shown that BDNF expression is regulated by MeCP2 through a complex interaction. A common polymorphism of BDNF (Val66Met [p.V66M]) has been found to correlate with severity and course of several neuropsychiatric disorders.
Methods:
We examined the association between disease severity score, assessed by the modified Percy score, and BDNF polymorphism, using regression methods, in 125 mutation-positive patients with RTT from the Australian Rett Syndrome Database and an Israeli cohort.
Results:
Those who were heterozygous (Val/Met) had slightly more severe disease than those who were homozygous for the wild-type (Val/Val) BDNF polymorphism (increased severity score 2.1, p = 0.09). In those with p.R168X, a commonly occurring MECP2 mutation in RTT, there was a 6-point increase in severity score for those who were heterozygous for the BDNF polymorphism, both unadjusted (p = 0.02) and adjusted for age (p = 0.03). Individuals with the p.R168X mutation and heterozygous for the BDNF polymorphism were also at an increased risk of seizure onset (hazard ratio 5.3, 95% confidence interval 1.6–17.7) compared with those homozygous for the wild-type BDNF allele.
Conclusions:
In addition to mutation type and degree of X-chromosome skewing, the common brain-derived neurotrophic factor (BDNF) polymorphism appears to be another genetic modifier of Rett syndrome (RTT) severity. This suggests that BDNF function may play a significant role in the pathogenesis of RTT.
GLOSSARY
- ARSD
= Australian Rett Syndrome Database;
- BDNF
= brain-derived neurotrophic factor;
- CI
= confidence interval;
- HR
= hazard ratios;
- MECP2
= methyl CpG-binding protein 2;
- Met
= methionine;
- NAA
= N-acetylaspartate;
- RTT
= Rett syndrome;
- Val
= valine.
Rett syndrome (RTT) is an X-linked dominant postnatal neurodevelopmental disorder primarily caused by mutations in the methyl CpG-binding protein 2 (MECP2) gene.1 MeCP2 protein may act as either a transcriptional repressor or activator depending on the target gene with which it associates.2,3 The severity of the RTT phenotype varies considerably depending on the MECP2 mutation type and location.4-8 The degree of X chromosome inactivation skewing has also been shown to affect phenotypic variability in Mecp2-null mice,9 and in RTT females.10 However, these two mechanisms only partially explain this variability.
Brain-derived neurotrophic factor (BDNF) is a neurotrophic factor that plays a major role in neuronal survival, neurogenesis, and neuronal plasticity.11-14 It has been identified as a MeCP2 target through a candidate gene approach,15,16 and abnormalities in BDNF homeostasis contribute to the neurologic phenotype in Mecp2-null mice.3,17
A relatively common single nucleotide polymorphism in the BDNF gene is a substitution of valine (Val) with methionine (Met) at codon 66 (p.V66M). This substitution is believed to disrupt folding, dimerization, and intracellular trafficking of the protein,12,18 decreased gray matter volume,19 and decreased dendritic arborization with neuronal loss.20 In the US population, the frequency of the Val/Val, Val/Met, and Met/Met genotypes is 70, 25, and 5%.12 Various studies have shown a relationship between the polymorphism type and the severity of clinical and imaging features in healthy subjects and in different neuropsychiatric and neurologic disorders.12,21-28
Because of the suspected role of BDNF in RTT pathogenesis, the dramatic wide-scale effect of BDNF in the CNS in general, the relative disadvantage of the p.V66M variant in culture, and clinical studies in other diseases, we decided to investigate the relationship between the presence of this BDNF variant and RTT clinical severity. In order to overcome the variability of severity between mutations, we also examined separately those patients with the two most common mutations, p.T158 M and p.R168X.
METHODS
Data for this study, based on RTT subjects with a confirmed MECP2 mutation, were ascertained from two sources. These were 1) cases in the Australian Rett Syndrome Database (ARSD) on whom DNA samples had been stored in the Westmead laboratory and available for analyses of BDNF polymorphisms and 2) cases seen at the Sheba Medical Centre, Israel. The study was approved by the Ethics Committees of the Princess Margaret Hospital, Western Australia, and the Sheba Medical Centre. Parental consent was provided for all subjects. Cases in the ARSD (n = 131) represent 42.8% of all cases known to the ARSD in December 2006. The ARSD is a population-based database of RTT subjects born since 1976,29 so that ages ranged from 2.9 to 28.9 years at the 2004 follow-up. Israeli cases were identified from those cases seen at Sheba Medical Centre, with ages ranging from 3.5–42 years.
Clinical severity was assessed using what we have coined the Percy scale,6,29 which takes into account early developmental characteristics as well as current clinical features and has been shown to be an appropriate measure. Severity scales provide quantitative estimates of clinical severity. Each scale is a summation of individual items related to RTT characteristics, which are graded on a discrete scale based on their specific severity or degree of abnormality, with the highest level corresponding to the most severe or abnormal presentation. We chose to use the Percy scale, which has 15 items with maximum possible score of 45, because of its reasonable balance between current functioning and developmental characteristics.29 Data used to determine the scores were derived from information for ARSD cases that were provided to the ARSD in the 2004 follow-up questionnaire and coded for previous analyses.30 Information for Israeli cases was extracted from case records and coded in the same way as the Australian cases. Age at onset of seizures (if present, or age at data collection if seizures were not present) was recorded for cases in both cohorts.
The BDNF polymorphism (p.V66M) was genotyped using TaqMan® SNP Genotyping Assays (assay ID c_11592758_10, Applied Biosystems) according to the manufacturer’s protocol.
Statistical analysis was performed on the combined cohort and relevant subsets using Stata version 9. Comparison of groups was done using analysis of variance or the Kruskal-Wallis test where normality assumptions did not hold. Regression models were linear with dummy variables for categorical factors (including BDNF polymorphism and cohort). The effect of BDNF polymorphism on clinical severity using the Percy score was assessed for all cases with and without adjusting for age as a continuous variable. The relative risk of seizures was estimated using Cox regression with age as the underlying time variable. Subjects were followed up from birth to their age of first seizure, or their age at data collection, if they had experienced no seizures. Results are presented as hazard ratios (HR) and seizure-free survival curves were plotted using Kaplan-Meier estimates. The Mann-Whitney test was used to compare median ages of seizure onset (in those who had seizures) for the BDNF polymorphisms. Analyses were repeated separately for each of the two MECP2 mutation groups, p.T158M and p.R168X.
RESULTS
There were a total of 182 cases in the study with 72.0% (131) from ARSD and 28.0% (51) from Israel. The mean age at data collection for the Australian cases (15.9) was greater than for the Israeli cases (13.8) (p = 0.02). Eight common mutations were found in 30/46 Israeli cases and 97/131 Australian cases, with the commonest mutations being p.T158M (15.8%) and p.R168X (11.9%) (table 1). Severity scores were available for 118/131 ARSD cases and 45/51 Israeli cases. The mean severity scores for Australian cases (25.3) and Israeli cases (27.2) were similar (p = 0.11). The overall mean Percy score was 25.8 and ranged from 8 to 43. There was a slight increase in severity by age group from 24.6 to 26.3, although severity was greatest in those aged 16–21 years (figure 1). Of the 178 cases with known seizure status, 136 (76.4%) had commenced seizures. The median age at onset for the entire cohort (using Kaplan-Meier estimates) was 5 years (95% confidence interval [CI] 4.5–6 years). Seizures were present in 82.8% of Australian cases compared with only 60% of Israeli cases (p = 0.04). However, in those cases with seizures, the median ages at diagnosis in Israeli (3 years) and Australian (4 years) cases were similar (p = 0.63).
Table 1 Distribution ofMECP2mutations by case source
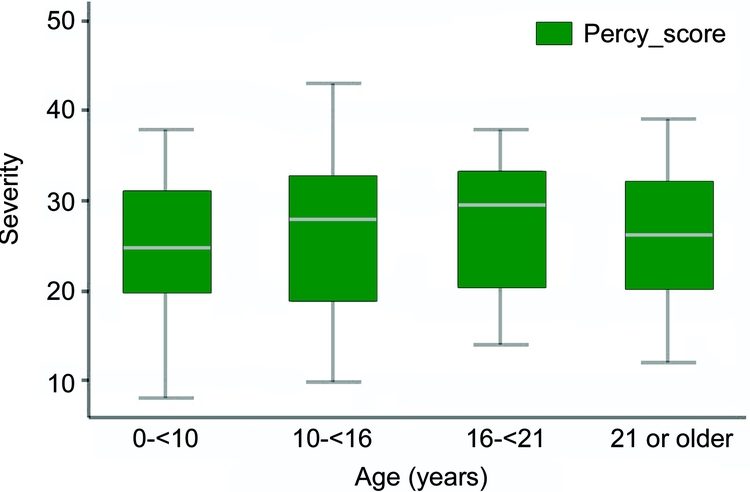
Figure 1 Severity by age group
For BDNF, 57.2% were homozygous for the wild-type (Val/Val) allele, 37.6% were heterozygous (Val/Met), and 5.2% were homozygous for the mutated (Met/Met) allele, demonstrating Hardy-Weinberg equilibrium (p = 0.84). Since only a small number of cases were homozygous for the Met/Met allele (4 from the Israeli cohort and 5 from the ARSD cohort), they were excluded from the severity and seizure comparisons. Overall, those heterozygous for the BDNF polymorphism were slightly more severe than those who were homozygous for the wild type (increased severity 2.1, p = 0.09). When severity was examined for those with the p.R168X mutation (n = 19 with scores), severity was associated with a 6-point increase in the severity score among heterozygotes for the BDNF polymorphism, both unadjusted (p = 0.02) and adjusted for age (p = 0.03). No such difference was seen with the p.T158M mutation (n = 23 with scores) (table 2, figure 2). Risk of seizure onset, after accounting for cohort effects, was not significantly affected by BDNF polymorphism (HR when heterozygous for the BDNF polymorphism 1.2, p value = 0.24, 95% CI 0.9–1.8). The median age at onset of seizures, in cases with seizures, was similar for heterozygous (4.2 years) and wild-type (3.5 years) cases (p = 0.53). However, for cases with the p.R168X mutation, heterozygous cases had a significantly increased risk of seizure onset (HR 5.3, p value 0.006, 95% CI 1.6–17.7), and had an earlier age at seizure onset (median = 2 years), than those who were homozygous for the wild-type BDNF allele (median = 7 years) (figure 3). Of the nine cases homozygous for the mutant BDNF allele who were not included in this analysis, six had commenced seizures (median age at onset 5 years) and three had not.
Table 2 Severity and brain-derived neurotrophic factor polymorphism
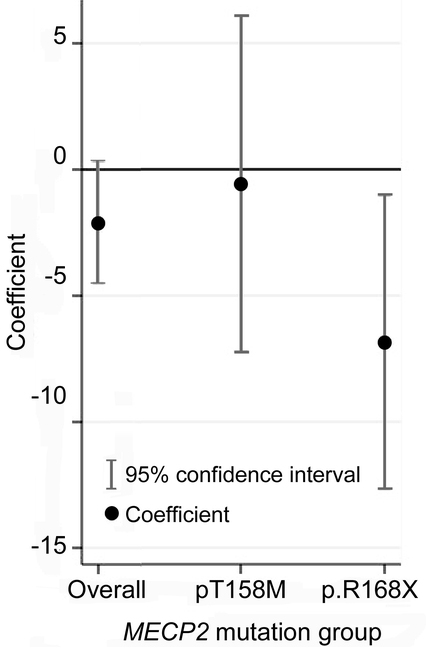
Figure 2 Change in Percy score for the brain-derived neurotrophic factor polymorphism compared with wild-type (valine/valine) brain-derived neurotrophic factor
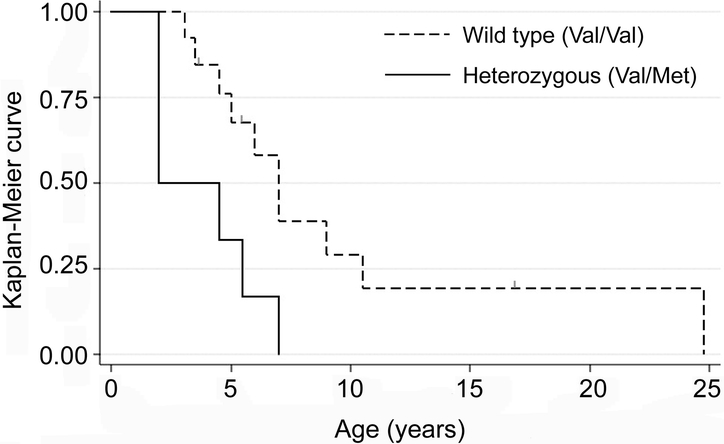
Figure 3 Kaplan-Meier curve for age at onset of seizures for cases with p.R168X (with censor points marked as small gray lines)
DISCUSSION
Among all these Rett patients, those who were heterozygous for the p.V66M variant were slightly more severe than those homozygous for the wild-type (Val/Val) BDNF allele. Patients with the p.R168X mutation who were heterozygous for the BDNF allele had a 6-point increase in the Percy severity score, as well as a fivefold increase in risk of seizures, after adjusting for age. Neither of these effects was seen in patients with the p.T158M mutation. The distribution of the BDNF polymorphism variant was found to be similar to what has been previously reported in the literature in Caucasian populations12 including a cohort of patients with Rett syndrome.31
Both the type of mutation and degree of skewing of X-chromosome inactivation have an effect on RTT severity, including pattern of seizure onset32 and frequency.33 There were some differences in the distribution of mutations between the two case sources, e.g., the p.R270X mutation was more common in the Australian cohort, and C-terminal deletions were more common in the Israeli cohort. The Australian cohort had also experienced more seizures. The Australian cohort was population-based, including older patients and those who had died, in whom the prevalence of seizures was likely to have been higher, so it is perhaps not surprising that the seizure prevalence was higher in the Australian than Israeli cohort. However, paroxysmal nonepileptic events, which mimic epilepsy, also occur in RTT and differentiating between epileptiform and nonepileptiform events can be difficult without video-electroencephalographic monitoring.34 This was not feasible in the Australian cohort. Therefore we, like others,35 relied on parental report of seizures for this cohort and accepted their assessment that events were epileptic. This could be further contributing to the difference in epilepsy diagnosis between the two countries. However, adjusting for the cohort effect in our analysis should have ensured that this difference did not affect the relationship with the BDNF polymorphism.
Information on the X-inactivation skewing was not available for the majority of our cohort, so that it could not be used as a variable in this analysis. This missing information could affect our results in both directions, especially as we have previously demonstrated the contribution of X-inactivation to the variability of phenotype in these specific mutations.10 Despite this shortcoming, the fact that we did find a relationship between the p.V66M polymorphism and severity, in keeping with the direction we hypothesized, is consistent with previous studies in normal individuals, and other neuropsychiatric and neurologic diseases. This would suggest a role for BDNF activity in the pathogenesis of RTT. This role may be nonspecific, as suggested in the other diseases, and may relate to the general role of BDNF in neuronal survival, and plasticity. It could also be explained by an as yet untested possibility that the two proteins (MeCP2 and BDNF) have certain overlapping effects. In addition, it could be that MeCP2 dysfunction reduces overall neuronal activity, thereby indirectly resulting in decreased BDNF,36 further accentuating possible adverse effects of a less efficient BDNF protein variant. On the other hand, it could also suggest a direct role of MeCP2 protein in BDNF gene expression, and as a consequence a significant role of Bdnf/BDNF protein activity in the pathogenesis of RTT in both the mouse model and the human.
In mice, BDNF has distinct effects if depleted during early development, including extreme impairment in learning and memory compared with depletion only in the adult brain, which results in diminished long-term potentiation in the hippocampus, a process believed to be the cellular mechanism of learning and memory. There is also a gender-specific effect. BDNF-deficient males are hyperactive but show no depression-related behaviors, whereas BDNF-deficient females show no motor changes but express depression-related behaviors.37
The existence of two distinct possible mechanisms by which BDNF may affect function in the developing and mature brain may suggest a different role and impact in infants and young children with evolving RTT and in adolescent and adult RTT patients. Investigating the effect of the common BDNF polymorphism on different aspects of the RTT phenotype which are age related would be worthwhile but would require a much larger dataset.
In a recent publication,31 the effect of the common BDNF functional polymorphism was investigated in a cohort of 81 girls with various mutations looking at both a general score (using the Kerr scoring system38) and also on 14 different clinical features separately (combining all mutations together). Although the authors did not find a significant relationship with the overall severity score, they did find that the BDNF genotype distribution tended to be different in the areas of hand skills and age at seizure onset (defined simply as a categorical variable, with early onset described as before 2 years of age). They suggested that the polymorphism might even be protective against seizures. In contrast to their findings, and consistent with our hypothesis regarding the relative dysfunction of the p.V66M polymorphism, we found that age at onset of seizures was earlier in those heterozygous for the BDNF polymorphism, particularly for people with the p.R168X mutation. We did not categorize age at seizure onset but instead we retained the actual ages at onset or ages at data collection in people without seizures and used survival analysis to take account of censoring. We also found that overall severity was greater in those who were heterozygous for the p.V66M allele compared with those with the wild-type BDNF sequence, particularly in those individuals with the p.R168X MECP2 mutation. It may be that both the earlier age at seizure onset and the additional severity we find with the Val/Met variant in subjects with the p.R168X mutation are manifestations relating to the general role of BDNF expression in RTT pathogenesis. Further research could investigate the effect of the common BDNF polymorphism on specific phenotypic characteristics, such as the degree and type of breathing abnormality and other abnormalities of autonomic function, based on the specific role on these functions of BDNF found in brainstem of the Mecp2-null mice.39
The exact role of BDNF in brain development and maturation and its contribution to the pathogenesis of RTT remain unclear, even in face of new data demonstrating that BDNF expression is specifically reduced in the Mecp2-null mouse model.3 However, we have been able to show in a large Australian/Israeli combined RTT dataset that the presence of the common BDNF functional polymorphism appears to confer some mutation-specific additional severity and increase the risk of seizure onset, particularly in those with the p.R168X mutation. Further international collaborative studies combining national data from individual countries (as already done through the infrastructure of an international Rett syndrome database7) and examining specific phenotypic and age-related features could be beneficial.
AUTHOR CONTRIBUTIONS
Statistical analysis was performed by Ami Bebbington, assisted by Nick de Klerk.
Address correspondence and reprint requests to Dr. Helen Leonard, Telethon Institute for Child Health Research, Centre for Child Health Research, University of Western Australia, Perth, Australia
hleonard@ichr.uwa.edu.au
Supported by the NIH (5R01HD043100-05) (major aspects of the Australian Rett Syndrome program) and National Medical and Health Research Council (NHMRC) project grant 303189 (certain clinical aspects). The international Rett Syndrome Research Program through which the Israeli data were accessed is funded by the International Rett Syndrome Foundation. H.L. is funded by a NHMRC program grant (353514) and J.C. is funded by NHMRC project grants (346603 and 457238) and the Rett Syndrome Australian Research Fund.
Disclosure: The authors report no disclosures.
Received July 31, 2008. Accepted in final form December 30, 2008.
REFERENCES
- 1.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185–188. [DOI] [PubMed] [Google Scholar]
- 2.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 2003;278:4035–4040. [DOI] [PubMed] [Google Scholar]
- 3.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008;320:1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huppke P, Held M, Handefeld F, Engel W, Laccone F. Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics 2002;33:63–68. [DOI] [PubMed] [Google Scholar]
- 5.Colvin L, Leonard H, de Klerk N, et al. Refining the phenotype of common mutations in Rett syndrome. J Med Genet 2004;41:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schanen C, Houwink EJ, Dorrani N, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am J Med Genet A 2004;126A:129–140. [DOI] [PubMed] [Google Scholar]
- 7.Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008;70:868–875. [DOI] [PubMed] [Google Scholar]
- 8.Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70:1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of Rett syndrome. Am J Hum Genet 2004;74:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Archer HL, Evans J, Leonard H, et al. Correlation between clinical severity in Rett syndrome patients with a p.R168X or p.T158M MECP2 mutation and the direction and degree of skewing of X chromosome inactivation. J Med Genet 2006;140A:691–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron 2007;56:422–437. [DOI] [PubMed] [Google Scholar]
- 12.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003;112:257–269. [DOI] [PubMed] [Google Scholar]
- 13.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron 1995;15:979–981. [DOI] [PubMed] [Google Scholar]
- 14.Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res 2000;128:183–191. [DOI] [PubMed] [Google Scholar]
- 15.Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003;302:890–893. [DOI] [PubMed] [Google Scholar]
- 16.Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003;302:885–889. [DOI] [PubMed] [Google Scholar]
- 17.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 2006;49:341–348. [DOI] [PubMed] [Google Scholar]
- 18.Chen ZY, Patel PD, Sant G, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci 2004;24:4401–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001;24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu B, Zang K, Ruff NL, et al. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 2000;26:233–245. [DOI] [PubMed] [Google Scholar]
- 21.Chao HM, Kao HT, Porton B. BDNF Val66Met variant and age of onset in schizophrenia. Am J Med Genet B Neuropsychiatr Genet 2007;4:505–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho BC, Andreasen NC, Dawson JD, Wassink TH. Association between brain-derived neurotrophic factor Val66Met gene polymorphism and progressive brain volume changes in schizophrenia. Am J Psychiatry 2007;164:1890–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntosh AM, Moorhead TW, McKirdy J, et al. Temporal grey matter reductions in bipolar disorder are associated with the BDNF Val66Met polymorphism. Mol Psychiatry 2007;12:902–903. [DOI] [PubMed] [Google Scholar]
- 24.Muller DJ, de Luca V, Sicard T, King N, Strauss J, Kennedy JL. Brain-derived neurotrophic factor (BDNF) gene and rapid-cycling bipolar disorder: family-based association study. Br J Psychiatry 2006;189:317–323. [DOI] [PubMed] [Google Scholar]
- 25.Borroni B, Archetti S, Costanzi C, et al. Role of BDNF Val66Met functional polymorphism in Alzheimer’s disease-related depression. Neurobiol Aging Epub 2008 Jan 5. [DOI] [PubMed]
- 26.Enoch MA, White KV, Waheed J, Goldman D. Neurophysiological and genetic distinctions between pure and comorbid anxiety disorders. Depress Anxiety 2007;25:383–392. [DOI] [PubMed] [Google Scholar]
- 27.Hemmings SM, Kinnear CJ, Van Der Merwe L, et al. Investigating the role of the brain-derived neurotrophic factor (BDNF) val66met variant in obsessive-compulsive disorder (OCD). World J Biol Psychiatry 2007:1–9. [DOI] [PubMed] [Google Scholar]
- 28.Mai M, Akkad AD, Wieczorek S, et al. No association between polymorphisms in the BDNF gene and age at onset in Huntington disease. BMC Med Genet 2006;7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colvin L, Fyfe S, Leonard S, et al. Describing the phenotype in Rett syndrome using a population database. Arch Dis Child 2003;88:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young DJ, Bebbington A, Anderson A, et al. The diagnosis of autism in a female: could it be Rett syndrome? Eur J Pediatr 2007;167:661–669. [DOI] [PubMed] [Google Scholar]
- 31.Nectoux J, Bahi-Buisson N, Guellec I, et al. The p.Val66Met polymorphism in the BDNF gene protects against early seizures in Rett syndrome. Neurology 2008;70:2145–2151. [DOI] [PubMed] [Google Scholar]
- 32.Jian L, Nagarajan L, de Klerk N, et al. Predictors of seizure onset in Rett syndrome. J Pediatr 2006;149:542–547. [DOI] [PubMed] [Google Scholar]
- 33.Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Paediatr Neurol 2007;11:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glaze DG, Schultz RJ, Frost JD. Rett syndrome: characterization of seizures versus non-seizures. Electroencephalogr Clin Neurophysiol 1998;106:79–83. [DOI] [PubMed] [Google Scholar]
- 35.Steffenburg U, Hagberg G, Hagberg B. Epilepsy in a representative series of Rett syndrome. Acta Paediatr 2001;90:34–39. [DOI] [PubMed] [Google Scholar]
- 36.Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA 2005;102:12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monteggia LM. Elucidating the role of brain-derived neurotrophic factor in the brain. Am J Psychiatry 2007;164:1790. [DOI] [PubMed] [Google Scholar]
- 38.Kerr AM, Nomura Y, Armstrong D, et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev 2001;23:208–211. [DOI] [PubMed] [Google Scholar]
- 39.Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci 2007;27:10912–10917. [DOI] [PMC free article] [PubMed] [Google Scholar]