

Abstract
Objective:
To establish phenotype–genotype correlations in early-onset Parkinson disease (EOPD), we performed neurologic, neuropsychological, and psychiatric evaluations in a series of patients with and without parkin mutations.
Background:
Parkin (PARK2) gene mutations are the major cause of autosomal recessive parkinsonism. The usual clinical features are early-onset typical PD with a slow clinical course, an excellent response to low doses of levodopa, frequent treatment-induced dyskinesias, and the absence of dementia.
Methods:
A total of 44 patients with EOPD (21 with and 23 without parkin mutations) and 9 unaffected single heterozygous carriers of parkin mutations underwent extensive clinical, neuropsychological, and psychiatric examinations.
Results:
The neurologic, neuropsychological, and psychiatric features were similar in all patients, except for significantly lower daily doses of dopaminergic treatment and greater delay in the development of levodopa-related fluctuations (p < 0.05) in parkin mutation carriers compared to noncarriers. There was no major difference between the two groups in terms of general cognitive efficiency. Psychiatric manifestations (depression) were more frequent in patients than in healthy single heterozygous parkin carriers but did not differ between the two groups of patients.
Conclusion:
Carriers of parkin mutations are clinically indistinguishable from other patients with young-onset Parkinson disease (PD) on an individual basis. Severe generalized loss of dopaminergic neurons in the substantia nigra pars compacta in these patients is associated with an excellent response to low doses of dopa-equivalent and delayed fluctuations, but cognitive impairment and special behavioral or psychiatric symptoms were not more severe than in other patients with early-onset PD.
GLOSSARY
- CPRS
= Comprehensive Psychopathological Rating Scale;
- EOPD
= early-onset Parkinson disease;
- FAB
= Frontal Assessment Battery;
- MADRS
= Montgomery-Asberg Depression Rating Scale;
- MDRS
= Mattis Dementia Rating Scale;
- MINI
= Mini International Neuropsychiatric Interview;
- MMSE
= Mini-Mental State Examination;
- TMT
= Trail Making Test;
- UPDRS
= Unified Parkinson's Disease Rating Scale;
- WCST
= Wisconsin Card Sorting Test.
Mutations in the parkin gene are considered to be the predominant cause of early-onset Parkinson disease (EOPD) particularly when the family history is compatible with autosomal recessive inheritance.1,2 Parkin-linked PD has a broad range of clinical phenotypes, some atypical, but is generally early-onset parkinsonism, with a slow clinical course, excellent response to low doses of levodopa, frequent treatment-induced dyskinesias, and no dementia.3,4 Cognitive function remains normal in the majority of patients,3–6 but behavioral disorders have been reported, including anxiety, psychosis, panic attacks, depression, and disturbed sexual, behavioral, and obsessive-compulsive disorders.4,5,7–9
Mutations have been found in the homozygous or compound heterozygous state, compatible with recessive transmission, but occasionally as single heterozygous mutations.2,5,10 In these patients, it is still not clear whether a second mutation outside the open reading frame of the gene may be overlooked or whether some heterozygous mutations are sufficient to cause the disease.11
To establish pheno–genotype correlations in EO parkinsonism, we performed a clinical study of 21 patients with and 23 patients without parkin mutations. We expected behavioral or psychiatric problems in parkin mutation carriers because neuropathologic studies of parkin patients show severe generalized loss of dopaminergic neurons in the substantia nigra pars compacta that would greatly decrease dopaminergic efferents to the limbic and sensory-motor systems. In addition, although it is generally assumed that parkin gene mutations carriers have no dementia, systematic assessment of cognitive functions in this population was not performed. We aim to fill this gap. Additionally, we evaluated 9 healthy sibs of our patients with single heterozygous parkin mutations.
METHODS
Forty-four patients with isolated or familial early-onset parkinsonism (<45 years) recruited in Paris (n = 25), Grenoble and Lyon (n = 19) underwent neurologic, neuropsychological, and psychiatric evaluation. The inclusion criteria for PD were at least two of the parkinsonian triad of signs (bradykinesia, rigidity, rest tremor) and at least 30% improvement under levodopa therapy, in familial or isolated cases. Exclusion criteria were the existence of extensor plantar reflexes, ophthalmoplegia, early dementia, or early autonomic failure. There were 13 women and 31 men; mean age at onset was 33.1 years ± 8.3 (12–44) and mean disease duration was 17.4 years ± 7.8. (5–34). Twenty-five patients had known family histories of PD. Among the 44 patients originating from France (n = 41), Asia (n = 2), and North Africa (n = 1), 21 had homozygous or compound heterozygous parkin mutations and 23 patients no parkin mutations. Five female and four male heterozygous parkin carriers originating from France (n = 7) and North Africa (n = 2), healthy sibs of the parkin patients, also participated in the study. Their age at examination was 47.9 years ± 12.6 (28–68).
A standardized form was used to assess the history of the disease in the patient and the family, the clinical signs, additional diseases, and the response to present treatment. All patients were evaluated with the Unified Parkinson's Disease Rating Scale (UPDRS) I to VI, with and without treatment (“on” and “off” state), except UPDRS I and IV, which were evaluated only in the “off” state. The “off” state was usually reached after interruption of antiparkinsonian medication for at least 12 hours, and the best “on” state was obtained after administration of a single suprathreshold dose of levodopa (50 mg higher than the usual effective morning dose). The response to treatment was calculated from the “off” and “on” values of UPDRS III. We used the conversion factors proposed by Thobois12 to calculate the daily levodopa dose-equivalent taken by the patients.
Neuropsychological and psychiatric tests were performed while the patients received their usual treatment. The Mini-Mental State Examination (MMSE) and the Mattis Dementia Rating Scale (MDRS) were used to assess global intellectual efficiency. Verbal episodic memory was investigated with the Grober and Buschke test to control for effective encoding and to facilitate retrieval with the same semantic cueing, providing a comparison between free and cued recall. Two parallel forms of the Grober and Buschke test were used to control test-retest effects. Executive functions were assessed with the simplified version of the Wisconsin Card Sorting Test (WCST), the category and literal fluency test, the Trail Making Test (TMT), the Frontal score, and the Frontal Assessment Battery (FAB). The psychiatric evaluation was based on the Montgomery-Asberg Depression Rating Scale (MADRS), the Comprehensive Psychopathological Rating Scale (CPRS), and a standardized Mini International Neuropsychiatric Interview (MINI).
Molecular analysis of the parkin gene was performed by denaturing high performance liquid chromatography and sequencing and semiquantitative multiplex PCR as described before.13 The G2019S mutation of the LRRK2 gene and mutations in the DJ-1 and Pink1 genes were excluded in all patients.14
Data are expressed as the mean ± SD (range) or percentage (n). Group comparisons were made using the Kruskal-Wallis test for quantitative variables and the Fisher exact test for qualitative variables. When significant differences were detected, post hoc comparisons were performed with a Bonferroni correction for multiple comparisons. For the delay before dyskinesia, dystonia, and fluctuations, Kaplan-Meier estimations were obtained. The Sidak correction for multiple comparisons was applied by discipline (clinical, psychiatric, neuropsychological). The SAS 8.1 statistical package (SAS Institute, Cary, NC) was used for the analyses.
The study was approved by the Ethical Committee of the Salpêtrière University Hospital Paris, France, and all patients gave their written informed consent.
RESULTS
Twenty-one out of 44 patients with EOPD had homozygous or compound heterozygous parkin mutations (table 1). Nine healthy sibs of the patients with parkin mutations had a single heterozygous mutation (table 1). Age at examination did not differ significantly among the three groups (47.5 years ± 9.1 for affected parkin mutation carriers, 48.0 years ± 8.0 for affected non-mutation carriers, and 47.9 years ± 12.6 for unaffected single heterozygous carriers). Age at onset (31.1 years ± 8.3 vs 34.9 years ± 6.2) and disease duration (17.4 years ± 7.8 vs 12.9 years ± 7.7) were similar in patients with and without parkin mutations (table 2). Clinical characteristics such as mentation, behavior, and mood (UPDRS I), activities of daily living (UPDRS II, “off” and “on” drug), parkinsonian motor disability (UPDRS III, “off” and “on” drug), Hoehn and Yahr Scale (UPDRS V), and the Schwab and England Scale (UPDRS VI) were also similar in patients with and without parkin mutations (table 2). However, the daily levodopa dose equivalents were significantly lower in patients with parkin mutations than in noncarriers (636 mg ± 462 vs 1,139 mg ± 451), although the duration of treatment was similar (13.2 years ± 6.5 vs 9.9 years ± 6.3). The UPDRS IV scores, which evaluate levodopa-related complications, did not differ between the two groups, but the delay before the appearance of levodopa-related fluctuations after treatment was significantly shorter in noncarriers than in patients with mutations (median: 5 years 95% CI [4–18] vs 14 years [9–28]). One affected non-mutation carrier but none of the patients with mutations had orthostatic hypotension, and none of the patients in either group had urinary incontinence. Patients from both groups had problems with sleep, such as sleep disruption, daytime sleepiness, or sleep behavior disorder (16/23 non-mutation carriers vs 10/21 parkin mutation carriers, data not shown), but the difference was not significant. Careful neurologic examination revealed no parkinsonism in the sibs with single heterozygous parkin mutations.
Table 1 Parkin mutations in affected and unaffected carriers
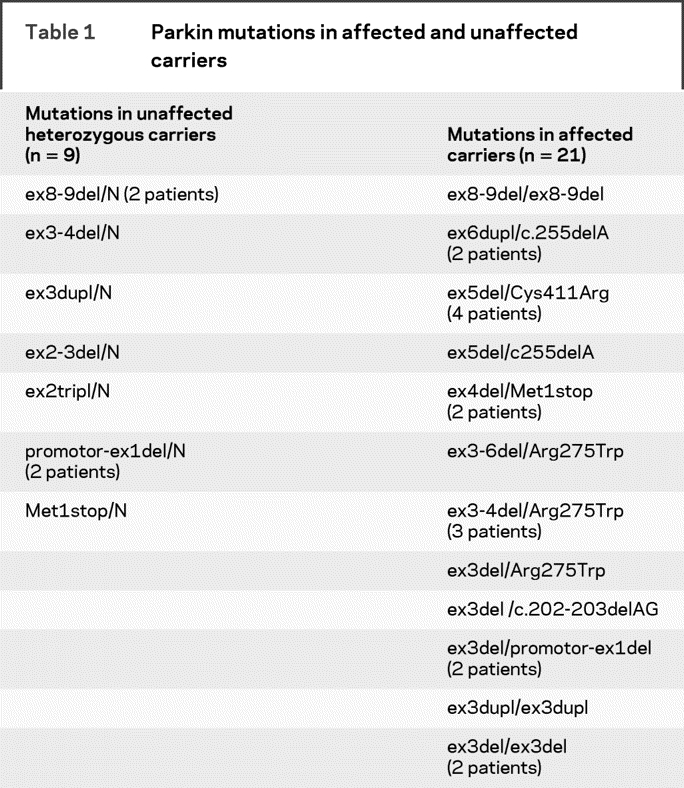
Table 2 Clinical characteristics of patients with Parkinson disease (PD) with parkin and without parkin mutations

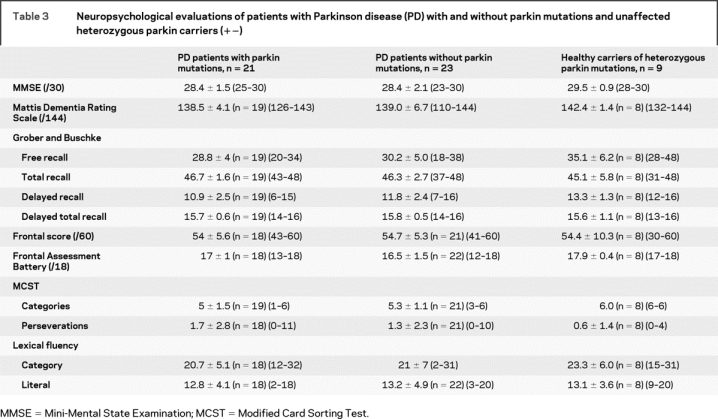
Detailed neuropsychological examinations, including the MMSE, MDRS, Grober and Buschke test, WCST, TMT, FAB, and frontal score, did not detect any significant differences among the three groups (table 3). The results of the MDRS and the FAB were significantly different among the three groups, but after the Sidak correction for multiple comparisons, the difference was no longer significant. Four patients carrying two parkin mutations, one healthy heterozygous mutation carrier and one non-mutation carrier, had MDRS scores below the threshold of 136/144. The patient without parkin mutations (SPD-150) was a 55-year-old man, who started PD at age 33 and developed epilepsy during the 24-year evolution of his disease. The low cognitive efficiency of the patient (MMSE: 24/30 and MDRS: 110/144) was attributed to either his epileptic status or his carbamazepine treatment.
Table 3 Neuropsychological evaluations of patients with Parkinson disease (PD) with and without parkin mutations and unaffected heterozygous parkin carriers (+−)
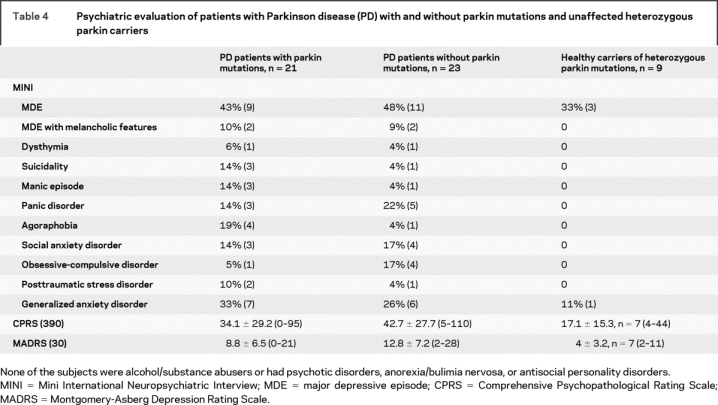
The psychiatric profiles obtained with the MADRS, CPRS, and MINI tests were similar in patients with and without parkin mutations. Interestingly, patients tended to experience more depressive episodes and had higher MADRS scores (p = 0.2) than unaffected heterozygous parkin carriers (table 4). Additionally, six patients without and three patients with parkin mutations had MADRS scores >15; five affected non-mutation carriers and three affected mutation carriers had scores >18. However, the mean scores for both groups were not significantly different.
Table 4 Psychiatric evaluation of patients with Parkinson disease (PD) with and without parkin mutations and unaffected heterozygous parkin carriers
DISCUSSION
This is one of the most detailed clinical studies to combine neurologic, neuropsychological, and psychiatric investigations to establish phenotype–genotype correlation in early-onset parkinsonism. We expected to find particular behavioral or psychiatric pattern and maybe cognitive disorders in parkin mutation carriers because neuropathologic studies of these patients have shown severe generalized loss of dopaminergic neurons in the substantia nigra pars compacta13 that would greatly decrease dopaminergic efferents to the limbic and subcortical-frontal and sensory-motor systems. The 21 patients with and 23 patients without parkin mutations were appropriately matched for age at onset (31.1 years ± 8.3 vs 34.9 years ± 6.2) and disease duration (17.4 years ± 7.8 vs 12.9 years ± 7.7). Detailed clinical evaluations including UPDRS I–VI, with and without treatment, did not detect any significant differences between the two groups. The daily doses of levodopa were also similar in parkin mutation and non-mutation carriers (528 mg ± 439 vs 778 mg ± 343), but after calculation of the daily levodopa dose equivalents,12 a significant difference between the groups was observed: parkin mutation carriers improved to the same extent as noncarriers (68.4% ± 13.1 vs 70.2% ± 18.6, data not shown) with significantly lower daily levodopa dose equivalents (636 mg ± 462 vs 1,139 mg ± 451).
Previous studies have already shown in large cohorts of patients that parkin carriers had more levodopa induced dyskinesias, brisk reflexes, onset with foot dystonia,3,15–17 and a better response to low doses of medication, even after a long evolution of the disease,4,5 although none of these studies compared patients matched for age and disease duration. Our study confirms that parkin patients have very good responses to low doses of antiparkinsonian treatment. Furthermore, our parkin mutation carriers did not have more levodopa-induced complications than noncarriers, but the delay before the appearance of fluctuations after initiation of treatment was significantly longer in parkin mutation carriers than in patients without parkin mutations (14 years ± 5.1 vs 5 years ± 1.4). This delay in the development of levodopa-related dyskinesia was probably a result of the significantly lower doses of medication, but, curiously, the delay between the development of levodopa-related dyskinesias and dystonia did not differ between the two groups (10 vs 12 years and 13 vs 17 years, table 2). Interestingly, we show that dysautonomia, orthostatic hypotension, and urinary incontinence, which are absent in patients with parkin mutations, are also rare in the other early-onset PD cases.
All unaffected single heterozygous mutation carriers, with ages ranging from 28 to 68, had normal detailed neurologic examinations. Statistically significant reductions in [18F]fluorodopa uptake have been observed in carriers of a single heterozygous parkin mutation15,18 compared to controls, and some were reported to have subtle extrapyramidal signs, such as resting tremor, reduced arm swing, or a mask-like face.3,4,15,18 It cannot be excluded that these healthy single heterozygous mutation carriers will develop parkinsonian symptoms in the future, but six of them are already more than 15 years older than the age at onset of their affected sibs. This observation supports the hypothesis that a single, even truncating, mutation may not be sufficient to trigger PD. These unaffected sibs of affected parkin mutation carriers are useful controls, however, for neuropsychological and psychiatric evaluations, because they share genetic and environmental factors but not the disease with their affected sib.
Neuropsychological examinations did not reveal major differences in general cognitive efficiency (MMSE, Mattis DRS, episodic memory, Grober and Buschke test) or executive functions (frontal score and FAB) in patients with and without parkin mutations and unaffected heterozygous parkin carriers. This is consistent with and confirms, with more detailed neuropsychological evaluations than in previous studies, that cognitive function remains normal in the majority of patients with parkin mutations,4,5,19 even after 45 years of evolution.4 Additionally, we showed that patients with early-onset PD without parkin mutations do not have cognitive decline even after more than 30 years of disease evolution. However, it is interesting to note that only one patient without a parkin mutation, who also has epilepsy, four patients with parkin mutations, and one healthy single heterozygous parkin carrier had abnormal results on the MDRS (<136), although the groups did not differ significantly after the Sidak correction for multiple testing. Functional or structural abnormalities in the caudate nucleus have been postulated to play a role in frontal-subcortical cognitive impairment or dementia in patients with basal ganglia disease. Interestingly, several studies3,15,17,18,20–22 reported that the decrease in [18F]fluorodopa uptake in nigrostriatal terminals in the caudate nucleus is generally greater in patients with parkin mutations than in patients without this mutation. This pattern of nigrostriatal dysfunction might result in a different neuropsychological profile. However, this discussion remains speculative and the results must be confirmed by further studies with larger patient groups.
Behavioral disorders, including anxiety and psychosis, panic attacks, depression, disturbed sexual behavior, and obsessive-compulsive disorders, have been reported with variable frequency in patients with parkin mutations,4,5,7–9 and were suggested to be a distinctive feature of parkin disease.4 Dopaminergic dysfunction in cortical areas which might contribute to the psychiatric disorders, as postulated for patients with idiopathic PD,23 has been demonstrated in 13 homozygous or compound heterozygous parkin mutation carriers using PET with 11C-raclopride.22 However, our detailed psychiatric examinations including MINI, MADRS, and CPRS did not detect any significant qualitative or quantitative differences between parkin mutation carriers and noncarriers. Psychiatric manifestations were present in both groups of patients, but at a similar rate. They were less frequent, however, in the unaffected heterozygous parkin carriers, supporting the hypothesis that dopaminergic dysfunction or antiparkinsonian drugs might account for their greater frequency in patients. Nevertheless, they do not appear to be more frequent in parkin-related parkinsonism than in other early-onset patients.
The large spectrum of parkin gene defects, which differ in their predicted consequences on the function of the protein, also raises the question of their role in the variability of the phenotype. Despite the large number of parkin mutation carriers included, the number of cases in each of the genotype specific groups was too small for a specific neurologic, neuropsychological, or psychiatric pattern to emerge.
The results of this detailed clinical study indicate that patients with PD with parkin mutations are clinically indistinguishable from other early-onset patients. Severe generalized loss of dopaminergic neurons in the substantia nigra pars compacta in these patients is associated with an excellent response to low doses of levodopa-equivalent and delayed fluctuations. Their neuropsychological performance is not distinctive. Interestingly, behavioral problems and psychiatric symptoms, which have been considered to be markers of parkin disease, are observed at similar rates in both groups of early-onset patients and are more frequent in these patients than in unaffected heterozygous parkin mutation carriers.
ACKNOWLEDGMENT
The authors thank the patients and their families; Céline Chamayou and Aurélie Funkiewiez for the neuropsychological data, Cécile Behar and Mircéa Polosan for psychiatric advice, Merle Ruberg for critical reading of the manuscript, and the DNA and Cell Bank of the IFR 070 for sample preparation; and the nurses of the Centre d'Investigation Clinique who provided care for the patients.
APPENDIX
The French Parkinson's Disease Genetics Study Group: Y. Agid, A.-M. Bonnet, M. Borg, A. Brice, E. Broussolle, Ph. Damier, A. Destée, A. Dürr, F. Durif, E. Lohmann, M. Martinez, C. Penet, P. Pollak, O. Rascol, F. Tison, C. Tranchant, M. Vérin, F. Viallet, M. Vidailhet, and J.-M. Warter (deceased).
Received March 10, 2008. Accepted in final form June 27, 2008.
Address correspondence and reprint requests to Prof. Alexis Brice, INSERM UMR S_679, Hôpital Pitié-Salpêtrière, 47 boulevard de l'Hôpital, F-75013 Paris, France alexis.brice@upmc.fr
Editorial, page 106
e-Pub ahead of print on November 5, 2008, at www.neurology.org.
*The French Parkinson's Disease Genetics Study Group members are listed in the appendix.
Authors' affiliations are listed at the end of the article.
Supported by INSERM/AP-HP (PCR02006-P011104), the NIH grant NS41723-01A1, and the European commission (EU Contract No.LSHM-CT-2003-503330/APOPIS).
Disclosure: The authors report no disclosures.
REFERENCES
- 1.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 2.Periquet M, Latouche M, Lohmann E, et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 2003;126:1271–1278. [DOI] [PubMed] [Google Scholar]
- 3.Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 2002;125:2248–2256. [DOI] [PubMed] [Google Scholar]
- 4.Khan NL, Graham E, Critchley P, et al. Parkin disease: a phenotypic study of a large case series. Brain 2003;126:1279–1292. [DOI] [PubMed] [Google Scholar]
- 5.Lohmann E, Periquet M, Bonifati V, et al. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 2003;54:176–185. [DOI] [PubMed] [Google Scholar]
- 6.Lucking CB, Brice A. Alpha-synuclein and Parkinson's disease. Cell Mol Life Sci 2000;57:1894–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tassin J, Durr A, de Broucker T, et al. Chromosome 6-linked autosomal recessive early-onset Parkinsonism: linkage in European and Algerian families, extension of the clinical spectrum, and evidence of a small homozygous deletion in one family. The French Parkinson's Disease Genetics Study Group, and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Am J Hum Genet 1998;63:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu RM, Shan DE, Sun CM, et al. Clinical, 18F-dopa PET, and genetic analysis of an ethnic Chinese kindred with early-onset parkinsonism and parkin gene mutations. Mov Disord 2002;17:670–675. [DOI] [PubMed] [Google Scholar]
- 9.Yamamura Y, Hattori N, Matsumine H, Kuzuhara S, Mizuno Y. Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev 2000;22 suppl 1:S87–91. [DOI] [PubMed] [Google Scholar]
- 10.West A, Periquet M, Lincoln S, et al. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 2002;114:584–591. [DOI] [PubMed] [Google Scholar]
- 11.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 2007;6:652–662. [DOI] [PubMed] [Google Scholar]
- 12.Thobois S. Proposed dose equivalence for rapid switch between dopamine receptor agonists in Parkinson's disease: a review of the literature. Clin Ther 2006;28:1–12. [DOI] [PubMed] [Google Scholar]
- 13.Lesage S, Magali P, Lohmann E, et al. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat 2007;28:27–32. [DOI] [PubMed] [Google Scholar]
- 14.Lesage S, Lohmann E, Tison F, Durif F, Durr A, Brice A. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J Med Genet 2008;45:43–46. [DOI] [PubMed] [Google Scholar]
- 15.Hilker R, Klein C, Ghaemi M, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol 2001;49:367–376. [PubMed] [Google Scholar]
- 16.Ishikawa A, Tsuji S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology 1996;47:160–166. [DOI] [PubMed] [Google Scholar]
- 17.Lucking CB, Chesneau V, Lohmann E, et al. Coding polymorphisms in the parkin gene and susceptibility to Parkinson disease. Arch Neurol 2003;60:1253–1256. [DOI] [PubMed] [Google Scholar]
- 18.Khan NL, Horta W, Eunson L, et al. Parkin disease in a Brazilian kindred: manifesting heterozygotes and clinical follow-up over 10 years. Mov Disord 2005;20:479–484. [DOI] [PubMed] [Google Scholar]
- 19.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 20.Portman AT, Giladi N, Leenders KL, et al. The nigrostriatal dopaminergic system in familial early onset parkinsonism with parkin mutations. Neurology 2001;56:1759–1762. [DOI] [PubMed] [Google Scholar]
- 21.Sawle GV, Leenders KL, Brooks DJ, et al. Dopa-responsive dystonia: [18F]dopa positron emission tomography. Ann Neurol 1991;30:24–30. [DOI] [PubMed] [Google Scholar]
- 22.Scherfler C, Khan NL, Pavese N, et al. Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain 2004;127:1332–1342. [DOI] [PubMed] [Google Scholar]
- 23.Williams-Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain 2007;130:1787–1798. [DOI] [PubMed] [Google Scholar]