

Abstract
Background:
Autopsy series have shown that some elderly people remain with normal cognitive function during life despite having high burdens of pathologic lesions associated with Alzheimer disease (AD) at death. Understanding why these individuals show no cognitive decline, despite high AD pathologic burdens, may be key to discovery of neuroprotective mechanisms.
Methods:
A total of 36 subjects who on autopsy had Braak stage V or VI and moderate or frequent neuritic plaque scores based on Consortium to Establish a Registry for Alzheimer's Disease (CERAD) standards were included. Twelve had normal cognitive function and 24 a diagnosis of AD before death. Demographic characteristics, clinical and pathologic data, as well as antemortem brain volumes were compared between the groups.
Results:
In multiple regression analysis, antemortem hippocampal and total brain volumes were significantly larger in the group with normal cognitive function after adjusting for gender, age at MRI, time from MRI to death, Braak stage, CERAD neuritic plaque score, and overall presence of vascular disease.
Conclusion:
Larger brain and hippocampal volumes were associated with preserved cognitive function during life despite a high burden of Alzheimer disease (AD) pathologic lesions at death. A better understanding of processes that lead to preservation of brain volume may provide important clues for the discovery of mechanisms that protect the elderly from AD.
GLOSSARY
- AD
= Alzheimer disease;
- CDR
= Clinical Dementia Rating Scale;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- CIRS
= Cumulative Illness Rating Scale;
- ICV
= intracranial volume;
- LB
= Lewy bodies;
- MMSE
= Mini-Mental State Examination;
- NCSE
= Neurobehavioral Cognitive Status Examination;
- NFT
= neurofibrillary tangle;
- NIA
= National Institute on Aging;
- NP
= neuritic plaques;
- OHSU
= Oregon Health & Science University;
- Ref
= reference;
- SES
= socioeconomic status;
- UPDRS
= Unified Parkinson's Disease Rating Scale.
Autopsy series of elderly subjects with normal cognitive function have consistently found high burdens of pathologic lesions associated with Alzheimer disease (AD).1-5 Some studies refer to this state as preclinical AD, suggesting that this is a precursor to AD.3 While some studies report subtle changes in neuropsychological measures in cognitively intact individuals with high AD pathology burdens compared to those with no or low AD pathology burdens, other studies have not found such changes.1,2,6 It remains unknown why these individuals do not show symptoms of overt dementia despite these pathologic changes. One explanation is that the high burdens of classic AD lesions found in these subjects may be necessary but not sufficient to cause cognitive impairment because of compensatory mechanisms or brain reserve.
Most previous studies are descriptive and report pathology results of autopsy series according to premorbid clinical state. In contrast to these studies, we specifically designed our study to investigate factors that may protect individuals with high AD pathology from having overt symptoms of dementia and cognitive decline. Using a case control design, subjects who remained with normal cognitive function despite a high burden of AD pathologic lesions were matched to a group of patients with AD with an equivalent burden of lesions. Clinical and pathologic characteristics as well as antemortem brain volumes obtained by MRI scans were compared between the two groups.
METHODS
The Oregon Health & Science University (OHSU) institutional review board approved this study.
Description of cohorts.
Subjects were from the longitudinal cohort studies conducted at the National Institute on Aging (NIA)-OHSU Layton Aging and AD Center. Recruitment, exclusion, and inclusion criteria for these studies and subject evaluations have been described previously.7 Briefly, these longitudinal aging studies ongoing since 1989 recruit cognitively intact elders who are 65 years of age and older from the community, are without conditions that impair cognition, and have no risk factors for vascular disease. Only those with no evidence of cognitive impairment, questionable dementia, or memory problems and no evidence of clinical depression are enrolled. Subjects with AD are recruited from the NIA-Layton Aging and AD Center clinic at OHSU.
Subject evaluations.
All subjects were evaluated semiannually with standardized clinical examinations. Cognition and functional status were assessed using the Clinical Dementia Rating Scale (CDR),8 Mini-Mental State Examination (MMSE),9 Neurobehavioral Cognitive Status Examination (NCSE),10 and a psychometric test battery that covers key domains.11,12 From this psychometric battery we used the animal fluency test and the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Word List Memory task since these were available for almost all of the study subjects. The CERAD Word List Memory task has three components12: word list memory (involves learning a list of 10 words over three trials; the maximum score is 30); word list delayed recall (involves recalling the words after a 3–5 minute delay; the maximum score is 10); and word list recognition (involves recognizing the 10 words of the Word List Memory task when presented among 10 distractor words; the maximum score is 20). Raw scores not adjusted for education were used.
Clinical assessment data related to general chronic disease burden was assessed with the Cumulative Illness Rating Scale (CIRS).13 Parkinsonism was assessed with a modified Unified Parkinson's Disease Rating Scale (UPDRS) motor subscale.14 Demographic data included socioeconomic status (SES),15 years of education, and the presence of family history of AD. A subgroup of subjects had annual MRI scans and some subjects donated their brains upon death. All subjects had APOE genotyped. For this study, we used the clinical assessments most proximal to death.
Subject selection.
From the autopsy case series of 477 subjects at the OHSU NIA-Layton Aging and AD Center, the subjects meeting the following criteria were included in the current study: having a diagnosis of probable or possible AD based on previously published criteria16 or remaining cognitively intact on last evaluation (cognitively intact was described as having a CDR score = 0); having a last clinical evaluation within 1 year of death; found on autopsy to have high AD pathology burden (high AD pathology was described for this study as having a Braak stage V or VI and moderate or frequent neuritic plaques [NP] based on CERAD criteria17,18); and having antemortem MRI brain volumes and neuropsychological tests.
Following these criteria, 24 AD and 12 cognitively intact subjects met the inclusion criteria.
MRI methods.
MRI scans measuring intracranial, total brain, ventricular, and hippocampal volumes were obtained before death. Scan protocols and analysis methods for MRI volumes have been described previously.19 Briefly, MR images were obtained using a 1.5-Tesla magnet. The protocol consisted of continuous-slice, multiecho, multiplanar image acquisition, with 4-mm-thick coronal slices and a 24-cm2 field of view using a 256 × 256 acquisition matrix with 0.5 excitations. Multiecho coronal sequence with repetition time 3,000 msec, echo time 30 and 80 msec was used to visualize the brain. To orient the coronal plane, T1-weighted sagittal images centered in the midsagittal plane were used.
MR images were analyzed using REGION, a semiautomated analysis program developed by our research team.19 Recursive regression was used to discriminate between different tissue types for ventricular and total brain volumes. Hippocampal volumes were manually traced. Interrater reliability for all regions assessed by intraclass correlation coefficient was ≥0.9. Time from MRI to death was calculated by subtracting age at MRI from age at death. All brain volumes of interest were divided by intracranial volume to adjust for differences in head size.
Neuropathologic methods.
Brains were examined for neurofibrillary tangle (NFT) and NP pathology and staged by Braak and Braak and CERAD systems.17,18 Neuropathologic evaluation of subjects has been described previously.20 Briefly, brains were fixed in neutral-buffered formaldehyde solution for at least 2 weeks and examined grossly as well as microscopically. For microscopic evaluation, tissue samples were taken from all cortical lobes bilaterally or unilaterally, frontal lobe white matter, anterior cingulate gyrus, hippocampus, amygdala, bilateral striatum and thalamus, midbrain, pons, medulla, and cerebellum. Six-micrometer sections were routinely stained with hematoxylin-eosin, Luxol fast blue, Congo red-gallocyanin, and by the modified Bielschowsky silver impregnation method. Selected sections of hippocampus and neocortical regions were immunostained with antibody to tau (tau2, Sigma, St. Louis, MO). Pathologic diagnoses were established using current consensus criteria.21 Information related to NP and NFT burdens, presence of ischemic, hemorrhagic, or vascular pathology, amyloid angiopathy, large vessel strokes, lacunes, presence of Lewy bodies (LB), hippocampal sclerosis, and degree of arteriosclerosis were summarized using the National Alzheimer's Coordinating Center Neuropathology Data Form.22
Statistical analysis.
Statistical analysis was performed by Dr. Erten-Lyons (from the Portland Veterans Affairs Medical Center and OHSU Neurology Department). The following characteristics were compared univariately between the AD and cognitively intact groups: 1) demographic characteristics: gender, age at last evaluation, age at death and at MRI, time from last evaluation to death, years followed, SES, years of education, presence of family history of AD and the APOE ɛ4 allele; 2) clinical characteristics: CIRS, UPDRS, MMSE, CERAD word list task, animal fluency scores; 3) neuropathologic characteristics: Braak NFT and CERAD NP scores, presence of ischemic, hemorrhagic, or vascular pathology, large vessel strokes, lacunes, hippocampal sclerosis, LBs, amyloid angiopathy, and degree of arteriosclerosis; 4) morphometric characteristics: brain weight and intracranial volume.
Univariate analyses were conducted by using χ2 or Fisher exact test for categorical variables and t test for continuous variables.
Multiple regression models were run for the outcomes hippocampal, total brain, and ventricular volume proportions with a group membership (AD vs cognitively intact) as an independent variable, adjusting for age at MRI, time from MRI to death, gender, Braak NFT and CERAD NP scores, and presence of ischemic, hemorrhagic, or vascular pathology. Multiple regression analysis was also performed for the outcome intracranial volume with a group membership as an independent variable, adjusting for gender. Significance was set at p ≤ 0.05. Statistical analysis was conducted using JMP 5.0.1a (SAS Institute, Cary, NC).
RESULTS
Univariate analyses.
Demographic and clinical characteristics.
Age at last evaluation and death, gender distribution, education, SES, time from last evaluation to death, presence of family history of AD, and presence of APOE ɛ4 allele were not different between the groups. The AD group was followed longer and time from last MRI to death was longer in this group (table 1).
Table 1 Selected demographic characteristics

No subjects had PD. UPDRS and CIRS scores were not different between the groups. The cognitively intact group scored higher on all cognitive tests (table 2).
Table 2 Selected clinical characteristics

Neuropsychological testing most proximal to death was not always at the last clinical evaluation before death. All cognitively intact subjects had neuropsychological tests within 1 year of death. For AD subjects, neuropsychological tests were available at a mean of 1.58 (±1.22) years before death (range 0.2–4.8 years).
Neuropathologic and morphometric characteristics.
The AD group had more individuals with Braak stage VI NFTs. The presence of frequent NPs, presence of ischemic, hemorrhagic, or vascular pathology, presence of amyloid angiopathy, and degree of arteriosclerosis were not significantly different between the groups. Seven subjects with AD had coexisting LB pathology, and none of the cognitively intact subjects had LBs. Five AD and two cognitively intact subjects had large vessel strokes; six AD and two cognitively intact subjects had lacunar infarcts. One subject with AD had hippocampal sclerosis (table 3). Brain weight and intracranial volume were not different between the groups.
Table 3 Selected pathologic characteristics

Multiple regression analysis.
In multiple regression analyses after adjusting for gender, age at MRI, time from MRI to death, Braak and CERAD scores, presence of ischemic, hemorrhagic, or vascular pathology, hippocampal and total brain volumes were significantly associated with group membership. These remained significant at p < 0.01 after Bonferroni correction for multiple comparisons. Ventricular volume did not show a significant association with group membership (table 4). Intracranial volume was not associated with group membership after adjusting for gender.
Table 4 Multiple regression models for brain volume proportions
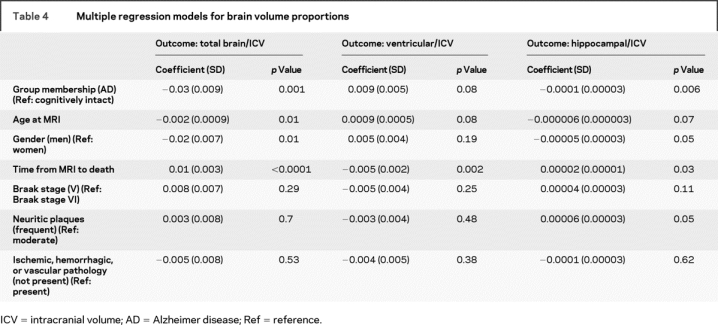
We repeated the above analysis by adding presence of lacunar infarcts and LBs as independent variables. Group membership remained significantly associated with total brain and hippocampal volume proportions, while presence of lacunes or LBs were not significantly associated with either of the brain volumes. The results did not change when excluding the subject with AD with hippocampal sclerosis. We repeated the above analysis in a subgroup of subjects with Braak stage V and frequent NPs (11 AD and 5 cognitively intact). Hippocampal volume remained significantly different between the two groups.
DISCUSSION
Our results suggest that having a larger hippocampal and total brain volume sets cognitively intact individuals with a high burden of AD pathology apart from individuals with overt dementia and a similar amount of AD pathologic changes. This may be interpreted in several ways. First, larger brain volumes may indicate a greater preexisting brain reserve. Second, the cognitively intact group may have other forms of compensation or protection from the pathologic processes that underlie AD-related changes. Third, the brain volume loss in the AD group is not directly caused by the NFTs and NPs. In this case NFTs and NPs are makers of other pathologic processes that actually lead to brain atrophy in AD.
The relationship between preexisting brain reserve and risk of AD has been investigated in several studies. While some studies have shown an association between indirect measures of brain size such as head circumference or intracranial volume and risk of AD, others have failed to show such an association.23-27 In our study, there was no difference in intracranial volume between the two groups. If we assume that intracranial volume is a crude measure of maximum brain size attained during life, then this argues that the larger brain volume we observed does not reflect a preexisting reserve. We also did not observe a difference in education or SES between our cases and controls. Both education and SES have been suggested to be an index of cognitive reserve providing protection against dementia.28
The second interpretation of our results is that there are other physiologic or molecular mechanisms providing protection against the pathologic changes associated with AD. An example of such mechanisms that may promote or protect cognitive function are the number of synapses.29 A study investigated the number of synapses in individuals with AD, mild cognitive impairment, and no cognitive impairment.29 The authors reported that synapse loss was higher in the AD group compared to the mild cognitive impairment and cognitively intact groups, and was a structural correlate of cognitive decline. The authors also investigated an association between total Braak scores, NIA Reagan scores, and synapse numbers, and these did not show an association. Lack of such an association further supports the notion that features such as synapse number may contribute to whether cognitive decline proceeds in the presence of AD pathology. Studying protein expression in the brains of cases and controls like ours with equivalent AD pathology may also improve our understanding of other mechanisms playing a role in AD pathophysiology.30
Apoptotic pathways represent another mechanism that may play a role in resistance to dementia in these individuals. Apoptosis has been suggested to be one of the main causes for the cell loss accompanying neurodegenerative diseases such as AD.31 Brain volume loss in patients with AD may be secondary to activation of apoptotic pathways by the NFT and plaques.32 Thus differences in regulators of apoptosis may lead to resistance to neurodegeneration and associated brain volume loss. For example, the FAS gene, which is a member of the tumor necrosis factor receptor superfamily, plays a role in apoptosis and has been associated with AD.33 It has also been shown to be associated with brain volumes obtained by MRI scans in patients with AD.34 Polymorphisms in genes such as FAS, which play a role in regulation of apoptosis, may mediate the relationship between plaques and tangles and the degree of neurodegeneration, brain volume loss, and presence of symptoms of AD.
Another interpretation of our finding is that the plaques and tangles do not directly cause loss of brain volume observed in patients with AD. Other mechanisms that are closely correlated with plaques and tangles may lead to brain volume loss in patients with AD. Several studies have shown a correlation between postmortem brain volumes and postmortem Braak stage, NFT measures, and neuron numbers.35-37 One study found that postmortem neocortical NFT and NP pathology correlated well with last ventricular volume prior to death and rate of ventricular volume increase in patients with AD while in cognitively intact individuals such a correlation did not exist.37 The authors also reported that the last hippocampal volume prior to death correlated well with hippocampal NFT pathology in patients with AD, while in the cognitively intact subjects the hippocampal NFT pathology did not correlate with antemortem hippocampal volume. Lack of an association between brain volumes and pathology in the cognitively intact subjects may mean that NFTs and NPs are markers of another process in AD, but do not lead to brain volume loss in the absence of these other processes related to AD. However, a correlation between brain volume and AD neuropathology in nondemented individuals has been reported in some other studies.35,36 This may be because the nondemented group in these studies may represent a more heterogeneous group with some having preclinical AD or some mild memory problems.
Our entry criteria and prospective clinical evaluations enhanced the likelihood that subjects in our study do not fall into the preclinical AD group and did not have subtle memory problems. When we compared several psychometric tests between the cognitively intact group with high AD pathology and another group of cognitively intact elderly who were found to have low AD neuropathology (Braak stage I or II and no or sparse NPs by CERAD criteria), we did not find significant differences between these two groups (table e-1 on the Neurology® Web site at www.neurology.org). Similarly, a study using Pittsburgh compound to image amyloid deposition reported no significant differences in cognitive performance between cognitively intact elderly with high vs low amyloid binding.38 Another study suggested that the relationship between postmortem brain volumes and cognitive function is more robust than the relationship between neuropathology and cognitive function.39 This further supports the notion that brain volume seems to play a major role whether cognitive decline occurs in the setting of AD neuropathology.
This study has several limitations. First, matching subjects with high AD pathologic lesion burden and selecting only those with antemortem MRI scans resulted in a restricted sample size. Nevertheless, we observed a statistically significant difference in the brain volumes between AD and cognitively intact subjects even with our relatively small sample size. Our results need replication in a larger sample. Second, since most patients with AD at the final stages of their disease become housebound, subjects in the AD group had a longer time between their MRI and death. This has an effect of minimizing the magnitude of difference in the brain volumes. Ideally one would prefer to have MRI scans within 1 year of death for both groups and we tried to correct for this confounder statistically. Finally, we matched the subjects based on widely used semiquantitative methods to assess the presence of tangles and plaques. Given the possibility of individual variation within the same Braak or CERAD scores, quantitative pathologic assessment methods may be needed in future studies to be able to match cases and controls more precisely.
Our results suggest that individuals with a high burden of AD pathologic lesions do not manifest overt cognitive impairment if they also have larger hippocampal and brain volumes. Identifying the mechanisms whereby larger hippocampal and brain volumes are protective, either by providing more brain reserve or as a result of other processes leading to resistance to neuronal loss traditionally attributed to NFT and NPs, warrants further investigation.
ACKNOWLEDGMENT
The authors thank Robin Guariglia, the volunteers and staff at the Layton Aging and Alzheimer's Disease Center, Dr. Robert Edwards, and the Merle West Center for Medical Research for their contribution to this research.
Supplementary Material
Supplemental data at www.neurology.org
Address correspondence and reprint requests to Dr. Deniz Erten-Lyons, Layton Aging and Alzheimer's Disease Center, 3181 SW Sam Jackson Park Road CR 131, Portland, OR 97239 ertenlyo@ohsu.edu
Supported by Merit Review Grant & Research Career Development Award, Office of Research and Development, Department of Veterans Affairs, National Institute on Aging, National Institutes of Health (AG08017, MO1 RR000334).
Disclosure: The authors report no disclosures.
Received May 30, 2008. Accepted in final form September 29, 2008.
REFERENCES
- 1.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 2.Goldman WP, Price JL, Storandt M, et al. Absence of cognitive impairment or decline in preclinical Alzheimer's disease. Neurology 2001;56:361–367. [DOI] [PubMed] [Google Scholar]
- 3.Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM. Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol 1998;57:1168–1174. [DOI] [PubMed] [Google Scholar]
- 4.Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988;23:138–144. [DOI] [PubMed] [Google Scholar]
- 5.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology 2000;55:370–376. [DOI] [PubMed] [Google Scholar]
- 7.Howieson DB, Holm LA, Kaye JA, Oken BS, Howieson J. Neurologic function in the optimally healthy oldest old: neuropsychological evaluation. Neurology 1993;43:1882–1886. [DOI] [PubMed] [Google Scholar]
- 8.Morris JC. Clinical Dementia Rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 1997;9 suppl 1:173–176; discussion 177–178. [DOI] [PubMed]
- 9.Folstein MF, Robins LN, Helzer JE. The Mini-Mental State Examination. Arch Gen Psychiatry 1983;40:812. [DOI] [PubMed] [Google Scholar]
- 10.Kiernan RJ, Mueller J, Langston JW, Van Dyke C. The Neurobehavioral Cognitive Status Examination: a brief but quantitative approach to cognitive assessment. Ann Intern Med 1987;107:481–485. [DOI] [PubMed] [Google Scholar]
- 11.Hickman SE, Howieson DB, Dame A, Sexton G, Kaye J. Longitudinal analysis of the effects of the aging process on neuropsychological test performance in the healthy young-old and oldest-old. Dev Neuropsychol 2000;17:323–337. [DOI] [PubMed] [Google Scholar]
- 12.Morris JC, Mohs RC, Rogers H, Fillenbaum G, Heyman A. Consortium to Establish a Registry for Alzheimer's Disease (CERAD) clinical and neuropsychological assessment of Alzheimer's disease. Psychopharmacol Bull 1988;24:641–652. [PubMed] [Google Scholar]
- 13.Miller MD, Paradis CF, Houck PR, et al. Rating chronic medical illness burden in geropsychiatric practice and research: application of the Cumulative Illness Rating Scale. Psychiatry Res 1992;41:237–248. [DOI] [PubMed] [Google Scholar]
- 14.Fahn S, Elton R. Unified Parkinson's Disease Rating Scale. In: Fahn S, Marsden CD, Goldstein M, Calne DB, eds. Recent Developments in Parkinson's Disease. Florham Park, NJ: MacMillan Healthcare Information; 1987:153–163. [Google Scholar]
- 15.Hollingshead A. Two-Factor Index of Social Position. New Haven, CT: A.B. Hollingshead; 1957. [Google Scholar]
- 16.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271–278; discussion 278–284. [DOI] [PubMed]
- 18.Mirra SS, Heyman A, McKeel D et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 19.Mueller EA, Moore MM, Kerr DC, et al. Brain volume preserved in healthy elderly through the eleventh decade. Neurology 1998;51:1555–1562. [DOI] [PubMed] [Google Scholar]
- 20.Green MS, Kaye JA, Ball MJ. The Oregon Brain Aging Study: neuropathology accompanying healthy aging in the oldest old. Neurology 2000;54:105–113. [DOI] [PubMed] [Google Scholar]
- 21.Consensus report of the Working Group on Molecular and Biochemical Markers of Alzheimer's Disease: The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Neurobiol Aging 1998;19:109–116. [PubMed] [Google Scholar]
- 22.Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord 2004;18:270–277. [PubMed] [Google Scholar]
- 23.Edland SD, Xu Y, Plevak M, et al. Total intracranial volume: normative values and lack of association with Alzheimer's disease. Neurology 2002;59:272–274. [DOI] [PubMed] [Google Scholar]
- 24.Graves AB, Mortimer JA, Larson EB, Wenzlow A, Bowen JD, McCormick WC. Head circumference as a measure of cognitive reserve. Association with severity of impairment in Alzheimer's disease. Br J Psychiatry 1996;169:86–92. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins R, Fox NC, Rossor AM, Harvey RJ, Rossor MN. Intracranial volume and Alzheimer disease: evidence against the cerebral reserve hypothesis. Arch Neurol 2000;57:220–224. [DOI] [PubMed] [Google Scholar]
- 26.MacLullich AM, Ferguson KJ, Deary IJ, Seckl JR, Starr JM, Wardlaw JM. Intracranial capacity and brain volumes are associated with cognition in healthy elderly men. Neurology 2002;59:169–174. [DOI] [PubMed] [Google Scholar]
- 27.Schofield PW, Logroscino G, Andrews HF, Albert S, Stern Y. An association between head circumference and Alzheimer's disease in a population-based study of aging and dementia. Neurology 1997;49:30–37. [DOI] [PubMed] [Google Scholar]
- 28.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 2002;8:448–460. [PubMed] [Google Scholar]
- 29.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007;68:1501–1508. [DOI] [PubMed] [Google Scholar]
- 30.Montine TJ, Woltjer RL, Pan C, Montine KS, Zhang J. Liquid chromatography with tandem mass spectrometry-based proteomic discovery in aging and Alzheimer's disease. NeuroRx 2006;3:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol 1996;6:493–506. [DOI] [PubMed] [Google Scholar]
- 32.Cotman CW, Anderson AJ. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol 1995;10:19–45. [DOI] [PubMed] [Google Scholar]
- 33.Su JH, Anderson AJ, Cribbs DH, et al. Fas and Fas ligand are associated with neuritic degeneration in the AD brain and participate in beta-amyloid-induced neuronal death. Neurobiol Dis 2003;12:182–193. [DOI] [PubMed] [Google Scholar]
- 34.Erten-Lyons D, Jacobson A, Kramer P, Grupe A, Kaye J. The FAS gene is associated with brain volume in mild cognitive impairment and Alzheimer's disease. Neurology 2008;70(suppl 1):A328. [Google Scholar]
- 35.Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology 2002;58:1476–1482. [DOI] [PubMed] [Google Scholar]
- 36.Huesgen CT, Burger PC, Crain BJ, Johnson GA. In vitro MR microscopy of the hippocampus in Alzheimer's disease. Neurology 1993;43:145–152. [DOI] [PubMed] [Google Scholar]
- 37.Silbert LC, Quinn JF, Moore MM, et al. Changes in premorbid brain volume predict Alzheimer's disease pathology. Neurology 2003;61:487–492. [DOI] [PubMed] [Google Scholar]
- 38.Jack CR Jr, Lowe, VJ Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortimer JA, Gosche KM, Riley KP, Markesbery WR, Snowdon DA. Delayed recall, hippocampal volume and Alzheimer neuropathology: findings from the Nun Study. Neurology 2004;62:428–432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.