

Abstract
Objective:
To characterize sequence variation within the glucocerebrosidase (GBA) gene in a select subset of our sample of patients with familial Parkinson disease (PD) and then to test in our full sample whether these sequence variants increased the risk for PD and were associated with an earlier onset of disease.
Methods:
We performed a comprehensive study of all GBA exons in one patient with PD from each of 96 PD families, selected based on the family-specific lod scores at the GBA locus. Identified GBA variants were subsequently screened in all 1325 PD cases from 566 multiplex PD families and in 359 controls.
Results:
Nine different GBA variants, five previously reported, were identified in 21 of the 96 PD cases sequenced. Screening for these variants in the full sample identified 161 variant carriers (12.2%) in 99 different PD families. An unbiased estimate of the frequency of the five previously reported GBA variants in the familial PD sample was 12.6% and in the control sample was 5.3% (odds ratio 2.6; 95% confidence interval 1.5–4.4). Presence of a GBA variant was associated with an earlier age at onset (p = 0.0001). On average, those patients carrying a GBA variant had onset with PD 6.04 years earlier than those without a GBA variant.
Conclusions:
This study suggests that GBA is a susceptibility gene for familial Parkinson disease (PD) and patients with GBA variants have an earlier age at onset than patients with PD without GBA variants.
GLOSSARY
- CI
= confidence interval;
- GD
= Gaucher disease;
- GDS
= Geriatric Depression Scale;
- MMSE
= Mini-Mental State Examination;
- NCRAD
= National Cell Repository for Alzheimer’s Disease;
- NPL
= nonparametric lod;
- OR
= odds ratio;
- PD
= Parkinson disease;
- UPDRS
= Unified Parkinson’s Disease Rating Scale.
Parkinson disease (PD) is the second most common neurodegenerative disease after Alzheimer disease. Mutations in SNCA, PRKN, DJ1, and PINK1 typically result in early onset PD1,2 while mutations in LRRK2 result in idiopathic PD with more typical, later onset.3,4 These mutations result in disease in fewer than 5% of patients with PD.
Gaucher disease (GD) is an inherited deficiency of lysosomal glucocerebrosidase arising from mutations in the gene encoding glucosidase beta acid (GBA), more commonly known as glucocerebrosidase.5–7 Over 200 different mutations have been identified. GD is most common in the Ashkenazi Jewish population. While patients with GD presenting with parkinsonian symptoms were reported as early as 1939, only recently has it been hypothesized that a deficiency of glucocerebrosidase might contribute to an increased susceptibility to parkinsonism.8,9 In a recent study, GBA variants were found in 21% of subjects with PD, a much higher estimate than would be expected based on the carrier frequency of GD in the general population.10 In addition, GBA variants were more frequent among younger patients.
Subsequent screenings of patients with PD has yielded contradictory results regarding the association of GBA in PD.10–23 In one study of Ashkenazi Jewish patients with PD, GBA variants were more frequently found in patients as compared with controls.11 A large study of both Jewish and non-Jewish samples found an association between GBA mutations and PD in the Jewish group only.15 Chinese patients with PD from Singapore demonstrated a significant association with GBA,21 whereas a similar study of Chinese patients from Taiwan did not.20 A survey of Italian patients with PD for the N370S and L444P mutations found a significant association of these variants with PD,22 while a Norwegian sample showed comparable frequencies of these two mutations in patients with PD and controls.17 Most recently, a study of Portuguese patients with PD detected a significant increase in GBA variants in patients as compared to controls.23 In some studies, patients with PD harboring GBA variants had earlier age at disease onset.12,15,19,21 Whether the discrepant results regarding the association of GBA with disease and age at onset result from true ethnic differences in GBA variant frequencies or from differences in the scope of the studies (i.e., only screening for certain variants as opposed to sequencing the entire coding region) remains to be determined.
Of the studies previously reported, none has examined the relationship of GBA to PD susceptibility in a largely familial cohort.10–23 The goals of this study were to characterize sequence variation within GBA in a select subset of our large sample of patients with familial PD and then to test in the entire sample whether these sequence variants increased the risk for PD or were associated with an earlier onset of disease.
METHODS
Subjects.
A total of 1,325 individuals with PD from 566 multiplex families were ascertained through a pair of siblings, both of whom were reported to have PD (PROGENI study). At the time of these analyses, 1325 individuals with PD from 566 multiplex PD families had been recruited. All available affected individuals were seen by a movement disorder specialist at one of 59 Parkinson Study Group sites located throughout North America (table 1). Each participant completed a uniform clinical assessment that included the Unified Parkinson’s Disease Rating Scale (UPDRS) Parts II (Activities of Daily Living) and III (Motor Exam),24,25 Schwab & England score,26 Hoehn & Yahr stage,27 the Mini-Mental State Examination (MMSE),28 the Geriatric Depression Scale (GDS),29 and the Blessed Functional Activity Scale (Blessed).30 In addition, a diagnostic checklist was used to classify individuals as having either verified PD (65%) or nonverified PD (35%).31 Peripheral blood was obtained after completion of appropriate written informed consent approved by each individual institution’s institutional review board.
Table 1 Patients with Parkinson disease (PD) and control sample demographics

Microsatellite markers closest to the GBA locus (D1S252, D1S498, D1S484, D1S2878) genotyped as part of a previous 10 cM genome screen31–33 were used to calculate a family-specific nonparametric lod (NPL) score and rank families based on their evidence of linkage to the GBA region. One affected individual from each of the 96 families with the highest NPL scores was selected for GBA sequencing.
The control sample consisted of 359 neurologically normal non-Hispanic Caucasians who provided appropriate written informed consent (see table 1). The control samples were obtained from three different sources: the National Cell Repository for Alzheimer’s Disease (NCRAD), the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center at the Coriell Cell Repositories (Camden, NJ; DNA panels NDPT002, NDPT006, NDPT009) and controls recruited as part of an ongoing PD study at Indiana University (PROGENI-CARES).34
Molecular genetic analysis.
PCR and sequencing primers were designed using the chromosome 1 genomic contig sequence NT_029419 enabling PCR/sequencing of all 11 coding exons and corresponding intron/exon boundaries of GBA (table e-1 on the Neurology® Web site at www.neurology.org). Primers were designed enabling preferential amplification of GBA over the GBA pseudogene also on chromosome 1. PCR products were purified and sequenced as previously described.34
TaqMan allelic-discrimination assays (Applied Biosystems, Foster City, CA) were developed to screen all 1,325 PD cases and 359 controls for the variants identified in the 96 sequenced samples (except L444P, A456P, V460V) as previously described.34,35 To screen for the L444P variant, exon 11 amplification products were digested to completion with HpaII (New England Biolabs, Beverly, MA) to assay for the L444P variant (gain of HpaII site). Digestion products were electrophoresed through 4% Metaphor Agarose (Cambrex, Rockland, ME). To screen for the RecNciI recombinant allele carrying variants A456P and V460V, PCR primers were synthesized as described previously.13 The gene-specific forward primer (5′-ggaaccatgattccctatcttc-3′) and the GBA pseudogene-specific reverse primer (5′-gtttaggacgaccacaacagg-3′) were used in a multiplex PCR reaction with an invariant primer set. The PCR products were electrophoresed through 2% agarose (Invitrogen, Carlsbad, CA) for detection of the RecNciI recombinant allele PCR fragment. Presence of the RecNciI recombinant allele was confirmed using long range PCR and sequencing of the entire GBA gene. Briefly, 200 ng of genomic DNA was amplified using the Invitrogen Elongase Enzyme Mix (Invitrogen, Carlsbad, CA) and primers 5′-cccattctccatgcaaatctgtgt-3′ (forward) and 5′-ccggaaccagatcctatctgtgc-3′ (reverse). Long range PCR products were purified and sequenced as above.
Statistical analysis.
Statistical analyses were limited to the subset of the PD sample that met the strictest diagnostic criteria of verified PD.31 This analytic sample consisted of 737 non-Hispanic, Caucasian individuals from 450 families (see table 1) and excluded those patients known to harbor a causative PD mutation (a single LRRK2 mutation or 2 PRKN mutations).
Two hypotheses were tested. The first was that presence of a GBA variant increased the risk of PD. A logistic model was employed with affection status as the dependent variable and presence or absence of a GBA variant as the independent variable (0 or 1). Age at examination and gender were included as covariates in the initial model; however, neither affected the magnitude or significance of the odds ratios (ORs) and were dropped from the final model. The second hypothesis was that those inheriting a GBA variant have earlier age at PD onset. A linear regression model was fitted with age at onset as the dependent variable and the presence or absence of a GBA variant as the independent variable. Education, gender, and smoking were considered as possible covariates; however, all were found to be nonsignificant and were dropped from the final model. Linear and logistic regression models were also used to test whether other measures relevant to PD (i.e., UPDRS subscores, MMSE, GDS, Hoehn & Yahr stage) differed based on the presence or absence of a GBA variant.
All analyses were carried out using SAS software (release 9.13; SAS Institute, Cary, NC). Our analytic sample consisted of families with multiple patients with PD. To ensure an unbiased analysis of the study hypotheses when using a sample of related individuals, we employed resampling techniques. Specifically, a single individual was sampled at random from each of the families. This was repeated 50,000 times, and common resampling techniques (bootstrapping) were employed to obtain a representative value. The median bootstrapped statistic was determined and the corresponding p values are reported for the tests of the two hypotheses.
RESULTS
Nine variants were identified by sequencing the entire coding region of GBA in 96 patients with PD (table 2). Four of the detected sequence variants were novel while the remaining five had been previously identified in patients with PD. The four novel variants were each found in additional affected family members of the PD subject in whom the variant was initially found. However, screening of all available PD cases did not identify the four novel variants in any additional families. Three of the variants (IVS6 589-2A>G, R262H, IVS10 1389-3C>G) were not identified in any of the control samples, making the estimated frequency of each <0.002 in the neurologically normal population. The frequency of the remaining novel variant, K303K, was not evaluated in the control samples as the designed TaqMan allelic discrimination assay failed. The remaining five variants had been previously reported in patients with PD and GD, as well as controls.8,10–15,17,19–21 An unbiased estimate (using resampling techniques as described in Methods) of the frequency of the five previously reported GBA variants in the subset of the familial PD sample that met our strictest diagnostic criteria of verified PD was 12.6% and in the control sample was 5.3 (table 3). The mean age at onset of the patients with PD harboring a GBA variant was 56.8 years (median: 58, range: 30–79).
Table 2GBA variants identified by sequencing in 96 familial patients with Parkinson disease (PD) and number of families identified to carry each by screening the full sample

Table 3 Odds ratios and proportions of cases and controls with a previously identified GBA variant permuted one per family

The presence of a previously described GBA variant significantly increased the risk for PD; 12.6% of verified PD cases carried a GBA mutation (permuted one per family as described in Methods), as compared with 5.3% of controls (OR 2.6, 95% confidence interval [CI] 1.5–4.4) (table 3). The analysis of individual variants showed nonsignificant ORs ranging from 1.7 (95% C.I. 0.4–6.8) for the N370S variant to 2.8 (95% C.I. 0.3–25.8) for the RecNciI recombinant allele variant.
Presence of a GBA variant was also associated with an earlier age at onset (p = 0.0001) (table 4). PD cases carrying a GBA variant were more likely to have onset ≤50 years as compared to those without a GBA variant (p = 0.0004). Among those with early onset disease, there was no difference in age at onset between the GBA variant carriers and the noncarriers; however, among those with onset >50 years, those with a GBA variant had earlier age at onset (61.59 years) as compared to late-onset cases without a GBA variant (65.37 years) (p = 0.001).
Table 4 Comparison of clinical features of GBA variant carriers and noncarriers with a diagnosis of verified Parkinson disease
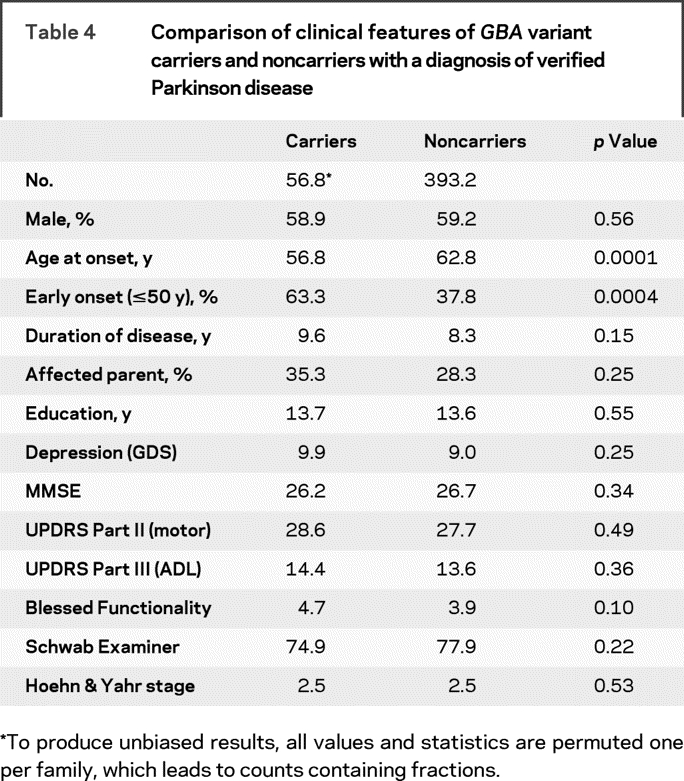
When a linear regression model was fitted to predict age at onset, presence of a GBA variant was associated with an earlier age at onset (p = 0.0001). On average, our model suggests that those patients carrying a GBA variant had onset with PD 6.04 years earlier than those without a GBA variant. The cumulative incidence of PD was higher in patients with GBA variants compared with noncarriers over nearly the entire age distribution (figure).
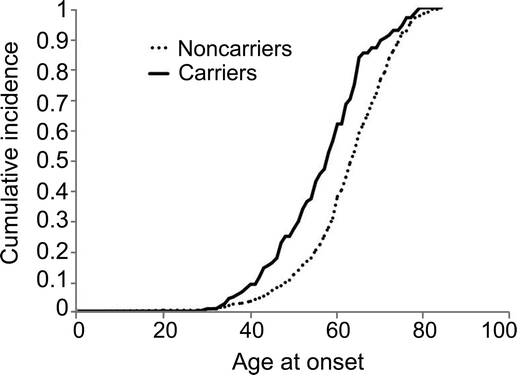
Figure Cumulative incidence rates of Parkinson disease among carriers and noncarriers of GBA variants
We also compared the clinical characteristics of the PD cases that carried a GBA variant with the PD cases that did not carry a GBA variant. There were no significant differences between GBA variant carriers and noncarriers for any of the clinical characteristics examined, including MMSE, UPDRS Parts II and III, Blessed, and Hoehn & Yahr (table 3).
DISCUSSION
The goals of this study were to characterize sequence variation within GBA in a select subset of our larger sample of familial patients with PD and to test in the larger sample whether these sequence variants increased the risk for PD and were associated with an earlier onset of disease. We identified four novel variants and five previously reported variants. In our sample, a subject was 2.6 times more likely to develop PD if they carried one of the five previously identified GBA variants (table 3). Furthermore, presence of a GBA variant was associated with an earlier onset of disease. Our results replicate the association of GBA with PD reported in other studies10–23; however, we extend the association to include later onset patients with PD as well as familial disease.
Our screening for these nine variants in 1,325 affected individuals from 566 families represents the largest study to date of GBA variants in patients with PD and PD families. In total, GBA variants were detected in 161 patients (12.2%) from 99 different PD families (17.5%). The frequency observed in our sample is intermediate with that in a recent study that reported 16.9% of Jewish PD cases carried a GBA variant, while only 8% of non-Jewish PD cases carried a variant.15 While the exact percentage of our sample which is of Jewish ancestry is not known, it is likely less than 10%.
As we did not sequence any of our control samples, our statistical analyses included only the five previously published GBA variants to avoid any ascertainment bias. While the analysis for these five variants combined yielded an OR of 2.6 (95% CI 1.5–4.4), individual ORs for each of the variants were not significant (table 3). For the N370S variant in our patients, the OR of 1.7 (95% CI 0.4–6.8) was less than the 5.6 (95% CI 1.3–24.3) in a recent report15; however, the CIs do overlap. This difference is likely due to the large number of Jewish PD cases (179/278) included in their study and the high frequency of the N370S variant in that population, in general. No other study has reported ORs for individual GBA variants as we have in this study. Since the variant observed most often in our verified patients with PD, E326K (table 3), has been described as a “mild” mutation or modifier allele,34,35 the analyses were also performed without those patients carrying this variant. Even when those patients are removed, the frequency of GBA variants in the cases remains greater than in the controls (p = 0.002) suggesting that the GBA effect is not solely due to E326K. In our study, a single patient not meeting our strictest diagnostic criteria of verified PD was shown to be homozygous for the E326K variant.
We have also demonstrated that those patients with PD carrying a GBA variant had onset with PD 6 years earlier than patients without a GBA variant. Among the patients with later onset disease (after age 50), those with a GBA variant had a significantly earlier age at onset (61.6 years) as compared to those without a GBA variant (65.4 years). However, the earlier age at onset in GBA variant carriers was not observed when limiting the analysis to those patients having earlier onset (≤ age 50). These results differ from those of a recent study that reported the presence of a GBA variant decreased age at onset by nearly 2 years among patients with early onset PD.15 Analysis of the later onset PD cases in this same study did not detect a significantly earlier age at onset in carriers of a GBA variant. It is unclear whether this results from a difference in the ethnicity of two sample populations studied or any bias in collection of the samples. When those with the E326K variant are removed from the analyses, patients with PD with a GBA variant continue to have earlier age at onset compared to those without a variant (p = 0.008).
Four novel variants were each identified in a single family. None of these variants have been reported in either patients with PD or GD, and none was identified in 359 normal control subjects. Two of the novel variants were intronic (IVS6 589-2A>G and IVS10 1389-3C>G), and their close proximity to the 3′ end of the intron (either −2 or −3 position) suggests that either could alter the splice donor site resulting in altered splicing of the GBA mRNA. One synonymous (K303K) and one nonsynonymous (R262H) variant were each identified in a single family. As the synonymous variant does not alter the amino acid sequence of glucocerebrosidase, it is not predicted to be pathogenic for either PD or GD. However, the possibility that the nucleotide substitution results in creation of either a cryptic splice donor or acceptor site cannot be ruled out. Any disease susceptibility attributable to the R262H variant could not be determined in this study. Thus, while none of these four GBA variants have been previously identified, each either alters or could potentially alter the glucocerebrosidase sequence and may contribute to disease pathogenesis.
In all, GBA variants were identified in 99 of 566 (17.5%) families. Discordance for GBA variant carrier status among affected individuals (both verified and nonverified PD) within the 99 families was common. Of the 99 GBA variant-carrying families, 63 demonstrated discordance for inheritance of the GBA variant among affected individuals. Among the discordant families, we compared age at onset between those PD cases who carried a GBA variant and those cases who did not. Those with a pathogenic variant (N370S, L444P, RecNciI) had a earlier age at onset (p = 0.01) than those without a variant (56.4 years vs 66.5 years). Interestingly, among the discordant families, carriers of a polymorphic variant (either E326K or T369M) also had lower age at onset than those without a variant (57.1 years vs 61.4 years; p = 0.03).
Our study is unique in that 96 unrelated familial patients with PD were selected for sequencing of the glucocerebrosidase gene based on their lod score in the GBA chromosomal region that exceeded 0.60. Previous reports focused primarily on sporadic or idiopathic PD except for that of Sato et al., for which 51% of patients with PD reported a positive family history.13 Nine GBA variants were identified in 21 of the 96 patients, making the yield for this lod score strategy 21.9%. The nine GBA variants were only identified in 16.5% of the remaining 470 families. Thus, while linkage methods have very limited specificity for identifying causative mutations when a majority of the families in a study contain only a single affected sibling pair, the apparent increase in sensitivity illustrates that this is a helpful strategy for prioritizing which individuals to sequence in the evaluation of potential PD susceptibility genes.
ACKNOWLEDGMENT
The authors thank the subjects for their participation in this research study.
APPENDIX
The following are members of the PROGENI Steering Committee: University of Tennessee Health Science Center: R.F. Pfeiffer; University of Rochester: F. Marshall, D. Oakes, A. Rudolph, A. Shinaman; Columbia University Medical Center: K. Marder; Indiana University School of Medicine: P.M. Conneally, T. Foroud, C. Halter; University of Kansas Medical Center: K. Lyons; Eli Lilly & Company: E. Siemers; Medical College of Ohio: L. Elmers; University of California, Irvine: N. Hermanowicz.
The following are Parkinson Study Group Investigators and Coordinators: Albany Medical College: S. Factor, D. Higgins, S. Evans; Barrow Neurological Institute: H. Shill, M. Stacy, J. Danielson, L. Marlor, K. Williamson; Baylor College of Medicine: J. Jankovic, C. Hunter; Beth Israel Deaconess Medical Center: D. Simon, P. Ryan, L. Scollins; Beth Israel Medical Center: R. Saunders-Pullman, K. Boyar, C. Costan-Toth, E. Ohmann; Brigham & Women’s Hospital: L. Sudarsky, C. Joubert; Brown University (Memorial Hospital of RI): J. Friedman, K. Chou, H. Fernandez, M. Lannon; Cleveland Clinic Florida-Weston: N. Galvez-Jimenez, A. Podichetty; Clinical Neuroscience Center: P. Lewitt, M. DeAngelis; Colorado Neurological Institute: C. O’Brien, L. Seeberger, C. Dingmann, D. Judd; Columbia University Medical Center: K. Marder, J. Fraser, J. Harris; Creighton University: J. Bertoni, C. Peterson; Evanston Northwestern Healthcare: M. Rezak, G. Medalle; Hotel-Dieu Hospital-Chum: S. Chouinard, M. Panisset, J. Hall, H. Poiffaut; Hunter Homes Mcguire Veterans Medical Center: V. Calabrese, P. Roberge; Indiana University School of Medicine: J. Wojcieszek, J. Belden, C. Halter; Institute for Neurodegenerative Disorders: D. Jennings, K. Marek, S. Mendick; Johns Hopkins University: S. Reich, B. Dunlop; London Health Sciences Centre: M. Jog, C. Horn; LSU Medical Center: J. Rao, M. Cook; Mayo Clinic Jacksonville: R. Uitti, M. Turk; Mcfarland Neurosciences: T. Ajax, J. Mannetter; McGill Centre for Studies in Aging: M. Panisset, J. Hall; Medical College of Georgia: K. Sethi, J. Carpenter, B. Dill, K. Ligon, S. Narayan, L. Woodward; Medical College of Wisconsin: K. Blindauer, K. Abou-Samra, J. Petit; Medical University of Ohio: L. Elmer, E. Aiken, K. Davis, C. Schell, S. Wilson; Mount Sinai School of Medicine New York: M. Velickovic, W. Koller (deceased), S. Phipps; North Shore-Long Island Jewish Health System: A. Feigin, M. Gordon, J. Hamann, E. Licari, M. Marotta-Kollarus, B. Shannon, R. Winnick; Northwestern University: T. Simuni, A. Kaczmarek, K. Williams, M. Wolff; Ochsner Clinic Foundation: J. Rao, M. Cook; Ohio State University: M. Fernandez, J. Hubble, S. Kostyk, A. Campbell, C. Reider, A. Seward; Oregon Health & Science University: J. Nutt, R. Camicioli, J. Carter, P. Andrews, S. Morehouse, C. Stone; Ottawa Hospital Civic Site: T. Mendis, C. Alcorn-Costa, D. Grimes, P. Gray, K. Haas, J. Vendette; Pacific Neuroscience Medical Group: J. Sutton, B. Hutchinson, J. Young; Saskatoon Dist Health Board Royal University Hospital: A. Rajput, A. Rajput, L. Klassen, T. Shirley; Scott & White Hospital/Texas A&M University: B. Manyam, P. Simpson, J. Whetteckey, B. Wulbrecht; The Parkinson’s & Movement Disorder Institute: D. Truong, M. Pathak, N. Luong, T. Tra, A. Tran, J. Vo; Toronto Western Hospital, University Health: A. Lang, L. Johnston, G. Kleiner-Fisman, A. Nieves, J. So; UMDNJ-School of Osteopathic Medicine: G. Podskalny, L. Giffin; University of Alabama at Birmingham: P. Atchison, C. Allen; University of Alberta: W. Martin, M. Wieler; University of Calgary: O. Suchowersky, M. Klimek; University of California Irvine: N. Hermanowicz, S. Niswonger; University of California San Diego: C. Shults (deceased), D. Fontaine; University of California San Francisco: M. Aminoff, C. Christine, M. Diminno, J. Hevezi; University of Chicago: A. Dalvi, U. Kang, J. Richman, S. Uy, J. Young; University of Cincinnati: A. Dalvi, M. Gartner, A. Sahay, D. Schwieterman, B. Wolthoff; University of Colorado Health Sciences Center: D. Hall, M. Leehey, S. Culver, T. Derian; University of Connecticut: T. Demarcaida, S. Belber; University of Iowa: R. Rodnitzky, J. Dobson; University of Kansas Medical Center: R. Pahwa, K. Lyons, T. Gales, S. Thomas; University of Maryland School of Medicine: L. Shulman, S. Reich, W. Weiner, K. Dustin; University of Miami: C. Singer, W. Koller, K. Lyons, W. Weiner, L. Zelaya; University of Minnesota: P. Tuite, V. Hagen, J. Kosowicz, S. Rolandelli, R. Schacherer; University of New Mexico: P. Gordon, J. Werner; University of Puerto Rico School of Medicine: C. Serrano, S. Roque; University of Rochester: R. Kurlan, D. Berry, I. Gardiner; University of South Florida: R. Hauser, J. Sanchez-Ramos, T. Zesiewicz, H. Delgado, K. Price, P. Rodriguez, S. Wolfrath; University of Tennessee Health Science Center: L. Davis, B. Pfeiffer; University of Texas Southwestern Medical Center: R. Dewey, B. Estes, B. Hayward, A. Johnson, M. Meacham; Wake Forest University School of Medicine: F. Walker, V. Hunt, C. O’neill; Washington University: B. Racette, L. Good, M. Rundle.
Biostatistics and Clinical Trials Coordination Centers Staff included A. Watts, A. Wang, T. Ross, S. Bennett, D. Kamp, E. Julian-Baros, S. Daigneault, and R. Doolan.
Supplementary Material
Address correspondence and reprint requests to Dr. William C. Nichols, Associate Professor of Pediatrics, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229 bill.nichols@cchmc.org
Supplemental data at www.neurology.org
e-Pub ahead of print on November 5, 2008, at www.neurology.org.
*The Parkinson Study Group–PROGENI Investigators are listed in the appendix.
Supported by R01 NS37167, MO1 RR-00750, and the National Cell Repository for Alzheimer’s Disease (U24 AG021886). This study used samples and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds).
Disclosure: The authors report no disclosures.
Received February 26, 2008. Accepted in final form July 2, 2008.
REFERENCES
- 1.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006;7:306–318. [DOI] [PubMed] [Google Scholar]
- 2.Gasser T. Genetics of Parkinson’s disease. Curr Opin Neurol 2005;18:363–369. [DOI] [PubMed] [Google Scholar]
- 3.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607. [DOI] [PubMed] [Google Scholar]
- 4.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44:595–600. [DOI] [PubMed] [Google Scholar]
- 5.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 2004;83:6–15. [DOI] [PubMed] [Google Scholar]
- 6.Butters TD. Gaucher disease. Curr Opin Chem Biol 2007;11:412–418. [DOI] [PubMed] [Google Scholar]
- 7.Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004;65:77–86. [DOI] [PubMed] [Google Scholar]
- 8.Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 2003;79:104–109. [DOI] [PubMed] [Google Scholar]
- 9.Tayebi N, Callahan M, Madike V, et al. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab 2001;73:313–321. [DOI] [PubMed] [Google Scholar]
- 10.Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004;81:70–73. [DOI] [PubMed] [Google Scholar]
- 11.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2004;351:1972–1977. [DOI] [PubMed] [Google Scholar]
- 12.Eblan MJ, Nguyen J, Ziegler SG, et al. Glucocerebrosidase mutations are also found in subjects with early-onset parkinsonism from Venezuela. Mov Disord 2006;21:282–283. [DOI] [PubMed] [Google Scholar]
- 13.Sato C, Morgan A, Lang AE, et al. Analysis of the glucocerebrosidase gene in Parkinson’s disease. Mov Disord 2005;20:367–370. [DOI] [PubMed] [Google Scholar]
- 14.Clark LN, Nicolai A, Afridi S, et al. Pilot association study of the beta-glucocerebrosidase N370S allele and Parkinson’s disease in subjects of Jewish ethnicity. Mov Disord 2005;20:100–103. [DOI] [PubMed] [Google Scholar]
- 15.Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007;69:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goker-Alpan O, Giasson BI, Eblan MJ, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006;67:908–910. [DOI] [PubMed] [Google Scholar]
- 17.Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ. Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 2006;66:415–417. [DOI] [PubMed] [Google Scholar]
- 18.Eblan MJ, Walker JM, Sidransky E. The glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2005;352:728–731. [DOI] [PubMed] [Google Scholar]
- 19.Tan EK, Tong J, Fook-Chong S, et al. Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 2007;64:1056–1058. [DOI] [PubMed] [Google Scholar]
- 20.Ziegler SG, Eblan MJ, Gutti U, et al. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol Genet Metab 2007;91:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu YR, Chen CM, Chao CY, et al. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry 2007;78:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Marco EV, Annesi G, Tarantino P, et al. Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov Disord 2008;23:460–463. [DOI] [PubMed] [Google Scholar]
- 23.Bras J, Paisan-Ruiz C, Guerreiro R, et al. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging EPub 2007 Dec 19. [DOI] [PMC free article] [PubMed]
- 24.Fahn S, Elton R, Committee UD. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden C, Goldstein M, eds. Recent Developments in Parkinson’s Disease. Florham Park, NY: Macmillan Healthcare Information, 1987:153–163. [Google Scholar]
- 25.Lang AE, Fahn S. Assessment of Parkinson’s disease. In: Munsat T, ed. Quantification of Neurologic Deficit. Boston: Butterworth, 1989:285–309. [Google Scholar]
- 26.Schwab R, England A. Projection technique for evaluating surgery in Parkinson’s disease. In: Gillingham F, Donaldson I, eds. Third Symposium on Parkinson’s Disease. Edinburgh: E&S Livingstone, 1969:152–157. [Google Scholar]
- 27.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 28.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 29.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 1982;17:37–49. [DOI] [PubMed] [Google Scholar]
- 30.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 1968;114:797–811. [DOI] [PubMed] [Google Scholar]
- 31.Pankratz N, Nichols WC, Uniacke SK, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet 2002;71:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pankratz N, Nichols WC, Uniacke SK, et al. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet 2003;12:2599–2608. [DOI] [PubMed] [Google Scholar]
- 33.Pankratz N, Uniacke SK, Halter CA, et al. Genes influencing Parkinson disease onset: replication of PARK3 and identification of novel loci. Neurology 2004;62:1616–1618. [DOI] [PubMed] [Google Scholar]
- 34.Nichols WC, Elsaesser VE, Pankratz N, et al. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology 2007;69:1737–1744. [DOI] [PubMed] [Google Scholar]
- 35.Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005;365:410–412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.