

Abstract
Background:
At cross-section, cognitively normal individuals (NL) with a maternal history of late-onset Alzheimer disease (AD) have reduced glucose metabolism (CMRglc) on FDG-PET in the same brain regions as patients with clinical AD as compared to those with a paternal and a negative family history (FH) of AD. This longitudinal FDG-PET study examines whether CMRglc reductions in NL subjects with a maternal history of AD are progressive.
Methods:
Seventy-five 50- to 82-year-old NL received 2-year follow-up clinical, neuropsychological, and FDG-PET examinations. These included 37 subjects with negative family history of AD (FH−), 9 with paternal (FHp), and 20 with maternal AD (FHm). Two subjects had parents with postmortem confirmed AD. Statistical parametric mapping was used to compare CMRglc across FH groups at baseline, follow-up, and longitudinally.
Results:
At both time points, the FH groups were comparable for demographic and neuropsychological characteristics. At baseline and at follow-up, FHm subjects showed CMRglc reductions in the parieto-temporal, posterior cingulate, and medial temporal cortices as compared to FH− and FHp (p < 0.001). Longitudinally, FHm had significant CMRglc declines in these regions, which were significantly greater than those in FH− and FHp (p < 0.05).
Conclusions:
A maternal history of Alzheimer disease (AD) predisposes normal individuals to progressive CMRglc reductions in AD-vulnerable brain regions, which may be related to a higher risk for developing AD.
GLOSSARY
- AD
= Alzheimer disease;
- ApoE-4
= apolipoprotein E-4 genotype;
- CMRglc
= cerebral metabolic rate for glucose;
- FDG-PET
= 2-[18F]fluoro-2-deoxy-d-glucose positron emission tomography;
- FH
= family history;
- FH−
= negative family history of AD;
- FHm
= maternal history of AD;
- FHp
= paternal history of AD;
- FWHM
= full-width at half maximum;
- GDS
= Global Deterioration Scale;
- GLM
= General Linear Model;
- MCI
= mild cognitive impairment;
- MMSE
= Mini-Mental State Examination;
- MNI
= McGill Neurologic Institute;
- mtDNA
= mitochondrial DNA;
- NL
= normal individuals;
- PCC
= posterior cingulate cortex;
- PHG
= parahippocampal gyrus;
- SMC
= subjective memory complaints.
Alzheimer disease (AD) is an age-dependent neurodegenerative disorder associated with progressive loss of cognitive function. After advanced age, having a first-degree family history of late-onset AD, especially when a parent is affected, is the major risk factor for developing AD among cognitively normal (NL) subjects.1 While the rare familial forms of early onset AD follow conventional patterns of autosomal dominant Mendelian inheritance, the vast majority of AD cases appear late in life, without clear nuclear genetic associations. However, first-degree relatives of affected probands are at 4- to 10-fold higher risk for AD as compared to individuals with no family history.2–4 The apolipoprotein E (ApoE) epsilon 4 genotype is an established genetic risk factor for late-onset AD, but it is found in less than 40% of AD cases,1 indicating that other factors contribute to the etiology and expression of disease. Although there is mixed evidence for parent of origin effects in late-onset AD families,5,6 epidemiology data indicate that having an AD-affected mother confers greater risk than having an AD-affected father,5 is associated with poorer cognitive performance in late life,7 and a more predictable age at dementia onset in the offspring.8 The biologic mechanisms through which a parental history of AD confers increased risk for developing dementia are unknown.
Our recent 2-[18F]fluoro-2-deoxy-d-glucose positron emission tomography (FDG-PET) study showed that NL individuals with a maternal history of AD had marked reductions in the cerebral metabolic rate for glucose (CMRglc) as compared to subjects with a paternal history and without a family history of AD.9 NL subjects with an AD-affected mother showed reduced CMRglc in the parieto-temporal, posterior cingulate, and medial temporal cortices, which are typically hypometabolic in patients with clinical AD.9 FDG-PET studies in AD have shown that CMRglc reductions in these regions occur years prior to symptoms onset10–13 and correlate with clinical progression.13,14
This 2-year longitudinal FDG-PET study examines whether CMRglc reductions in NL individuals with a maternal history of AD are progressive.
METHODS
Subjects.
We retrospectively examined 75 clinically and cognitively NL individuals who completed a 2-year follow-up FDG-PET examination and thorough family history (FH) evaluations. Subjects were recruited at NYU School of Medicine to participate as volunteers for longitudinal FDG-PET studies, including individuals interested in research participation and risk consultation; self-referred individuals with cognitive complaints; and spouses, family members, and caregivers of patients participating in other studies. Informed consent was obtained from all subjects. The study was approved by the NYU and Brookhaven National Laboratory (BNL, Upton, NY) IRB.
At baseline and follow-up, subjects received a standard diagnostic evaluation that included medical, psychiatric, neuropsychological, clinical MRI, and FDG-PET examinations within 2 months. Individuals with medical conditions or history of conditions that may affect brain structure or function, i.e., stroke, diabetes, head trauma, any neurodegenerative diseases, depression, hydrocephalus, intracranial mass, and infarcts on MRI, and use of psychoactive medications were excluded. All subjects had normal fasting blood glucose levels, blood pressure, cholesterol levels, and Modified Hachinski Ischemia Scale scores <4.15 ApoE genotype was determined using standard PCR procedures.
Subjects were 50–82 years of age at baseline, had education ≥12 years, Global Deterioration Scale (GDS) scores ≤2,16 and Mini-Mental State Examination ≥28. All subjects had normal cognitive test performance relative to normative values on the immediate and delayed recall of a paragraph and of paired associates, the digit-symbol substitution, designs, object naming, and WAIS-vocabulary tests.13 A FH of dementia that included at least one first-degree relative with dementia onset between 65 and 80 years was elicited by using the NYU Brain Aging Family History questionnaire (appendix e-1 on the Neurology® Web site at www.neurology.org). Participants were asked to fill in information of affected family members, which was confirmed by other family members in the interview with the examining neurologist. Subjects were not included if their parents had not lived to the age at risk of late-onset AD (i.e., 65 years). Subjects with maternal (FHm; i.e., only the mother was affected with AD), paternal (FHp; i.e., only the father was affected with AD), and negative FH of AD (FH−) were included in the study. The AD-affected parents of two subjects (one FHm and one FHp) received an autopsy and the postmortem diagnosis of AD.17
Brain imaging.
Subjects received a standardized whole-brain MRI scan protocol on a 1.5 T Signa imager (General Electric, Milwaukee, MI), including a contiguous 3 mm axial T2-weighted and a T1-weighted fast-gradient-echo image (25 cm field of view [FOV], number of excitations = 1, 256 × 128 matrix, 35 msec relaxation time, 9 msec excitation time, 1.2 mm sections, and 60º flip angle). These scans were used to rule out MRI evidence of hydrocephalus, intracranial mass, cortical strokes, subcortical gray matter lacunes, and moderate to severe white matter disease.18
All subjects received a PET scan at BNL on an ECAT 931 scanner (Siemens, Knoxville, TN; 6.2 mm full-width at half maximum [FWHM]; 6.75 mm slice thickness, 10 cm axial FOV). Subjects received 5–8 mCi of FDG IV while lying supine in a dimly lit room. PET images were obtained 35 minutes after injection over 20 minutes. Scans were acquired as two interleaved 15-slice PET volumes that overlapped by a half-slice thickness (∼3.4 mm) over two 10-minute frames.19 Arterial blood samples were drawn at standard intervals throughout the study and absolute CMRglc (μmol/100 g/min) was calculated using Sokoloff model with standard kinetic constants.20 Data were reconstructed using filtered back-projection (Fourier rebinning/2D back-projection, Hanning filter with a frequency cutoff of 0.5 cycles/pixel) and corrected for attenuation, scatter, and radioactive decay, yielding 128 × 128 matrix with a pixel size of 1.56 mm.
Image analysis.
FDG-PET scans were processed using Statistical Parametric Mapping (SPM2, Wellcome Department of Cognitive Neurology, London, UK).21 Scans were realigned, spatially normalized to an elderly brain template22 in the McGill Neurologic Institute (MNI) space, which approximates the Talairach and Tournoux space,23 by estimating the least squares 12-parameter affine transformation followed by an iterative estimate of local alignment, and smoothed with a 12 mm FWHM gaussian filter.21 Only voxels with values greater than 80% of the whole brain CMRglc were included in the analysis and only clusters exceeding an extent threshold of 30 voxels were considered significant. Anatomic location of brain regions showing significant effects was described using the Talairach and Tournoux coordinates using Talairach Daemon 12.0 (http://ric.uthscsa.edu/projects/talairachdaemon.html), after coordinates conversion to the Talairach space.23 CMRglc measures were extracted from the clusters of voxels showing significant effects using the Marsbar tool (http://www.mrc-cbu.cam.ac.uk/Imaging/marsbar.html) to be examined in further analyses.
Statistical analysis.
Analyses were done with SPSS 12.0 (SPSS Inc., Chicago, IL) and SPM2. Differences in demographic and neuropsychological measures between the study groups were examined with χ2 tests, Fisher exact test, and the General Linear Model (GLM) with post hoc LSD tests, as appropriate. For all analyses, results were considered significant at p < 0.05.
The GLM/univariate analysis with post hoc t tests was used to test for CMRglc differences across FH groups at baseline and at the 2-year follow-up. The GLM/repeated measures analysis with post hoc t tests was used to test for differential effects across groups over time, and to examine longitudinal CMRglc changes within each FH group. All analyses were performed using absolute CMRglc values and controlling for the individual’s global CMRglc, as done in previous FDG-PET study with similar subject groups to highlight regional differences.9,24 Prior to analysis, we confirmed that there were no cross-sectional (F[2,63] < 0.6, p > 0.55) or longitudinal (F[2,63] = 1.26, p = 0.20) differences in global CMRglc across groups (baseline: FH− = 33.1 ± 6.5, FHp = 34.2 ± 6.3, FHm = 33.7 ± 6.7 μmol/100 g/min; follow-up: FH− = 37.7 ± 7.4, FHp = 32.0 ± 8.5, FHm = 30.8 ± 5.2 μmol/100 g/min). Since we previously identified the brain regions showing CMRglc differences across FH groups,9 results were considered significant at p < 0.001, uncorrected for multiple comparisons.
Results were re-examined controlling for other potential risk factors for late-onset AD, such as age, female gender, education, ApoE-4 genotype, and presence of subjective memory complaints.
In addition, due to the small sample of FHp, we created three groups of nine subjects each, matched for age, gender, education, and ApoE genotype, and re-examined CMRglc for group effects using the GLM with post hoc LSD tests, as well as nonparametric Mann-Whitney rank sum tests (α = 0.05, exact significance, one-tailed).
Linear regressions were used to estimate the number of years prior to baseline when statistical differentiation across groups was possible. This was done for the brain regions showing baseline and longitudinal group effects by estimating the mean CMRglc for each group every 0.5 years prior to baseline, using the baseline CMRglc and the annual rate of CMRglc decline in each group.13 At each time point, CMRglc was compared between groups using independent-sample t tests at p < 0.05 (one-sided).
The spatially normalized FDG-PET scans of the two subjects with parents with pathology-verified AD were processed using NEUROSTAT25 to highlight CMRglc abnormalities in each subject as compared to an FDG-PET database of healthy controls.26 Results were examined at p ≤ 0.01 (one-sided) and three-dimensional stereotactic surface projections of the Z scores generated to visualize CMRglc deviations from controls.25,26
RESULTS
Of the 75 baseline subjects, 66 were examined in this study including 37 FH−, 9 FHp, and 20 FHm subjects. Of the remaining nine subjects, one had both parents affected, two had only siblings affected, three had only second-degree relatives affected, and three had a family history of an unspecified dementia and were conservatively excluded.
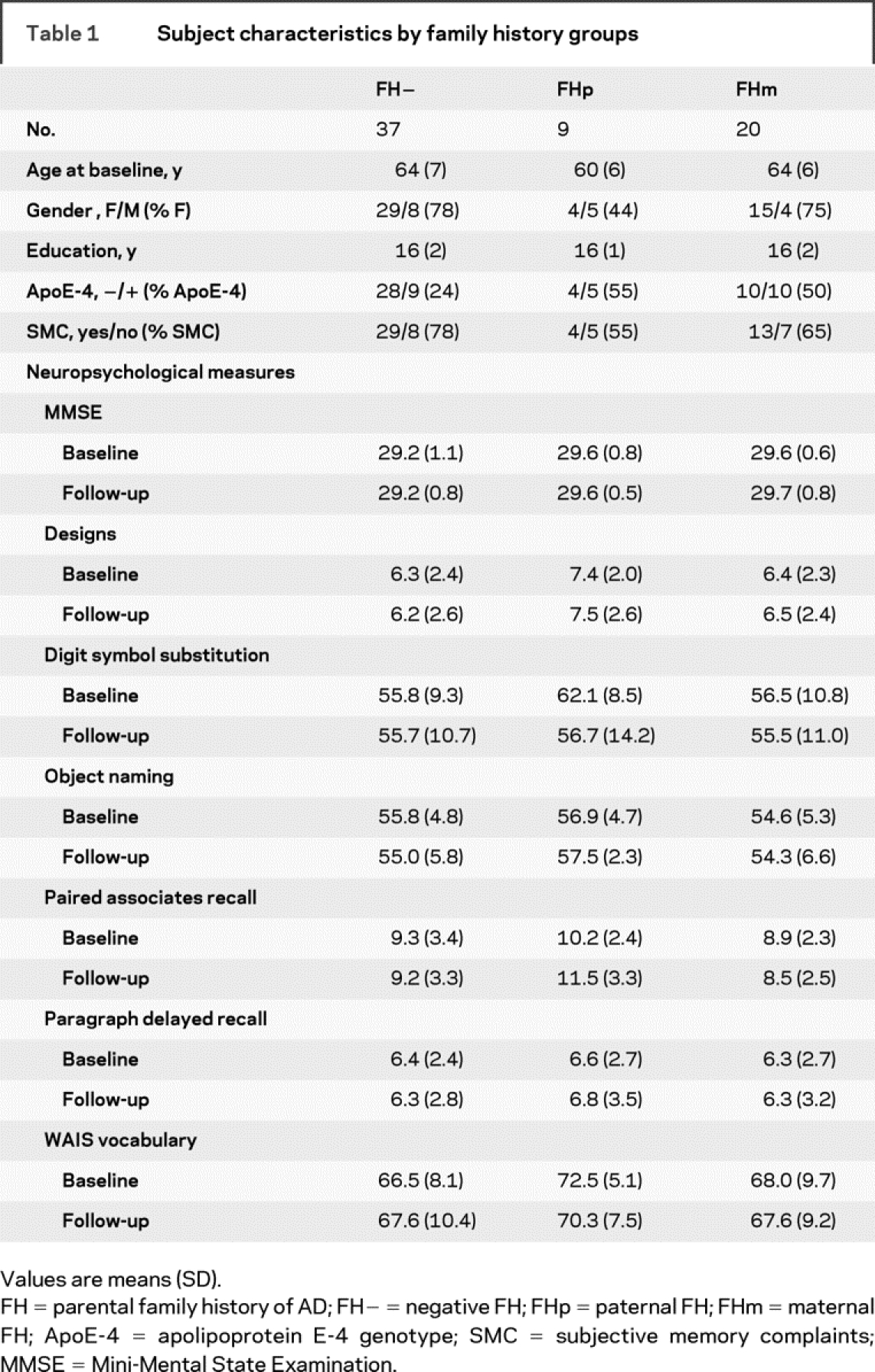
There were no significant differences between FH groups for age, education, and prevalence of subjective memory complaints (SMC) (table 1). The prevalence of females was lower in FHp (44%) as compared to FH− and FHm (78% and 75%; χ2(2) = 8.3, p = 0.01). The prevalence of ApoE-4 carriers was slightly lower in FH− (24%) as compared to FHp and FHm (55% and 50%; χ2(2) = 5.3, p = 0.07). There were no differences in neuropsychological scores across groups at baseline, follow-up, or longitudinally (table 1).
Table 1 Subject characteristics by family history groups
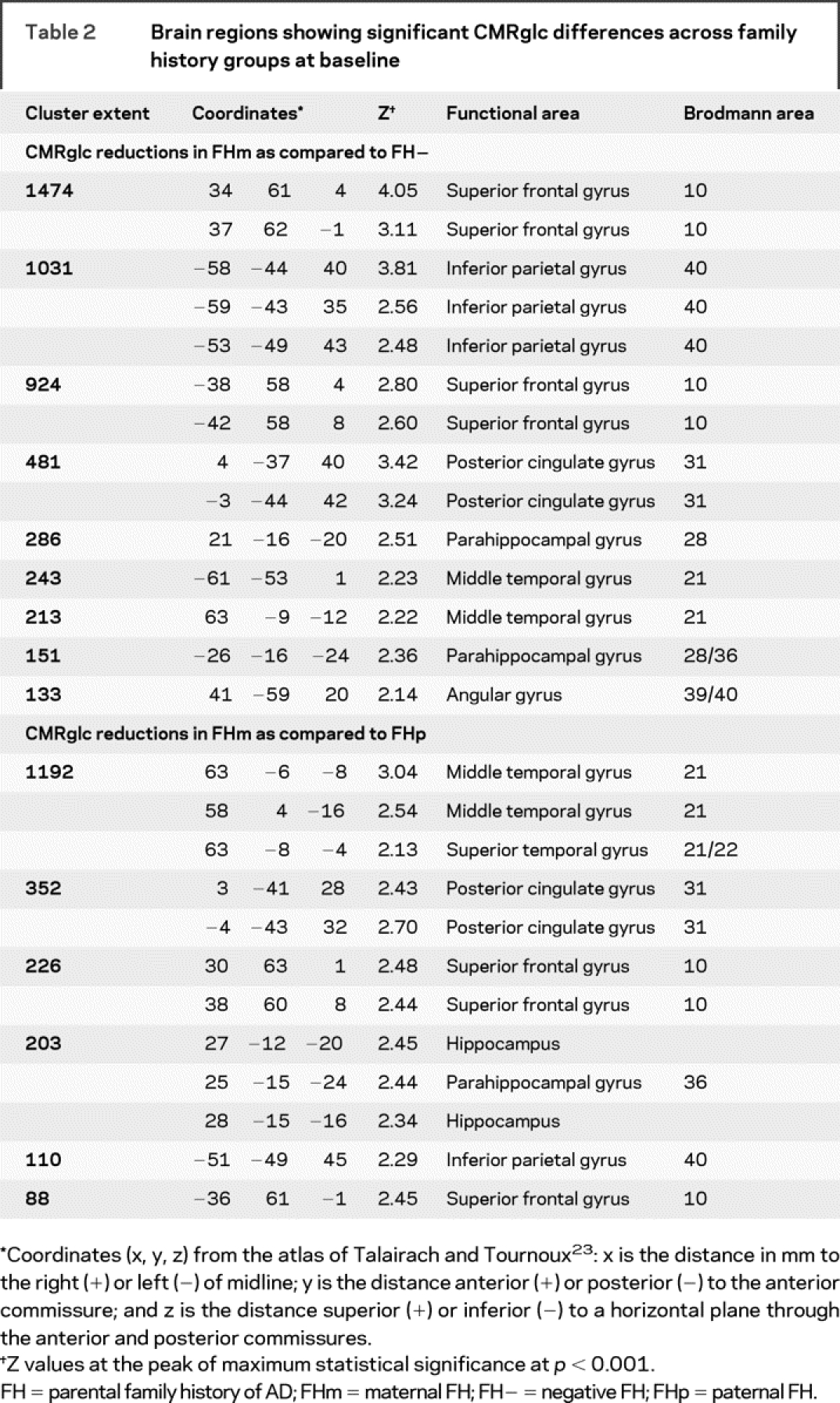
At baseline, FHm subjects showed CMRglc reductions as compared to FH− and FHp (figure 1). As compared to FH−, FHm subjects showed CMRglc reductions in the parietal, temporal, posterior cingulate (PCC), and frontal cortices, and parahippocampal gyrus (PHG), bilaterally (p < 0.001, table 2). As compared to FHp, FHm showed reduced CMRglc in the PCC and frontal cortices, bilaterally, in the right temporal cortex, PHG, and hippocampus, and in the left parietal cortex (p < 0.001, table 2). These results remained significant after accounting for age, gender, education, ApoE genotype, and SMC. There were no differences between FH− and FHp groups.

Figure 1 Maternal history of Alzheimer disease (FHm) subjects as compared to negative family history (FH−) (in blue) and paternal history (FHp) (in red) groups, and to both FH− and FHp (in purple)
Anatomic location and description of brain regions are found in table 2. Areas of hypometabolism are displayed onto the superior, anterior, and inferior views of a volume-rendered spatially normalized MRI.
Table 2 Brain regions showing significant CMRglc differences across family history groups at baseline
At the 2-year follow-up, as compared to FH− and FHp groups, FHm subjects showed CMRglc reductions in the same brain regions as at baseline (p < 0.001). The clusters of hypometabolism were slightly more extended with respect to the baseline results, but the topography of CMRglc reductions remained unchanged (data not shown). As with the baseline, there were no differences between FH− and FHp groups.
At both time points, there were no regions showing higher CMRglc in FHm as compared to FH− and FHp.
Longitudinal group by time interaction effects were detected in the PCC/precuneus, inferior parietal, and superior temporal cortex, bilaterally (p < 0.001). Post hoc comparisons showed that the longitudinal effects in the PCC and parieto-temporal cortices were driven by FHm subjects, who showed CMRglc reductions at follow-up as compared to baseline, whereas FH− and FHp groups did not show CMRglc reductions in these regions over time (p < 0.001, table 3). In FHm subjects, CMRglc within AD-related regions (i.e., the average of the parieto-temporal and PCC CMRglc) was reduced 13% at baseline and 23% at follow-up (p < 0.001), and the CMRglc reductions over time were more severe (F[2,63] = 17.9; p < 0.001) as compared to FH− and FHp (figure 2). The mean annual CMRglc decline in AD regions in FHm (−3.14 ± 1.3 μmol/100 g/min) was greater than that observed in FH− (−0.75 ± 1.5 μmol/100 g/min) and in FHp (−0.56 ± 0.91 μmol/100 g/min) (p < 0.001). This corresponded to higher annual rates of CMRglc reductions in AD regions in FHm (−5.8%) than in FHp and FH− (−0.9% and −1.4%, p < 0.05).
Table 3 Brain regions showing significant longitudinal CMRglc effects within FH groups
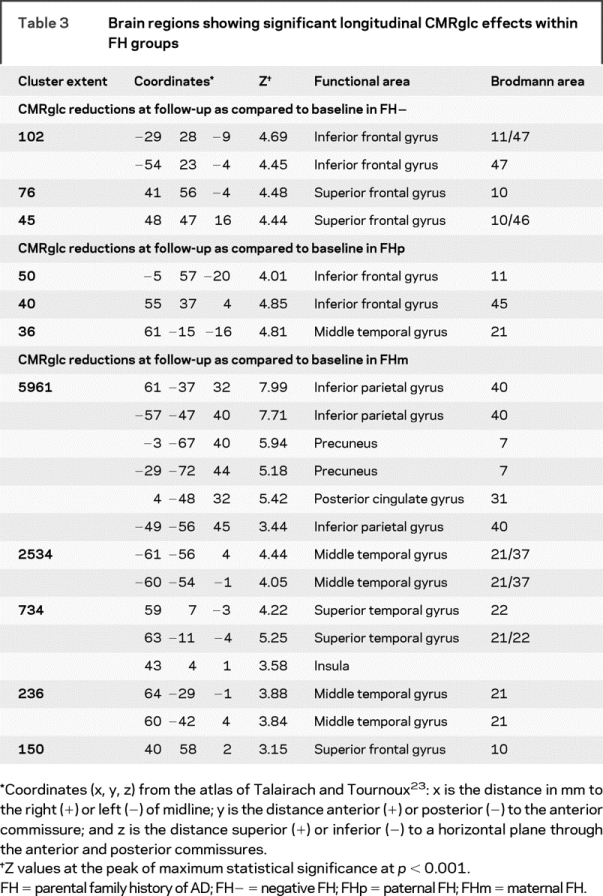
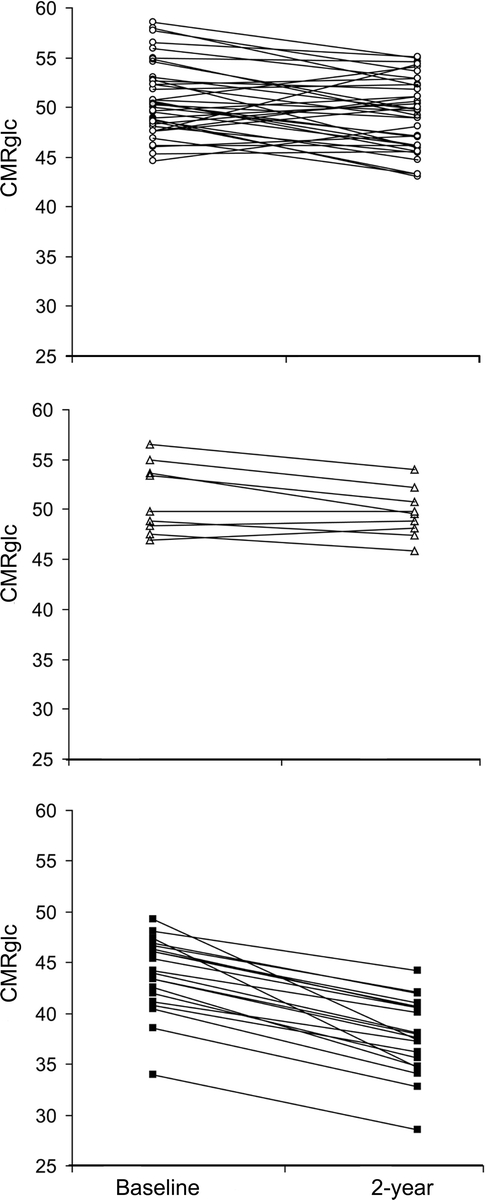
Figure 2 Longitudinal CMRglc changes in Alzheimer disease (AD)-vulnerable brain regions (parieto-temporal and posterior cingulate cortex) in negative family history (FH−) (top, white circles), paternal history (FHp) (middle, white triangles), and maternal history (FHm) subjects (bottom, black squares)
CMRglc are μmol/100 g/min.
Additionally, all FH groups showed modest longitudinal CMRglc reductions in the frontal cortex (p < 0.001, table 3), but there were no interaction effects in this region. There were no regions showing CMRglc increases over time in any FH group.
Analysis of three demographically and size matched FH groups confirmed the above CMRglc effects in AD brain regions. As compared to FH−, the FHm group showed CMRglc reduced 17% at baseline CMRglc and 25% at follow-up (Mann Whitney p = 0.001 and p = 0.002, respectively), as well as greater CMRglc decreases over time (FHm: −2.8 ± 1.1 μmol/100 g/min per year vs FH−: −0.54 ± 2.2 μmol/100 g/min per year, Mann Whitney p = 0.005). This corresponded to higher annual rates of CMRglc reductions in AD regions in FHm (−5.0% per year) than in FH− (−1.1% per year, p < 0.05).
Assuming a linear progression of CMRglc reductions and constant SEM, CMRglc reductions in FHm were estimated to reach significance 2 ± 0.5 years before baseline as compared to FH−, and 1.5 ± 0.5 years before baseline as compared to FHp (figure e-1).
Examination of the FDG-PET scans of the offspring of patients with postmortem verified AD showed that, as compared to controls, a 68-year-old NL son of an AD-affected father did not show hypometabolism, whereas a 58-year-old NL daughter of an AD-affected mother showed significant CMRglc reductions in the PCC and medial temporal cortex, bilaterally (Z scores range: 2.3–4), and to a lesser extent in the parieto-temporal cortex (p < 0.01, figure e-2).
DISCUSSION
We previously reported that, in cross-section, NL FHm individuals show CMRglc reductions in the same brain regions as patients with clinical AD as compared to FHp and FH−. The present longitudinal FDG-PET study replicates previous findings in a larger cohort, and shows that regional CMRglc continued to decline in FHm individuals during the 2-year follow-up. The decline in CMRglc preceded any evidence of cognitive deterioration and was significantly greater than in FH− and FHp subjects. These effects remained significant after accounting for potential risk factors for late-onset AD such as age, female gender, education, ApoE-4 genotype, and SMC.
Longitudinally, all FH groups showed CMRglc declines in the frontal regions, in agreement with previous age-related FDG-PET findings in NL elderly.12,13,27 However, the FHm subjects uniquely showed CMRglc declines also in the same AD-vulnerable brain regions that were hypometabolic at baseline, the parieto-temporal and PCC cortices, in which the rates of CMRglc decline were significantly higher in FHm as compared to the other groups. Regression analyses showed that CMRglc reductions in AD regions in FHm originated a few years prior to the baseline PET, suggesting that hypometabolism in FHm subjects may be a late-life phenomenon. Other studies with longer follow-ups and younger individuals are needed to replicate these findings.
Examination of two persons with parents with pathologically confirmed AD revealed that while the FHp subject did not show CMRglc abnormalities, the FHm subject showed a pattern of hypometabolism involving the PCC and medial temporal cortices, and to a lesser extent, the parieto-temporal regions as compared to controls. A similar pattern is consistently found in patients with mild cognitive impairment (MCI), a condition that places patients at very high risk for developing AD.14,22,28
Hypometabolism in these brain regions is known to precede the onset of cognitive symptoms in AD by many years,10,12,13 and to correlate with disease progression in NL elderly12,13 and patients with MCI14 declining to AD. Our FHm subjects showed a similar regional pattern of longitudinal CMRglc deficits, suggesting that progressive hypometabolism may be one of the biologic mechanisms that confer increased vulnerability to AD. Continued follow-up examination of our subjects and replication studies are necessary to determine whether the observed CMRglc reductions are predictive of AD.
The causes of the early CMRglc abnormalities in FHm subjects are not known. Although Mendelian inheritance is not evident in late-onset AD, the fact that children of affected individuals have an increased risk of developing the disease suggests a genetic component.1 Moreover, the fact that only children of affected mothers show CMRglc reductions consistent with AD suggests maternally inherited predisposition to brain “energetic” failure.
FDG-PET studies of NL ApoE-4 carriers have shown longitudinal CMRglc reductions similar to those observed in our FHm subjects.24 However, our results remained significant after controlling for ApoE genotype, which indicates that other factors contribute to the CMRglc abnormalities observed in FHm. With all that is known about the molecular processes involved in glucose metabolism, hypometabolism in FHm may be due to a combination of defective mitochondrial function, increased oxidative stress, and possible mitochondrial DNA (mtDNA) mutations, leading to CMRglc alterations in brain tissue.29 The fact that mtDNA is entirely maternally inherited in humans, and diseases associated with mtDNA mutations often present as sporadic disorders,29 lends support to this hypothesis. mtDNA abnormalities in AD correlate with increased reactive oxygen species production, mitochondrial respiratory enzymes defects, decreased ATP production, and enhanced amyloid-beta toxicity.30,31 A deficient energy metabolism could change the oxidative microenvironment for neurons during the pathogenesis of AD, rendering synapses more vulnerable to degeneration.29 Oxidative stress is strongly associated with neuronal loss in AD, which mainly affects the medial temporal, PCC, and parieto-temporal cortices.32,33 These regions were progressively hypometabolic in our FHm subjects.
Despite showing CMRglc reductions, FHm subjects did not show cognitive deficits, indicating that they may be compensating for advancing brain damage. Although no CMRglc increases were observed in FHm, resting-state FDG-PET is not the ideal tool to probe functional compensatory mechanisms, which may be better detected by techniques such as fMRI, using specific challenges. Hypometabolism on FDG-PET is known to precede cognitive deficits in NL elderly,12,13 suggesting that the metabolic declines in FHm may be related to a pathologic process. Our results are consistent with epidemiologic observations that a maternal history of AD negatively influences AD risk5,7,8 and offer a biologic substrate that may account for the clinical findings. Confirmation of this hypothesis awaits further longitudinal studies examining clinical decline in these subjects.
Our determination of parental AD in the absence of neuropathologic confirmation is vulnerable to error. We relied on a consensus diagnostic conference to review FH medical records, diagnoses were based on established clinical diagnostic criteria for AD,34,35 and FH questionnaires are known to have good agreement with neuropathologic findings.36 Nonetheless, our affected FH cohort may have included subjects whose parents did not have AD but another dementia. This would lead to erroneous inclusion of subjects in FH groups, with the potential effect of conservatively reducing the power to detect group differences.
Supplementary Material
Address correspondence and reprint requests to Dr. Lisa Mosconi, Department of Psychiatry, NYU School of Medicine, 550 First Avenue, New York, NY 10016 lisa.mosconi@med.nyu.edu; or Dr. Mony de Leon, Department of Psychiatry, NYU School of Medicine, 560 First Avenue, New York, NY 10016 mony.deleon@med.nyu.edu
Supplemental data at www.neurology.org
Editorial, page 486
e-Pub ahead of print on November 12, 2008, at www.neurology.org.
Supported by NIH-NIA AG13616, AG12101, AG08051, AG022374, NIH-NCRR MO1RR0096, and the Alzheimer’s Association.
Disclosure: The authors report no disclosures.
Received April 30, 2008. Accepted in final form July 24, 2008.
REFERENCES
- 1.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 2.Cupples LA, Farrer LA, Sadovnik AD, Relkin N, Whitehouse P, Green P. Estimating risk curves for first-degree relatives of patients with Alzheimer’s disease: the REVEAL study. Genet Med 2004;6:192–196. [DOI] [PubMed] [Google Scholar]
- 3.Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002;287:329–336. [DOI] [PubMed] [Google Scholar]
- 4.Silverman JM, Ciresi G, Smith CJ, Marin DB, Schnaider-Beeri M. Variability of familial risk of Alzheimer disease across the late life span. Arch Gen Psychiatry 2005;62:565–573. [DOI] [PubMed] [Google Scholar]
- 5.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology 1996;47:254–256. [DOI] [PubMed] [Google Scholar]
- 6.Ehrenkrantz D, Silverman JM, Smith CJ, et al. Genetic epidemiological study of maternal and paternal transmission of Alzheimer’s disease. Am J Med Genet 1999;88:378–382. [DOI] [PubMed] [Google Scholar]
- 7.Au R, Seshadri S, Wolf PA, et al. New norms for a new generation: cognitive performance in the Framingham offspring cohort. Exp Aging Res 2004;30:333–358. [DOI] [PubMed] [Google Scholar]
- 8.Gomez-Tortosa E, Barquero MS, Baron M, et al. Variability of age at onset in siblings with familial Alzheimer disease. Arch Neurol 2007;64:1743–1748. [DOI] [PubMed] [Google Scholar]
- 9.Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci USA 2007;104:19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosconi L, Sorbi S, de Leon MJ, et al. Hypometabolism exceeds atrophy in presymptomatic early-onset Familial Alzheimer’s disease. J Nucl Med 2006;47:1778–1786. [PubMed] [Google Scholar]
- 11.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol 1997;42:85–94. [DOI] [PubMed] [Google Scholar]
- 12.de Leon MJ, Convit A, Wolf OT, et al. Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-D-glucose/positron-emission tomography (FDG/PET). Proc Natl Acad Sci USA 2001;98:10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosconi L, De Santi S, Li J, et al. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging 2008;29:676–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drzezga A, Lautenschlager N, Siebner H, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging 2003;30:1104–1113. [DOI] [PubMed] [Google Scholar]
- 15.Hachinski VC, Lassen NA, Marshall J. Multi-infarct dementia, a cause of mental deterioration in the elderly. Lancet 1974;2:207–210. [DOI] [PubMed] [Google Scholar]
- 16.Reisberg B, Ferris SH, de Leon MJ, Crook T. The global deterioration scale for assessment of primary degenerative dementia. Am J Psychiatry 1982;139:1136–1139. [DOI] [PubMed] [Google Scholar]
- 17.The National Institute on Aging, the Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging 1997;18:S1–S2. [PubMed] [Google Scholar]
- 18.George AE, de Leon MJ, Kalnin A, Rosner L, Goodgold A, Chase N. Leukoencephalopathy in normal and pathologic aging: 2. MRI and brain lucencies. Am J Neuroradiol 1986;7:567–570. [PMC free article] [PubMed] [Google Scholar]
- 19.Bendriem B, Dewey SL, Schlyer DJ, Wolf AP, Volkow ND. Quantitation of the human basal ganglia with positron emission tomography: a phantom study of the effect of contrast and axial positioning. IEEE Trans Nucl Sci 1991;10:216–222. [DOI] [PubMed] [Google Scholar]
- 20.Reivich M, Alavi A, Wolf A, et al. Glucose metabolic rate kinetic model parameter determination in humans: The lumped constants and rate constants for [18F]fluorodeoxyglucose and [11C]deoxyglucose. J Cereb Blood Flow Metab 1985;5:179–192. [DOI] [PubMed] [Google Scholar]
- 21.Friston KJ, Frith CD, Liddle PF, Frackowiak RSJ. Comparing functional (PET) images: the assessment of significant change. J Cereb Blood Flow Metab 1991;11:690–699. [DOI] [PubMed] [Google Scholar]
- 22.Mosconi L, Tsui WH, De Santi S, et al. Reduced hippocampal metabolism in mild cognitive impairment and Alzheimer’s disease: automated FDG-PET image analysis. Neurol 2005;64:1860–1867. [DOI] [PubMed] [Google Scholar]
- 23.Talairach J, Tournoux P. Co-Planar Stereotaxic Atlas of the Human Brain. Stuttgart: Thieme; 1988. [Google Scholar]
- 24.Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci USA 2001;98:3334–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of Fluorine-18-FDG PET. J Nucl Med 1995;36:1238–1248. [PubMed] [Google Scholar]
- 26.Mosconi L, Tsui WH, Pupi A, et al. (18)F-FDG PET database of longitudinally confirmed healthy elderly improves detection of MCI and AD. J Nucl Med 2007;48:1129–1134. [DOI] [PubMed] [Google Scholar]
- 27.Herholz K, Salmon E, Perani D, et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage 2002;17:302–316. [DOI] [PubMed] [Google Scholar]
- 28.Nestor PJ, Fryer TD, Smielewski P, Hodges JR. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann Neurol 2003;54:343–351. [DOI] [PubMed] [Google Scholar]
- 29.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–795. [DOI] [PubMed] [Google Scholar]
- 30.Trimmer PA, Swerdlow RH, Parks JK, et al. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 2000;162:37–50. [DOI] [PubMed] [Google Scholar]
- 31.Swerdlow RH, Parks JK, Cassarino DS, et al. Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurol 1997;49:918–925. [DOI] [PubMed] [Google Scholar]
- 32.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 2001;21:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valla J, Berndt JD, Gonzales-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J Neurosci 2001;21:4923–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 35.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition). 4 ed. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 36.Kawas C, Segal J, Stewart WF, Corrada M, Thal LJ. A validation study of the Dementia Questionnaire. Arch Neurol 1994;51:901–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.