

Abstract
Acrolein is a ubiquitous component of environmental pollutants such as automobile exhaust, cigarette, wood, and coal smoke. It is also a natural constituent of several foods and is generated endogenously during inflammation or oxidation of unsaturated lipids. Because increased inflammation and episodic exposure to acrolein-rich pollutants such as traffic emissions or cigarette smoke have been linked to acute myocardial infarction, we examined the effects of acrolein on matrix metalloproteinases (MMPs), which destabilize atherosclerotic plaques. Our studies show that exposure to acrolein resulted in the secretion of MMP-9 from differentiated THP-1 macrophages. Acrolein-treatment of macrophages also led to an increase in reactive oxygen species (ROS), free intracellular calcium ([Ca2+]i), and xanthine oxidase (XO) activity. ROS production was prevented by allopurinol, but not by rotenone or apocynin and by buffering changes in [Ca2+]I with BAPTA-AM. The increase in MMP production was abolished by pre-treatment with the antioxidants Tiron and N-acetyl cysteine (NAC) or with the xanthine oxidase inhibitors allopurinol or oxypurinol. Finally, MMP activity was significantly stimulated in aortic sections from apoE-null mice containing advanced atherosclerotic lesions after exposure to acrolein ex vivo. These observations suggest that acrolein exposure results in MMP secretion from macrophages via a mechanism that involves an increase in [Ca2+]I, leading to xanthine oxidase activation and an increase in ROS production. ROS-dependent activation of MMPs by acrolein could destabilize atherosclerotic lesions during brief episodes of inflammation or pollutant exposure.
Keywords: matrix metalloproteinase, reactive oxygen species, acrolein, atherosclerosis, plaque destabilization, apoE-null mouse
INTRODUCTION
Atherosclerotic disease leading to myocardial infarction (MI) and stroke is the leading cause of morbidity and mortality in the Western world (Rosamond et al., 2008). It is currently believed that atherosclerotic lesions develop and grow as a result of vascular inflammation that leads to monocyte infiltration into the vessel wall. Once lodged in the sub-intimal space, the monocytes differentiate into macrophages which take up modified lipoproteins and as a result are transformed into foam cells (Glass et al., 2001; Reiss et al., 2006). These processes lead to the formation of distinct lesions (plaque) which are composed of foam cells, a lipid-rich-core and a fibrous, matrix-rich cap. Gradual erosion of this cap or its acute rupture exposes platelets to the underlying matrix proteins and pro-thrombotic molecules, activating them and initiating a thrombotic response. Vascular occlusion at these sites initiates ischemic episodes associated with myocardial and cerebral infarction.
Although physiological events contributing to plaque erosion and rupture are complex, matrix metalloproteinases (MMPs) have been suggested to be play an important role (Galis et al., 1994; Galis et al., 1995; Herman et al., 2001; Sukhova et al., 1999). MMP-mediated degradation of the extracellular matrix (ECM) is vital for several physiological functions including development, morphogenesis, angiogenesis, and tissue repair. Several pathological conditions, such as arthritis, cancer, nephritis, chronic ulcers, and fibrosis (Nagase et al., 2006) are, however, associated with excessive or unregulated MMP activity. MMP dysregulation is also a characteristic feature of cardiovascular abnormalities and increased levels of MMPs have indeed been found in atherosclerotic plaques (Galis et al., 1994; Galis et al., 1995; Halpert et al., 1996; Rajavashisth et al., 1999) and in patients with unstable angina or acute myocardial infarction (Kai et al., 1998; Tziakas et al., 2004).
Acrolein is an aldehydic compound that has been linked in epidemiological studies to cardiovascular pathology (Bhatnagar 2004; Feron et al., 1991). It is a particularly abundant component of air-borne particulate matter (PM) that arises during the burning of fossil fuels, cigarettes, or other organic material. In addition, acrolein is generated during the cooking or frying of food, is present in the effluent of industrial waste, and exists naturally in vegetables, fruits, and herbs (Feron et al., 1991). Acrolein is also an end product of the metabolism of certain pharmaceuticals (Ludeman 1999) and can likewise be produced by myeloperoxidase-catalyzed oxidation. It is therefore generated in high amounts at sites of inflammation (Anderson et al., 1997) or lipoprotein oxidation (Anderson et al., 1997; Burke et al., 2001; Feron et al., 1991; Steinberg 1997), particularly within vascular lesions (Shao et al., 2005)). The toxicity of acrolein is a consequence of its strongly reactive, electrophilic, carbonyl group which can react with cellular nucleophiles such as thiols or amines (Esterbauer et al., 1991). Thus acrolein can form adducts with proteins, disrupting cellular signaling or function, or nucleic acids, eliciting mutagenic or carcinogenic effects. In addition, the toxic effects of acrolein can result from indirect means. In particular, chronic acrolein exposure could deplete cellular antioxidants such as glutathione, rendering the cell prone to damage from free radicals. Cardiovascular tissue seems to be particularly sensitive to the toxic effects of acrolein. Epidemiological and animal studies have linked acrolein exposure to arrhythmia (Bhatnagar 1995b), hypertension (Feron et al., 1991), atherogenesis (Steinbrecher et al., 1990), dyslipidemia (McCall et al., 1995), and myocardial infarction (Alfredsson et al., 1993; Levine et al., 1984; Stewart et al., 1990). Despite these associations however, the precise mechanisms whereby acrolein contributes to acute cardiovascular pathology is unknown.
Given the extensive evidence implicating oxidative stress in MMP activation (Nelson et al., 2004), we tested the hypothesis that acrolein exposure results in MMP activation and thus contributes to acute plaque rupture and vascular occlusion. Using both a cell culture model and murine atherosclerotic tissue, we did indeed demonstrate that one consequence of acrolein exposure is MMP secretion. Furthermore we show that this was dependent upon increased intracellular calcium and increased ROS generation by xanthine oxidase. Our findings are likely to be of significance in understanding the acute inflammatory responses to acrolein generated endogenously or delivered from the environment.
MATERIALS AND METHODS
Reagents and Cells
The fluorescent reagents H2DCFDA, fluo-4 AM and DQ-gelatin were purchased from Invitrogen (Carlsbad, CA) while BAPTA-AM and apocynin were from Calbiochem (Gibbstown, NJ). Cell culture media (RPMI 1640) was obtained from Mediatech Inc. (Manassas, VA) and additional media components, fetal calf serum, glutamine, and penicillin/streptomycin were from Clonetics (Allendale, NJ). The MMP-9 antibody, low melting temperature agarose and all other chemicals and reagents were obtained from Sigma (St. Louis MO).
THP-1 cells were obtained from ATCC (Manassas, VA) and maintained in RPMI 1640 media supplemented with 10% fetal calf serum, 1% glutamine, and 1% penicillin/streptomycin. For differentiation into macrophages, 4 × 104 cells were harvested, resuspended in normal media and plated into a 96 well plate in the presence of 100 nM phorbol ester (PMA) for 72 h. For larger scale experiments, 2–3 × 105 cells were differentiated in a 24 well plate in the presence of PMA.
Biochemical Assays and Western blotting
ROS
Levels of ROS were measured in untreated or acrolein-treated THP-1 cells using the H2DCFDA reporter. For this procedure, the cells were incubated overnight in media containing 0.1% FCS, washed and then loaded with 5 μM of H2DCFDA in HBSS for 1h. After washing, the cells were returned to HBSS in the presence or absence of acrolein and changes in fluorescence (ex: 493nm; em: 525nm) were measured on a Perkin Elmer Fusion fluorescence plate reader. Rate of ROS production over a 40 min period was determined and normalized to untreated cells. To delineate the contribution of individual pathways, the cells were treated with metabolic inhibitors at concentrations as previously published or as recommended by the manufacturer. These inhibitors include allopurinol (100 μM), apocynin (100 μM), rotenone (100 μM), NAC (10 mM) or the calcium chelator BAPTA-AM (20 μM) and were done for 30 min before acrolein exposure.
Xanthine oxidase
Xanthine oxidase activity was directly determined in cell lysates using the Amplex Red reagent (Invitrogen; Carlsbad, CA) (Mohanty, et al., 1997). In brief, differentiated THP-1 cells were left untreated or were treated with acrolein for 30 min, harvested and pelleted by centrifugation. The cells were then lysed by sonication, the supernatants collected after centrifugation, and protein concentration determined with the Bradford reagent (BioRad; Hercules, CA). Equal protein amounts were then incubated in the presence of 100 μM xanthine, 0.2 U/ml horseradish peroxidase, and 50 μM Amplex Red for 30 min at 37°C and fluorescence properties (ex: 560nm; em: 590nm) determined on a Shimadzu RF-5301 PC spectrofluorometer. Specificity was demonstrated by inclusion of 10 μM allopurinol in the reaction mixture.
Intracellular calcium
Relative amounts of intracellular calcium were determined in untreated cells or in cells treated with acrolein. This was accomplished by first pre-loading the cells with the Fluo 4-AM dye for 30 min, washing, and then recording baseline absorbance at 514 nm using a Perkin Elmer Fusion fluorescence plate reader. The cells were then treated with acrolein or the vehicle and the absorbance increase monitored for an additional 6 min. Initial rates of fluorescence increase were determined for a 2 time period. Pre-incubation with 20 mM BAPTA for 30 min was used to establish the Ca+2-specificity of the fluorescence signal.
MMP-9 Western blotting
The levels of MMP-9 were determined in culture media from untreated cells or those treated with acrolein. For this, aliquots were resolved on 8% polyacrylamide gels, transferred to PVDF membranes (BioRad; Hercules, CA) and probed with a 1:1000 dilution the anti-MMP-9 antibody (Sigma, St. Louis, MO). Specific bands were detected after secondary antibody incubation and development using the ECL reagent (GE Healthcare; Piscataway NJ). Images were collected on an Amersham Typhoon Variable Imager and densitometric values determined with NIH Image software.
Zymography
Gel zymography
Levels of MMPs in conditioned media from untreated or acrolein-treated cells were determined by gel zymography. For this, differentiated THP-1 cells were incubated overnight in serum-free media. The following day, cells were treated with vehicle or acrolein and media collected 10 and 30 min after treatment. Aliquots of these samples were then resolved on 8% polyacrylamide gels containing 1 mg/ml gelatin. After washing in 2.5% Triton, the gels were incubated overnight in 50 mM Tris, pH 7.4, 5mM CaCl2, 150 mM NaCl, stained with Coomassie Blue and destained. Clear areas amongst the blue background are indicative of MMP activity.
In situ zymography
To determine whether acrolein-exposed tissue also demonstrated an increase in MMPs, we used in situ zymography. apoE−/− mice on a high fat diet for 15 weeks were euthanized and aortas isolated and incubated in complete DMEM for 2h with two changes of media. At this time these aortic segments were washed with PBS, incubated in serum-free media for 2 h and then exposed to new media containing acrolein (25 μM) or vehicle for an additional 2 h. The aortas were then frozen in OCT and sectioned on a cryostat. Prior to zymography, lesion-containing aortic sections were identified by oil red O staining. Adjacent sections were then overlaid with a solution of 1% low melting agarose containing 0.1mg/ml DQ-gelatin and incubated at 37°C for 4 – 8 h. To establish MMP specificity, control sections were incubated with an overlay solution also containing EGTA (20mM). Fluorescence in the stained sections, indicative of MMP activity, was detected by microscopy and quantified by digital image analysis using the MetaMorph software.
Statistical analysis
Multi-group analyses were done with one way analysis of variance, parametric or ranks where appropriate, using the Bonferroni or Dunnett’s sub test as required. Single group analyses were done with a paired Student’s t-test. All statistical analyses were done with Sigma stat software.
RESULTS
Exposure to acrolein stimulates MMP-9 and ROS in differentiated THP-1 cells
To begin to examine the effect of acrolein on MMPs, we used a macrophage cell line. Macrophages are a major cell component of atherosclerotic lesions (Gerrity 1981a; Gerrity 1981b). They express high levels of MMPs that are activated in response to oxidative, pathogenic, and inflammatory stimuli. Hence, we examined whether acrolein also affects MMP production by macrophages. For this, we utilized PMA-differentiated THP-1 cells, a widely used macrophage model. After differentiation to macrophages, these cells were treated either with the vehicle or with various doses of acrolein for 10 and 30 min upon which the culture media were collected and aliquots subjected to gel zymography. At both time points, we observed a single band of approximately 90 kDa that was increased relative to untreated cells (0 μM acrolein) (Figure 1A). Levels of MMP activity at the highest dose of acrolein (25 μM) were similar to those observed in the media from cells treated with 20 ng/ml TNFα, a positive control. As a molecular mass of 90 kDa is indicative of MMP-9, we next performed Western blotting with an antibody specific for this isoform. Consistent with gel zymography, these data showed an up-regulation of MMP-9 in acrolein-treated cells at both time points (Figure 1B). A quantitative analysis of the zymograms indicated that 25uM acrolein increased MMP-9 levels 1.2 ± 0.9 fold after 30 minutes of exposure (Figure 1C).
Figure 1. Acrolein induces MMP-9 in cultured macrophages.
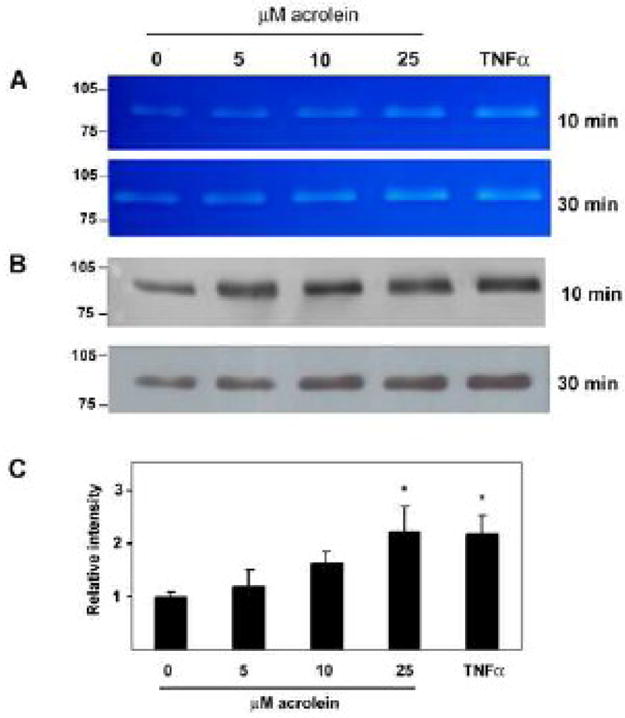
Differentiated THP-1 cells were serum-starved overnight and then exposed to the listed concentrations of acrolein for 10 min and 30 min as indicated. The medium was then collected and aliquots subjected to (A) gel zymography and (B) Western blot analysis using an anti-MMP-9 antibody. Cells treated with TNFα-were used as positive control. Illustrated are the zymograms and blots from a representative experiment. (C) Quantitative analysis of MMP-9 activity measured by zymography. Data are presented as mean +/− S.E. * p<0.05 versus 0 μM acrolein (n = 4 experiments).
As a key player in host defense, macrophages generate and secrete ROS in response bacterial endotoxins, secondary messengers, and mechanical forces (Droge 2002). ROS have also been implicated in the transcriptional and zymogenic regulation of MMPs (Nelson et al., 2004). Thus, we next determined if macrophages would also produce ROS upon exposure to acrolein in a fluorescence-based assay. Treatment with two doses of acrolein induced a statistically significant increase in the rate of ROS production compared to untreated cells (Figure 2). Exposure to 25 μM acrolein increased ROS to a similar extent as cells treated with 100 μM H2O2, a positive control. There was only minor inhibition in the rate of ROS production when the cells were treated with acrolein in the presence of L-NAME (data not shown) indicating that the contribution of NO to the observed changes in the fluorescence signal was minimal. On the other hand, when cells were pre-incubated with the thiol agent, NAC (10mM), prior to acrolein stimulation, the levels of ROS produced were not significantly increased over those found in untreated cells (Figure 2). These data indicate that in addition to MMP activation, acrolein exposure increases ROS levels in macrophages.
Figure 2. Acrolein increases ROS levels in cultured macrophages.
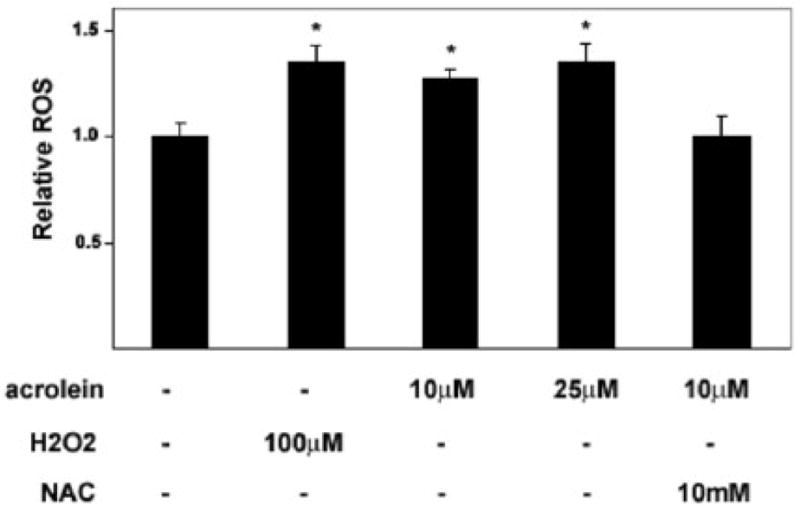
THP-1 cells differentiated with PMA for 72 h were starved in 0.1% serum overnight, loaded with H2DCFDA in HBSS and exposed to the indicated concentrations of acrolein or H2O2. Oxidation-dependent fluorescence of H2DCFDA was determined in a plate reader at 525nm and rates of fluorescence increase were determined over 40 min and normalized to untreated, control cells. Data are presented as mean +/− S.E. * p<0.05 versus untreated cells (n =4 experiments).
Acrolein-stimulated ROS production is mediated by intracellular calcium and xanthine oxidase
Several enzymatic reactions could contribute to ROS generation in macrophages. ROS could be generated directly by the NADPH-oxidase complex or as products of mitochondrial electron transport or the XO-catalyzed degradative oxidation of purine bases. To identify the source of acrolein-induced increase in ROS production, we examined each of these processes. For this, the cells were pre-incubated with inhibitors of NADPH-oxidase (apocynin), mitochondrial complex 1 (rotenone), and XO (allopurinol) prior to acrolein exposure and rates of ROS production were measured as described above. While apocynin- and rotenone-treated cells demonstrated little reduction in the rate of ROS production compared with untreated cells, allopurinol significantly decreased levels of ROS (Figure 3A). This inhibition of ROS generation could not be attributed to non-specific effects because allopurinol did not significantly attenuate ROS production in H2O2–treated cells (not shown). As an additional confirmation of the involvement of XO in acrolein-stimulated ROS production, we directly measured its activity in a fluorescence-based assay. Exposure to acrolein caused an approximate 50% increase in XO activity compared to untreated cells (Figure 3B). This increase was specific to XO as it was abolished when the cells were treated with acrolein in the presence of allopurinol.
Figure 3. Acrolein-stimulated ROS is xanthine oxidase-dependent.
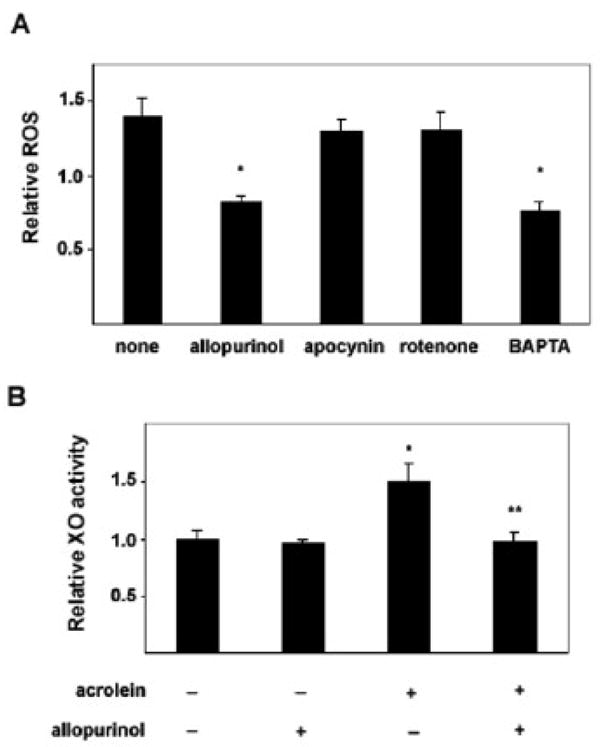
Serum-starved differentiated THP-1 cells were either left untreated (“none”) or were pre-incubated with allopurinol (100μM), apocynin (100μM), rotenone (100μM) or the calcium chelator BAPTA-AM (20μM) for 30 min prior to acrolein treatment. Relative rates of ROS production over 30min were measured and normalized to unexposed cells. (* p<0.05 vs “none”). B) Untreated cells or those pre-treated with allopurinol were variably exposed to acrolein for 30min as indicated. The cells were then lysed and xanthine oxidase activity was measured as described under Methods. Activity levels were normalized to untreated, unexposed (control) cells (* p<0.05 vs control; ** p<0.05 vs acrolein only). Each bar represents mean ± S.E. of 3 different measurements.
XO catalyzes the enzymatic degradation of xanthine and hypoxanthine to uric acid and in doing so transfers electrons to molecular oxygen. An alternative, larger molecular weight form of the same gene product, xanthine dehydrogenase (XDH), catalyzes the same reaction except that it uses nicotine adenine dinucleotide (NAD) as an electron acceptor (Pritsos 2000). One way by which XDH is converted into XO is by limited calcium-dependent proteolysis (Greene et al., 1994; Hirata et al., 1993; McCord et al., 1982). Therefore, we next examined the possibility that acrolein induces XO activity by increasing levels of intracellular calcium and consequently, the conversion of XDH to XO. Using the fluo-4 dye as a reporter, we observed an immediate increase in intracellular calcium upon acrolein exposure (Figure 4A). Intracellular calcium levels increased approximately 1.5 fold over those observed in untreated cells (Figure 4B). Importantly this fluorescence increase was attenuated when acrolein-treated cells were pre-loaded with the calcium chelator, BAPTA (Figure 4B). Furthermore, blocking the acrolein-induced increase in intracellular calcium with BAPTA also attenuated ROS production by these cells (Figure 3A). These observations suggest that the increase in ROS levels in acrolein-treated macrophages is mediated by XO and is dependent upon an increase in intracellular calcium levels.
Figure 4. Acrolein induces an increase in intracellular calcium.

(A) Differentiated THP-1 cells were loaded with the Fluo-4-AM dye for 30 min, washed and then exposed to acrolein (arrow). The change in fluorescence at 516nm was then recorded for an additional 6 min. (B) Group data showing increases in [Ca2+]i normalized to control, untreated cells as mean ± S.E. (* p<0.05 vs. acrolein treatment, n = 4 experiments.
Acrolein-mediated upregulation of macrophage MMP-9 is ROS dependent
The regulation of MMP expression or activity is tightly controlled and ROS may be one important regulator of these proteases (Nelson et al., 2004). Thus, we next used an inhibitor-based approach to determine if there was a causal relationship between acrolein-induced ROS generation and MMP activation. For this, the THP-1 cells were pre-incubated with two XO inhibitors- allopurinol and oxypurinol (100μM each), the calcium chelator BAPTA (20μM), and two antioxidants - NAC (10 mM) and Tiron (5 mM). After incubation with the indicated additives, the cells were treated with acrolein for 30 min and the levels of MMP-9 were measured in the medium. Similar to earlier experiments, acrolein-induced an increase in MMP-9 that was approximately 70% greater than that found untreated cells (Figure 5). This increase was, however, significantly attenuated by the XO inhibitors, BAPTA, and antioxidants. From these data we infer that stimulation of MMP-9 production by acrolein is mediated by XO-derived ROS.
Figure 5. Acrolein-induced MMP-9 is mediated by ROS, xanthine oxidase, and changes in intracellular calcium.

Differentiated THP-1 cells were pre-treated with vehicle (−), allopurinol (allo), oxypurinol (oxy), BAPTA-AM, Tiron, or NAC for 30 min and then stimulated with 25 μM acrolein as indicated. Additional cell samples pre-treated with vehicle were left unstimulated (control) or were stimulated with TNFα. A) After 30 min, aliquots of culture media were analyzed for the presence of MMP-9 by Western blotting. A representative Western blot is shown. B) Acrolein stimulated increases in MMP-9 which were reversed with xanthine oxidase inhibitors, a calcium chelator, and ROS scavengers. Data are presented as mean ± S.E (* p<0.05 versus acrolein only; **: p<0.05 versus control; n = 5 experiments).
Acrolein stimulates MMPs in atherosclerotic lesions
To assess whether acrolein also activates MMP ex vivo, we examined changes in MMPs in atherosclerotic lesions. Previous studies have shown that MMPs are expressed at high levels in advanced atherosclerotic lesions (Galis et al., 1994; Galis et al., 1995; Halpert et al., 1996; Rajavashisth et al., 1999), 1995) and that they are activated by an increase in oxidative stress within arterial plaques. Accordingly we hypothesized that acute acrolein exposure stimulates MMP activity in atherosclerotic lesions. To test this, we examined the effects of acrolein on MMP activity in aortic explants from apoE−/− mice. These mice, maintained on a high-fat diet for 15 weeks, demonstrated abundant, well-formed lesions as identified by oil red O staining (Figure 6A, B). When these explants were treated with 25 μM acrolein and then subjected to in situ zymography, a robust increase in MMP activity was observed (Figure 6B). The increase in fluorescence in acrolein-treated lesions appeared to be MMP-specific as it could be blocked by inclusion of 20 mM EGTA during zymography. In contrast, untreated aortic explants demonstrated minimal increase in fluorescence during this assay (Figure 6A). A quantitative analysis of these images demonstrates that MMP activity was approximately 10-fold greater in acrolein-exposed explants versus untreated explants (Figure 6C). We conclude, based on these observations, that exposure to acrolein results in acute activation of MMPs in the advanced atherosclerotic lesions of apoE−/− mice.
Figure 6. Acrolein stimulates MMP in atherosclerotic lesions.

Aortic explants isolated from apoE−/− mice on a high-fat diet for 15 weeks were treated with vehicle (A) or 25 μM acrolein (B) for 2h, frozen in OCT and sectioned. Lesion-containing sections were identified by oil red O (ORO) staining and adjacent sections were then subjected to in situ zymography with DQ-gelatin for 4 h. Representative ORO and fluorescent images are shown. Attenuation by 20mM EGTA during the zymography demonstrated that the fluorescence was specific for MMP activity. (C) Group data quantifying changes in fluorescence in untreated and acrolein-treated arterial lesions. Data are presented as mean ± S.E. * p < 0.05, (n = 3 experiments).
DISCUSSION
The major findings of this study are that exposure to acrolein increases intracellular calcium which in turn activates XO in human macrophages. These increases were furthermore associated with an increase in ROS generation and MMP-9 secretion (Figure 7). Conversely, we found that inhibition of XO prevented ROS generation and decreased MMP-9 secretion from acrolein-treated macrophages. Finally, our experiments with advanced arterial lesions of apoE-null mice showed that treatment with acrolein leads to an increase in MMP activity, indicating that acrolein stimulates MMPs both in isolated macrophages as well as in advanced atherosclerotic plaques. These results may be of significance in understanding how acrolein and oxidized lipids induce cardiovascular toxicity and how acute exposure to or increased generation of acrolein could destabilize arterial lesions and trigger clinical events.
Figure 7. Activation of MMPs by acrolein.

Exposure to acrolein results in an increase of free intracellular calcium levels through a mechanism that has not yet been established (?). This rise in calcium activates xanthine oxidase thereby generating ROS. These free radicals stimulate macrophage MMP secretion potentiating matrix degradation and enhancing the likelihood of atherosclerotic plaque rupture.
Humans are exposed to acrolein from a variety of exogenous and endogenous sources. Estimates of acrolein generation from measurement of the urinary metabolite S-(3-hydroxypropyl)mercapturic acid (HPMA) suggest that its basal concentration in healthy adults may be between 1 and 2 μM (Carmella et al., 1920; Roethig et al., 2007). However these levels may increase with exogenous exposure or endogenous acrolein production due to disease or inflammation. Urine from smokers or cyclophosphamide-treated patients, for instance, contains 2–4 μM acrolein (Carmella et al., 1920; Takamoto et al., 2004). Local concentrations of acrolein within inflammatory lesions may be higher still. Excessive generation of acrolein (10 fold) has also been observed in the sputum of chronic obstructive pulmonary disease (COPD) patients. (Deshmukh et al., 2008). Hence, the concentrations of acrolein found to activate MMPs (5 to 25 μM) in the present study appears to be within the range of acrolein levels achieved during pollutant exposure or during endogenous tissue inflammation.
Our results suggest that acrolein-induced an increase in MMP-9 through a mechanism involving an increase in intracellular calcium levels, activation of XO, and stimulation of ROS generation This is so because MMP-9 levels could be reversed with the antioxidants NAC and Tiron, the XO inhibitors allopurinol and oxypurinol, and the calcium chelator, BAPTA. Increased ROS generation could activate MMPs by a variety of mechanisms. Perhaps least likely are effects at the transcriptional level. Although ROS can stimulate MMP gene expression (Nelson et al., 2004), the rapid increase of MMP-9 in the media, within minutes of acrolein exposure, is inconsistent with transcriptional activation. More likely, ROS stimulate mechanisms directly affecting MMP catalytic function or secretion. MMPs are expressed as inactive zymogens, a consequence of Zn+2-coordination by histidines in their catalytic domains and cysteines in their pro-peptide regions. Activation is achieved upon pro-peptide cleavage by additional tissue or plasma proteinases or by other MMPs (Chakraborti et al., 2003; Nelson et al., 2004). In addition, ROS have been implicated in zymogen activation through sulfenic acid production with the cysteine in the MMP pro-peptide, thereby allowing conformational change and autocatalysis (Nelson et al., 2004). Furthermore, prior studies have shown that the secretion of MMPs was dependent upon various PKC isoforms (Chakrabarti et al., 2006; Hussain et al., 2002; Lee et al., 2004; O’Toole et al., 2008; Park et al., 2003). Given that the activity of the conventional PKCs (α,β,γ) is calcium-dependent, it is conceivable that the acrolein-induced increase in intracellular calcium, observed in this study, could result in MMP release through the stimulated activity of one of these PKC isoforms. Additional studies are required to distinguish between these possibilities.
That XO is the major source of ROS generation in acrolein-stimulated cells is suggested by the observations that its catalytic activity increased upon exposure and that its inhibition by allopurinol attenuated ROS production (Figure 3). The stimulation of XO-derived ROS in response to exogenous stimuli may be a common response of vascular or hematopoietic tissues. XO is expressed in high abundance in macrophages (Takao et al., 1996) and increased levels of this enzyme have been detected in human atherosclerotic lesions (Swain et al., 1995). In adenosine-stimulated macrophages, XO was found to be responsible for over 10 times the superoxide production attributable to the NADPH oxidase system (Tritsch et al., 1983). Also, it has previously been demonstrated that XO-derived ROS are major players in cardiovascular pathology, associated with hypertension (Nakazono et al., 1991; Swei et al., 1999), ischemia/repurfusion injury (McCord et al., 1985), atherosclerosis (Harrison et al., 2003; Madamanchi et al., 2005), and decreased myocardial contractility (Ferdinandy et al., 1999). Some of these in vivo complications could be reversed by treatment with XO inhibitors (Doehner et al., 2002; Farquharson et al., 2002;Gimpel et al., 1919; Guthikonda et al., 2003; Kogler et al., 2003), supporting the idea that XO is a frequent source of ROS is diseased cardiovascular tissue.
Calcium fluxes likewise appear to play a central role in acrolein-mediated pathophysiology. Our results showed that the acrolein-induced increase in XO activity was associated with a concomitant rise in intracellular calcium levels. Importantly, chelation of intracellular calcium with BAPTA attenuated the acrolein-mediated up-regulation of ROS and MMP-9. These results suggest that the disruption of calcium homeostasis may be general outcome of toxic aldehyde exposure in general and acrolein exposure in particular. Consistent with this idea, acrolein has been previously implicated in the calcium-dependent induction of Hsp72 in HUVECs (Misonou et al., 2005) and can also influence calcium signaling pathways in airway smooth muscle cells (Hyvelin et al., 2000). Exposure to another α,β unsaturated aldehyde and lipid oxidation product, 4-HNE, also resulted in a calcium influx in rat hepatocytes (Carini et al., 1996) and disruption of calcium homeostasis in cardiac myocytes (Bhatnagar 1995a).
The observation that exposure to acrolein activates MMPs in advanced atherosclerotic plaques suggests that exposure to acrolein-containing pollutants or increased production of endogenous acrolein could trigger plaque rupture. Although the normal vessel wall displays low MMP activity, atheromatous plaques with high levels of inflammation are associated with elevated MMP activity (Choudhary et al., 2006) and increased MMP activity is believed to promote the progression of stable atherosclerotic lesions to an unstable phenotype that is more likely to rupture (Newby 2007). Several studies have shown that inhibition of MMP activity can stabilize plaques (Johnson et al., 2006; Rouis et al., 1999) and that overexpression of MMP-9 destabilizes advanced atherosclerotic lesions of apoE−/− mice (Gough et al., 2006). Hence, MMP activation by acrolein in advanced lesions may lead to an imbalance between ECM deposition and degradation. In addition, MMP-mediated degradation of the ECM might result in the loss of cell adhesion and trigger the apoptosis of resident smooth muscle and endothelial cells. Thus, enhanced MMP activation may contribute to plaque instability and rupture, triggering thrombus formation and ischemic outcomes. It is significant that myeloperoxidase and oxidized LDL, both of which generate high levels of acrolein, co-localize to lipid-laden macrophages and are associated with MMP activation (Fu et al., 2001; Huang et al., 1919). Hence, based on the data presented here, it appears likely the increased acrolein formation during inflammation and the accumulation of oxidized LDL within the lesion may be a significant determinant of MMP activity and in turn plaque stability. It is possible that endogenous generation of acrolein during inflammation may also be a significant feature of other conditions associated with increased MMP activation such as COPD and emphysema (Foronjy et al., 2008; Tetley 2002), rheumatoid arthritis or cancer cell invasion and metastasis (Bjorklund et al., 2005;Egeblad et al., 2002). Interestingly, in a related study, α,β-unsaturated aldehydes from cigarette smoke also stimulated the release of IL-8 and TNF-α from macrophages, potentially contributing to the development of COPD (Facchinetti, et al., 2007)
The plaque-destabilizing effects of acrolein may also be of relevance in understanding the cardiovascular effects of environmental pollutants. Several epidemiological studies have shown that an episodic increase in air pollution is associated with an increase in cardiopulmonary mortality, particularly that associated with acute myocardial infraction (AMI) (Bhatnagar 2006; Pope, III et al., 2004). Although further studies are required to establish whether the presence of acrolein in ambient pollutants contributes to the increased risk of cardiovascular events in exposed populations, it has been reported that exposure to acrolein-rich pollutants such as automobile emissions (Peters et al., 2004) and cigarette smoke (Pell et al., 2008) could acutely and significantly increase AMI risk. Our observation that acrolein activates MMP supports the possibility that exposure to oxidants or acute inflammatory events could trigger plaque rupture and thereby precipitate myocardial infarction. Hence, measures taken to limit oxidant exposure, inflammation, and MMP production in individuals sensitive to environmental pollutant exposure may lessen the probability of adverse cardiovascular outcomes.
Acknowledgments
This work was supported in part by a grant from EPA, NIH grants ES11860, HL55477, HL59378, and a grant from Philip Morris.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alfredsson L, Hammar N, Hogstedt C. Incidence of myocardial infarction and mortality from specific causes among bus drivers in Sweden. Int J Epid. 1993;22:57–61. doi: 10.1093/ije/22.1.57. [DOI] [PubMed] [Google Scholar]
- Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha, beta-unsaturated aldehydes by phagocytes at sites of inflammation. J Clin Invest. 1997;99:424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ Res. 1995b;76:293–304. doi: 10.1161/01.res.76.2.293. [DOI] [PubMed] [Google Scholar]
- Bhatnagar A. Cardiovascular pathophysiology of environmental pollutants. Am J Phy - Hrt & Circ Phys. 2004;286:H479–85. doi: 10.1152/ajpheart.00817.2003. [DOI] [PubMed] [Google Scholar]
- Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ Res. 1995a;76:293–304. doi: 10.1161/01.res.76.2.293. [DOI] [PubMed] [Google Scholar]
- Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 9292006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophy Acta. 2005;1755:37–69. doi: 10.1016/j.bbcan.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Zahm DS, Macarthur H, Kolo LL, Westfall TC, Anwar M, Glickstein SB, Ruggiero DA. Catecholamine monoamine oxidase a metabolite in adrenergic neurons is cytotoxic in vivo. Brain Res. 2001;891:218–227. doi: 10.1016/s0006-8993(00)03199-1. [DOI] [PubMed] [Google Scholar]
- Carini R, Bellomo G, Paradisi L, Dianzani MU, Albano E. 4-Hydroxynonenal triggers Ca2+ influx in isolated rat hepatocytes. Biochem Biophys Res Comm. 1996;218:772–776. doi: 10.1006/bbrc.1996.0137. [DOI] [PubMed] [Google Scholar]
- Carmella SG, Chen M, Zhang Y, Zhang S, Hatsukami DK, Hecht SS. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: effects of cigarette smoking. Chem Res Tox. 2007;20:986–990. doi: 10.1021/tx700075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: novel pathways for tertiary granule release. J Leuk Bio. 2006;79:214–222. doi: 10.1189/jlb.0605353. [DOI] [PubMed] [Google Scholar]
- Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- Choudhary S, Higgins CL, Chen IY, Reardon M, Lawrie G, Vick GW, III, Karmonik C, Via DP, Morrisett JD. Quantitation and localization of matrix metalloproteinases and their inhibitors in human carotid endarterectomy tissues. Art, Throm Vasc Biol. 2006;26:2351–2358. doi: 10.1161/01.ATV.0000239461.87113.0b. [DOI] [PubMed] [Google Scholar]
- Deshmukh HS, Shaver C, Case LM, Dietsch M, Wesselkamper SC, Hardie WD, Korfhagen TR, Corradi M, Nadel JA, Borchers MT, Leikauf GD. Acrolein-activated matrix metalloproteinase 9 contributes to persistent mucin production. Am J Resp Cell Mol Biol. 2008;38:446–454. doi: 10.1165/rcmb.2006-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehner W, Schoene N, Rauchhaus M, Leyva-Leon F, Pavitt DV, Reaveley DA, Schuler G, Coats AJ, Anker SD, Hambrecht R. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circ. 2002;105:2619–2624. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Phys Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Rad Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Facchinetti F, Amadei F, Geppetti P, Tarantini F, DiSerio C, Dragotto A, Gigli PM, Catinella S, Civelli M, Patacchini R. α,β-unsaturated aldehydes in cigarette smoke release inflammatory mediators from human macrophages. Am J Respir Cell Mol Biol. 37:617–623. doi: 10.1165/rcmb.2007-0130OC. [DOI] [PubMed] [Google Scholar]
- Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circ. 2002;106:221–226. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Panas D, Schulz R. Peroxynitrite contributes to spontaneous loss of cardiac efficiency in isolated working rat hearts. Am J Phys. 1999;276:H1861–1867. doi: 10.1152/ajpheart.1999.276.6.H1861. [DOI] [PubMed] [Google Scholar]
- Feron VJ, Til HP, de VF, Woutersen RA, Cassee FR, van Bladeren PJ. Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mut Res. 1991;259:363–385. doi: 10.1016/0165-1218(91)90128-9. [DOI] [PubMed] [Google Scholar]
- Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, D’Armiento J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Phys - Lung Cell Mol Phys. 2008;294:L1149–1157. doi: 10.1152/ajplung.00481.2007. [DOI] [PubMed] [Google Scholar]
- Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Kranzhofer R, Clark S, Libby P. Macrophage foam cells from experimental atheroma constitutively produce matrix-degrading proteinases. Proc Nat Acad Sci USA. 1995;92:402–406. doi: 10.1073/pnas.92.2.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Path. 1981a;103:181–190. [PMC free article] [PubMed] [Google Scholar]
- Gerrity RG. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Path. 1981b;103:191–200. [PMC free article] [PubMed] [Google Scholar]
- Gimpel JA, Lahpor JR, van der Molen AJ, Damen J, Hitchcock JF. Reduction of reperfusion injury of human myocardium by allopurinol: a clinical study. Free Rad Biol Med. 1995;19:251–255. doi: 10.1016/0891-5849(94)00242-c. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EL, Paller MS. Calcium and free radicals in hypoxia/reoxygenation injury of renal epithelial cells. Am J Phys. 1994;266:F13–20. doi: 10.1152/ajprenal.1994.266.1.F13. [DOI] [PubMed] [Google Scholar]
- Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circ. 2003;107:416–421. doi: 10.1161/01.cir.0000046448.26751.58. [DOI] [PubMed] [Google Scholar]
- Halpert I, Sires UI, Roby JD, Potter-Perigo S, Wight TN, Shapiro SD, Welgus HG, Wickline SA, Parks WC. Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc Nat Acad Sci USA. 1996;93:9748–9753. doi: 10.1073/pnas.93.18.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Card. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M, Schonbeck U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circ. 2001;104:1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- Hirata Y, Ishii K, Taguchi T, Suita S, Takeshige K. Conversion of xanthine dehydrogenase to xanthine oxidase during ischemia of the rat small intestine and the effect of trifluoperazine on the conversion. J Ped Surg. 1993;28:597–600. doi: 10.1016/0022-3468(93)90668-b. [DOI] [PubMed] [Google Scholar]
- Huang Y, Mironova M, Lopes-Virella MF. Oxidized LDL stimulates matrix metalloproteinase-1 expression in human vascular endothelial cells. Art, Thromb Vas Biol. 1999;19:2640–2647. doi: 10.1161/01.atv.19.11.2640. [DOI] [PubMed] [Google Scholar]
- Hussain S, Assender JW, Bond M, Wong LF, Murphy D, Newby AC. Activation of protein kinase Czeta is essential for cytokine-induced metalloproteinase-1, -3, and -9 secretion from rabbit smooth muscle cells and inhibits proliferation. J Biol Chem. 2002;277:27345–27352. doi: 10.1074/jbc.M111890200. [DOI] [PubMed] [Google Scholar]
- Hyvelin JM, Roux E, Prevost MC, Savineau JP, Marthan R. Cellular mechanisms of acrolein-induced alteration in calcium signaling in airway smooth muscle. Tox Appl Pharm. 2000;164:176–183. doi: 10.1006/taap.1999.8879. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Baker AH, Oka K, Chan L, Newby AC, Jackson CL, George SJ. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circ. 2006;113:2435–2444. doi: 10.1161/CIRCULATIONAHA.106.613281. [DOI] [PubMed] [Google Scholar]
- Kai H, Ikeda H, Yasukawa H, Kai M, Seki Y, Kuwahara F, Ueno T, Sugi K, Imaizumi T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Card. 1998;32:368–372. doi: 10.1016/s0735-1097(98)00250-2. [DOI] [PubMed] [Google Scholar]
- Kogler H, Fraser H, McCune S, Altschuld R, Marban E. Disproportionate enhancement of myocardial contractility by the xanthine oxidase inhibitor oxypurinol in failing rat myocardium. Card Res. 2003;59:582–592. doi: 10.1016/s0008-6363(03)00512-1. [DOI] [PubMed] [Google Scholar]
- Lee HS, Park SY, Lee HW, Choi HS. Secretions of MMP-9 by soluble glucocorticoid-induced tumor necrosis factor receptor (sGITR) mediated by protein kinase C (PKC)delta and phospholipase D (PLD) in murine macrophage. J Cell Biochem. 2004;92:481–490. doi: 10.1002/jcb.20099. [DOI] [PubMed] [Google Scholar]
- Levine RJ, Andjelkovich DA, Shaw LK. The mortality of Ontario undertakers and a review of formaldehyde-related mortality studies. J Occ Med. 1984;26:740–746. doi: 10.1097/00043764-198410000-00014. [DOI] [PubMed] [Google Scholar]
- Ludeman SM. The chemistry of the metabolites of cyclophosphamide. Curr Pharm Design. 1999;5:627–643. [PubMed] [Google Scholar]
- Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Imm Meth. 1997;202:133–141. doi: 10.1016/s0022-1759(96)00244-x. [DOI] [PubMed] [Google Scholar]
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Art, Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- McCall MR, Tang JY, Bielicki JK, Forte TM. Inhibition of lecithin-cholesterol acyltransferase and modification of HDL apolipoproteins by aldehydes. Art, Thromb Vasc Biol. 1995;15:1599–1606. doi: 10.1161/01.atv.15.10.1599. [DOI] [PubMed] [Google Scholar]
- McCord JM, Roy RS. The pathophysiology of superoxide: roles in inflammation and ischemia. Can J Phys Pharm. 1982;60:1346–1352. doi: 10.1139/y82-201. [DOI] [PubMed] [Google Scholar]
- McCord JM, Roy RS, Schaffer SW. Free radicals and myocardial ischemia. The role of xanthine oxidase. Adv Myocard. 1985;5:183–189. [PubMed] [Google Scholar]
- Misonou Y, Takahashi M, Park YS, Asahi M, Miyamoto Y, Sakiyama H, Cheng X, Taniguchi N. Acrolein induces Hsp72 via both PKCdelta/JNK and calcium signaling pathways in human umbilical vein endothelial cells. Free Rad Res. 2005;39:507–512. doi: 10.1080/10715760500072255. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Card Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc Nat Acad Sci USA. 1991;88:10045–10048. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Rad Biol Med. 2004;37:768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Card Med. 2007;17:253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole EA, van KR, Chen M, Woodley DT. Hypoxia induces epidermal keratinocyte matrix metalloproteinase-9 secretion via the protein kinase C pathway. J Cell Phys. 2008;214:47–55. doi: 10.1002/jcp.21160. [DOI] [PubMed] [Google Scholar]
- Parent RA, Caravello HE, Sharp DE. Metabolism and distribution of [2,3-14C]acrolein in Sprague-Dawley rats. J App Tox. 1996;16:449–457. doi: 10.1002/(SICI)1099-1263(199609)16:5<449::AID-JAT369>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Park MJ, Park IC, Lee HC, Woo SH, Lee JY, Hong YJ, Rhee CH, Lee YS, Lee SH, Shim BS, Kuroki T, Hong SI. Protein kinase C-alpha activation by phorbol ester induces secretion of gelatinase B/MMP-9 through ERK 1/2 pathway in capillary endothelial cells. Int J Onc. 2003;22:137–143. [PubMed] [Google Scholar]
- Pell JP, Haw S, Cobbe S, Newby DE, Pell AC, Fischbacher C, McConnachie A, Pringle S, Murdoch D, Dunn F, Oldroyd K, Macintyre P, O’Rourke B, Borland W. Smoke-free legislation and hospitalizations for acute coronary syndrome. New Eng J Med. 2008;359:482–491. doi: 10.1056/NEJMsa0706740. [DOI] [PubMed] [Google Scholar]
- Peters A, von KS, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Lowel H Cooperative Health Research in the Region of Augsburg Study Group. Exposure to traffic and the onset of myocardial infarction. New Eng J Med. 2004;351:1721–1730. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- Pope CA, III, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circ. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- Pritsos CA. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem-Biol Int. 2000;129:195–208. doi: 10.1016/s0009-2797(00)00203-9. [DOI] [PubMed] [Google Scholar]
- Rajavashisth TB, Xu XP, Jovinge S, Meisel S, Xu XO, Chai NN, Fishbein MC, Kaul S, Cercek B, Sharifi B, Shah PK. Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediators. Circ. 1999;99:3103–3109. doi: 10.1161/01.cir.99.24.3103. [DOI] [PubMed] [Google Scholar]
- Reiss AB, Glass AD. Atherosclerosis: immune and inflammatory aspects. J Invest Med. 2006;54:123–131. doi: 10.2310/6650.2006.05051. [DOI] [PubMed] [Google Scholar]
- Roethig HJ, Zedler BK, Kinser RD, Feng S, Nelson BL, Liang Q. Short-term clinical exposure evaluation of a second-generation electrically heated cigarette smoking system. J Clin Pharm. 2007;47:518–530. doi: 10.1177/0091270006297686. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circ. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- Rouis M, Adamy C, Duverger N, Lesnik P, Horellou P, Moreau M, Emmanuel F, Caillaud JM, Laplaud PM, Dachet C, Chapman MJ. Adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-1 reduces atherosclerotic lesions in apolipoprotein E-deficient mice. Circ. 1999;100:533–540. doi: 10.1161/01.cir.100.5.533. [DOI] [PubMed] [Google Scholar]
- Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280:36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- Steinbrecher UP, Zhang HF, Lougheed M. Role of oxidatively modified LDL in atherosclerosis. Free Rad Biol Med. 1990;9:155–168. doi: 10.1016/0891-5849(90)90119-4. [DOI] [PubMed] [Google Scholar]
- Stewart PA, Schairer C, Blair A. Comparison of jobs, exposures, and mortality risks for short-term and long-term workers. J Occ Med. 1990;32:703–708. [PubMed] [Google Scholar]
- Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circ. 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- Swain J, Gutteridge JM. Prooxidant iron and copper, with ferroxidase and xanthine oxidase activities in human atherosclerotic material. FEBS Letters. 1995;368:513–515. doi: 10.1016/0014-5793(95)00726-p. [DOI] [PubMed] [Google Scholar]
- Swei A, Lacy F, Delano FA, Parks DA, Schmid-Schonbein GW. A mechanism of oxygen free radical production in the Dahl hypertensive rat. Microcirc. 1999;6:179–187. [PubMed] [Google Scholar]
- Takamoto S, Sakura N, Namera A, Yashiki M. Monitoring of urinary acrolein concentration in patients receiving cyclophosphamide and ifosphamide. J Chrom B: Anal Tech Biomed Life Sci. 2004;806:59–63. doi: 10.1016/j.jchromb.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Takao S, Smith EH, Wang D, Chan CK, Bulkley GB, Klein AS. Role of reactive oxygen metabolites in murine peritoneal macrophage phagocytosis and phagocytic killing. A J Phys. 1996;271:C1278–1284. doi: 10.1152/ajpcell.1996.271.4.C1278. [DOI] [PubMed] [Google Scholar]
- Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121:156S–159S. doi: 10.1378/chest.121.5_suppl.156s. [DOI] [PubMed] [Google Scholar]
- Tritsch GL, Niswander PW. Modulation of macrophage superoxide release by purine metabolism. Life Sci. 1983;32:1359–1362. doi: 10.1016/0024-3205(83)90811-1. [DOI] [PubMed] [Google Scholar]
- Tziakas DN, Chalikias GK, Parissis JT, Hatzinikolaou EI, Papadopoulos ED, Tripsiannis GA, Papadopoulou EG, Tentes IK, Karas SM, Chatseras DI. Serum profiles of matrix metalloproteinases and their tissue inhibitor in patients with acute coronary syndromes. The effects of short-term atorvastatin administration. Int J Card. 2004;94:269–277. doi: 10.1016/j.ijcard.2003.05.013. [DOI] [PubMed] [Google Scholar]