

Abstract
Objectives:
To determine the involvement of the midbrain and hindbrain (MHB) in the groups of classic (cLIS), variant (vLIS), and cobblestone complex (CBSC) lissencephalies and to determine whether a correlation exists between the cerebral malformation and the MHB abnormalities.
Methods:
MRI scans of 111 patients (aged 1 day to 32 years; mean 5 years 4 months) were retrospectively reviewed. After reviewing the brain involvement on MRI, the cases were reclassified according to known mutation (LIS1, DCX, ARX, VLDLR, RELN, MEB, WWS) or mutation phenotype (LIS1-P, DCX-P, RELN-P, ARX-P, VLDLR-P) determined on the basis of characteristic MRI features. Abnormalities in the MHB were then recorded. For each structure, a score was assigned, ranging from 0 (normal) to 3 (severely abnormal). The differences between defined groups and the correlation between the extent of brain agyria/pachygyria and MHB involvement were assessed using Kruskal–Wallis and χ2 McNemar tests.
Results:
There was a significant difference in MHB appearance among the three major groups of cLIS, vLIS, and CBSC. The overall score showed a severity gradient of MHB involvement: cLIS (0 or 1), vLIS (7), and CBSC (11 or 12). The extent of cerebral lissencephaly was significantly correlated with the severity of MHB abnormalities (p = 0.0029).
Conclusion:
Our study focused on posterior fossa anomalies, which are an integral part of cobblestone complex lissencephalies but previously have not been well categorized for other lissencephalies. According to our results and the review of the literature, we propose a new classification of human lissencephalies.
GLOSSARY
- A
= autosomal;
- ACC
= agenesis of corpus callosum;
- AD
= autosomal dominant;
- AP
= anteroposterior;
- AR
= autosomal recessive;
- CBL
= cerebellar;
- CBSC
= cobblestone complex;
- cLIS
= classic lissencephaly;
- CMD
= congenital muscular dystrophy;
- CSZ
= cell-sparse zone;
- DV
= dorsal–ventral;
- FCMD
= Fukuyama congenital muscular dystrophy;
- IVH
= inferior vermis hypoplasia;
- LV
= lateral ventricle;
- m
= medulla;
- M
= midbrain;
- MDS
= Miller–Dieker syndrome;
- MEB
= muscle–eye–brain;
- MHB
= midbrain and hindbrain;
- MR
= magnetic resonance;
- ND
= not determined;
- P
= pons;
- RC
= rostrocaudal;
- SCBH
= subcortical band heterotopia;
- SELH
= subependymal linear heterotopia;
- V
= vermis;
- vLIS
= variant lissencephaly;
- WI
= weighted image;
- WWS
= Walker–Warburg syndrome;
- XLD
= X-linked dominant;
- XLR
= X-linked recessive.
The term lissencephaly (smooth outer brain surface) refers to a paucity of gyral and sulcal development.1 It encompasses a spectrum of gyral malformations ranging from complete agyria to regional pachygyria and includes subcortical band heterotopia. Lissencephaly has been traditionally classified in two distinct groups: classic (cLIS, formerly called lissencephaly type 1) and cobblestone complex (CBSC, formerly called lissencephaly type 2) based on both brain imaging and pathology. To date, five genes have been identified as causing or contributing to human cLIS: LIS1, 14-3-3ɛ in Miller–Dieker syndrome (MDS), DCX, RELN, and ARX.1–6 A new classification has recently been proposed defining four groups of cLIS differing from each other by genetics and neuropathologic features of the cerebral cortex, cerebellum, and brainstem.3 1) LIS1 mutations and MDS show a four-layered cortex predominantly posterior with white matter heterotopia in the cerebellum, a normal three-layered cerebellar cortex, and normal to small pons. 2) DCX mutations show a four-layered cortex similar to LIS1 but predominantly anterior; cerebellar and pontine abnormalities are similar to those in LIS1 mutations. 3) ARX mutations have a three-layered cortex; the cerebellum is normal, but the pons is small due to hypoplastic corticospinal tracts. 4) Lissencephaly with a two-layered cortex, diffuse involvement of the brain surface with no gradient; the cerebellum is extremely hypoplastic with pontine and medullary disorganization. No RELN-like mutations were studied. The authors divided these lissencephalies into two groups: LIS1 and DCX were classified as cLIS, whereas ARX and two-layered lissencephalies were classified as variant lissencephaly (vLIS).3
Numerous genes have been identified in association with the CBSC (also called dystroglycanopathies).7–12 Most affected patients have symptoms of congenital muscular dystrophies (CMDs) with CNS involvement. Among these disorders, three phenotypes are well defined: Fukuyama congenital muscular dystrophy (FCMD), Walker–Warburg syndrome (WWS), and muscle–eye–brain disease (MEB).1,7–11 These phenotypes have been associated with mutations of multiple genes involved in O-glycosylation of α-dystroglycan, including FCMD, FKRP, POMT1, POMT2, LARGE, and POMGnT1.12
The aim of our study was to determine the involvement of the midbrain and hindbrain (MHB) in these disorders, and to determine whether a correlation exists between the cerebral malformation and the MHB characteristics as determined by MRI.
METHODS
MRI scans or portions of MRI scans of 120 patients were retrospectively reviewed. The scans were acquired from the private teaching collection of the senior author (acquired over 22 years), from the teaching file of the radiology department at our institution (searching using lissencephaly, agyria, pachygyria, band heterotopia, cobblestone malformation, and congenital muscular dystrophy as key words), and from MRIs reviewed for a study of the genetics of epilepsy. MRIs were only included if good quality images of the cerebrum (to assess the type and severity of the cortical malformation) and posterior fossa were available as determined by a consensus of the authors. Most studies included T1-weighted and T2-weighted images with scans performed in at least two planes. Four cases were excluded because only CT images were available and, therefore, assessment of the posterior fossa content was suboptimal. Five others were excluded because the posterior fossa structures were not assessed on magnetic resonance (MR) images available. The remaining 111 MRI studies were analyzed. Patients were aged 1 day to 32 years (mean 5 years 4 months; median 2 years) at the time of MRI, including 54 men, 41 women, and 16 with unavailable sex information. The MRIs included sagittal and axial T1-weighted, axial, and coronal T2-weighted images with slices of 3- to 5-mm thickness. Cases were classified as “lissencephalies” in the database according to the diagnosis based on brain MRI features or known genetics. Clinical information was not often available; when available, it was usually brief, reporting mental retardation and seizures and, sometimes, family history. After reviewing the brain involvement, the cases were reclassified according to known mutation (LIS1, DCX, ARX, VLDLR, RELN, MEB, WWS) or mutation phenotype (LIS1-P, DCX-P, RELN-P, ARX-P, VLDLR-P) according to characteristic MR features1; many of the patients were imaged before testing for specific mutations was available and were not able to be found; therefore, the number with a confirmed genetic diagnosis is low. Phenotypes were determined as follows:
LIS1-P: agyria (absence of gyri) or pachygyria (few broad and flat gyri) more severe posteriorly (parietal and occipital lobes) than anteriorly with a thick cortex (10–15 mm), smooth brain surface, reduced white matter, and cell-sparse zone between a thin outer layer cortex and a thick inner layer of gray matter (figure 1A).
DCX-P: the same features as in LIS1-P but more severe in anterior brain (frontal or frontocentral regions). Band heterotopia that was more severe in the frontal and frontoparietal regions was included in this group (figure 1C).
RELN-P: thickened cortex (8–10 mm) with too few sulci, decreased hippocampal rotation, no cell-sparse zone.
ARX-P: moderately thick cortex (5–7 mm) with undersulcation most severe in temporal and occipital lobes, absence of corpus callosum, and small, dysplastic basal ganglia. No ARX-P were identified in our database.
Not determined (ND): no definite genetic diagnosis and features too non specific to propose a “probable” genetic diagnosis.

Figure 1 Classic and variant lissencephalies
(A and B) Patient 2 with LIS1 mosaicism. Axial T2-weighted image (WI) (A) shows a pachygyria with a gradient that is worse posteriorly. The inner cortex is thick and separated from the outer thin layer (black arrowheads) by a cell-sparse zone (white arrowheads) that has signal intensity of myelinated white matter with some areas of hyperintensity. Sagittal T1WI (B) shows the smooth surface of the brain involving the posterior frontal, parietal, and occipital cortices (white arrowheads). In the posterior fossa, midbrain and hindbrain appear normal. The vermis is normal with visible primary fissure (short arrow) and prepyramidal fissure (long arrow) defining three parts in equal proportions. (C and D) Patient 34 with DCX mutation. Axial T1WI (C) shows a thick cortex with few broad gyri (white arrowheads) that are worse anteriorly. On the sagittal T1WI (D), the inferior vermis (arrowheads) appears small compared with the anterior vermis (limited posteriorly by the primary fissure [arrow]). The other posterior fossa structures are normal. (E) Patient 61 with presumed RELN mutation having the same brain magnetic resonance features as his female sibling (patient 59) in whom RELN mutation was found. On sagittal T1WI, pachygyria is worse anteriorly, the midbrain (M) is small, and the pons (P) is hypoplastic, resulting in the flattening of its ventral aspect (double arrows). The medulla (m) is mildly hypoplastic, whereas the vermis is severely hypoplastic. (F) Patient 54 with ARX mutation. Sagittal T1WI shows agenesis of the corpus callosum with prominent anterior commissure (arrowheads), small basal ganglia (arrow), hypoplastic midbrain (M), pons (P) and medulla (m), and hypoplastic and dysplastic vermis (V). (G) Patient 89, from a Hutterite family, having dysequilibrium syndrome and VLDLR mutation. Sagittal TIWI shows same features of midbrain and hindbrain involvement as in RELN mutation (A), including a severe hypoplasia of the vermis (V) and cerebellar hemispheres.
The VLDLR mutation was proposed (VDLR-P) for patients having the so-called dysequilibrium syndrome.13,14 Patients with cLIS (LIS1, LIS1-P, DCX, DCX-P) were designated as cLIS, and those with ARX, RELN, RELN-P, and ND were designated as vLIS. All cases with defined mutations of O-glycosylation genes (FKRP, POMGnT1) and those without defined mutation (CMD-P) but features of CBSC on brain MRI were classified as CBSC. Cases not fitting the previously defined groups were considered to be a new group. We did not include CMD with Merosin deficiency in this study, because it does not manifest brain features of CBSC and is not of the group of disorders previously called type 2 lissencephaly.15
After this initial classification, the posterior fossa in each case was carefully reviewed with separate attention to the midbrain, the pons, the medulla, the cerebellar vermis, and the cerebellar hemispheres. Morphologic features were then recorded according to a systematic visual analysis as follows:
Midbrain: dorsal–ventral (DV) and rostrocaudal (RC) size, tectal size, and collicular morphology.
Pons and medulla: DV and RC size. Hypoplasia refers to loss of pontine anterior curvature and small ventral pons.
Cerebellar vermis: Anteroposterior (AP) and RC size. Hypoplasia refers to a small vermis showing a few or no fissures and no identifiable prepyramidal fissure; dysplasia refers to abnormal foliation or disorientation of fissures. The presence or absence of cysts was noted.
Cerebellar hemispheres: overall size and fissures. Hypoplasia, dysplasia, and cysts were defined with same criteria as for the vermis.
The degree of involvement was further characterized as mild or severe, based on consensus by the authors.
For statistics, a score was assigned for each main feature: normal = 0; mildly hypoplastic (including inferior vermis hypoplasia) = 1; severely hypoplastic (including vermian hypoplasia) = 2; dysplastic (including cerebellar and vermian cysts as well as pontomedullary “kink”) = 3. To assess the differences between defined groups of patients based on MHB features and to evaluate the correlation between the brain agyria/pachygyria and MHB involvement, nonparametric statistical tests were used, Kruskal–Wallis test for multiple group comparison and χ2 McNemar test (Statistica version 7.1, StatSoft France, 2005, www.statsoft.fr). A p value less than 0.05 was considered significant.
RESULTS
Table e-1 (on the Neurology® Web site at www.neurology.org) shows all patients included in the study listed by number, sex, and age, and classified according to lissencephaly groups and genotype/phenotype. For each patient, corresponding forebrain, midbrain, and hindbrain involvement are reported.
Table 1 summarizes the results of the statistical tests. Only significant p values are reported to show group differences. The discriminating MR feature makes the difference between groups for each MHB structure. Significant differences were found between cLIS and vLIS, and between cLIS and CBSC. Moreover, according to the predefined score assignment, we found a severity gradient of MHB involvement.
Table 1 Statistical analyses

Normal mid-hindbrain was found in 85% of cLIS patients, including all LIS1, 60% of LIS1-P, 50% of DCX, and 58% of DCX-P (figure 1, A and B). Moderate mid-hindbrain involvement corresponded to isolated inferior vermis hypoplasia (IVH), small or hypoplastic vermis, cerebellar hemispheres, midbrain, pons, or medulla. Among the cLIS patients with IVH, 75% were either DCX or DCX-P (in equal proportions) (figure 1, C and D). Severe mid-hindbrain involvement included severe hypoplasia or dysplasia of the vermis and cerebellar hemispheres, cysts within the vermis and cerebellar hemispheres, midbrain hypoplasia or dysplasia, severe hypoplasia of the pons and medulla, pontomedullary kink, and pontine midline cleft. These features were remarkably predominant among the vLIS (figure 1, E–G) and CBSC groups, differentiating them significantly from cLIS groups (table 1). Interestingly, all patients with WWS phenotype had a ventral midline pontine cleft associated with severe hindbrain hypoplasia (figure 2).

Figure 2 Cobblestone complex in patient 107 with WWS phenotype
(A) Coronal T2-weighted image (WI) showing typical magnetic resonance appearance of cobblestone complex with irregular inner layer of ectopic neurons (black arrowheads) and overmigrated neurons at the surface of the cortex (white arrowheads) and between both fibroglial bundles crossing the white matter radially, hydrocephalus, and profound hypomyelination. Midbrain and hindbrain (white stars on cerebellar hemispheres) are severely hypoplastic with near total absence of the vermis and pontine midline cleft (black arrow). (B) Sagittal T1WI showing the characteristic dysplastic large tectum (arrowheads), deformity of the mid-hindbrain junction (pontomedullary “kink,” white line), and near total absence of the vermis with very hypoplastic cerebellar hemisphere (white star).
We found an association between agyria/pachygyria of the entire brain (with or without gradient) and severe MHB involvement (p = 0.0029).
Agenesis of corpus callosum (ACC) (1.27% of all lissencephalies) was always associated with severe involvement of the vermis; it was found in vLIS with no defined genotype, as expected in all ARX mutations, and in two patients with complete agyria and undulating band heterotopia (figure 3, A and B). In addition, periventricular nodular heterotopia was seen in patients with normal to mild MHB involvement and subependymal linear heterotopia (SELH) in two patients with near total absence of the vermis and hypoplastic pons (figure 3, C and D).

Figure 3 Not-determined lissencephalies
(A and B) Patient 67 classified as not determined (ND). Axial T2-weighted image (WI) (A) shows entire brain agyria with very thin outer cortex layer (black arrows) and undulating gray matter heterotopia (white arrows) surrounding the enlarged lateral ventricles (LVs). Sagittal T1WI (B) displays callosal hypogenesis with small posterior genu (white arrows) but absent body (arrowheads), with small midbrain (M), pons (P), and medulla (m), and enlarged dysplastic vermis. (C and D) Patient 78 classified as ND. Axial T2WI (C) shows agyria of the entire brain with subependymal linear gray matter heterotopia bilaterally (white arrowheads) and marked ventriculomegaly in this microcephalic patient. Sagittal T1WI (D) displays callosal agenesis and extremely hypoplastic midbrain (M), pons (P), and medulla (m). The entire cerebellum is nearly absent.
DISCUSSION
This study used a database including many patients with lissencephalies to perform a systematic review of MHB structures in a large series including many types of lissencephalies. More than a simple, exploratory approach to the distribution of cerebellar and brainstem abnormalities in lissencephalies, this study tested whether a relationship existed between the type and extent of supratentorial involvement and the involvement of the mid-hindbrain, and whether, as hypothesized, the distribution among different defined groups could help to predict the mutation by associating the pattern of cerebral involvement with mid-hindbrain involvement.
The patterns of cerebral involvement have been well documented in human lissencephalies and have helped to classify them and to correlate them with histology and genetics.1–10 Consistent differences were found in the gyral patterns among cLISs, with the malformation more severe posteriorly in individuals with LIS1 mutations and more severe anteriorly in individuals with DCX mutations.4 These observations were later correlated with neuropathologic findings as mentioned in the introduction.3 The pattern of cortical brain involvement in cLIS is typically different from what is seen in “cobblestone malformations” associated with defects in O-glycosylation of α-dystroglycan.7–11,16 The agyria/pachygyria complex seen in cLIS and vLIS is mainly the result of incomplete migration of neurons,2–6 whereas CBSC is primarily a result of overmigration of neurons, many of which pass through gaps in the glial limiting membrane.7–11 Involvement of the cerebellum and the pons has been described in cLISs that were classified according to mutation analysis. Some authors found hypoplasia of the cerebellar vermis to be more common in patients with DCX mutations, whereas both DCX and LIS1 sometimes had normal cerebellum and brainstem.4 The new pathologic classification recently proposed included cerebellum and brainstem abnormalities associated with cLIS (LIS1 and DCX) and vLIS (ARX and two-layered cortex).3 They found the most severe involvement of the hindbrain (hypoplasia) in vLIS group and, most notably, in two-layered cortex. In our series, the mid-hindbrain in cLIS ranged from normal to small (more frequently among DCX and DCX-P patients) in agreement with previous studies. Our observations were similar, revealing an association between the extent of brain involvement and severity of MHB abnormalities (p = 0.0029). Marked to severe MHB hypoplasia was most often found in patients with pachygyria involving the entire cerebrum, relatively thin cortex (5 mm or less), and no cell-sparse zone (vLIS-ND, likely Forman’s two-layered cortex lissencephaly). As discussed by previously cited authors,3 this association could be related to extensive impairment of cortical fibers projecting to the pons as well as hypoplasia of mid-hindbrain nuclei.
Of interest, we found a high rate of isolated inferior vermis hypoplasia associated with DCX. This sign is probably to be considered as the smallest degree of MHB involvement and would give an interesting clue for differential diagnosis if the finding is substantiated on subsequent studies. MHB was severely hypoplastic in ARX patients (with dysmorphic features of the vermis) and in patients with RELN mutations. Different phenotypes of lissencephaly with cerebellar hypoplasia (LCH) are associated with LIS1, DCX, and RELN genes.6,17
All patients initially classified as VLDLR and VLDLR-P had MHB features similar to patients with RELN mutation. Consequently, the two groups had the same overall score for MHB involvement (table 2). Severe hypoplasia of the vermis was found in all RELN (two cases) and in five of eight VLDLR and VLDLR-P patients. These were grouped together as vLIS. The VLDLR mutation has been described as responsible for the so-called dysequilibrium syndrome and can therefore be presumed in any patient with characteristic clinical and brain MRI features.18
Table 2 New classification

Table 2Continued
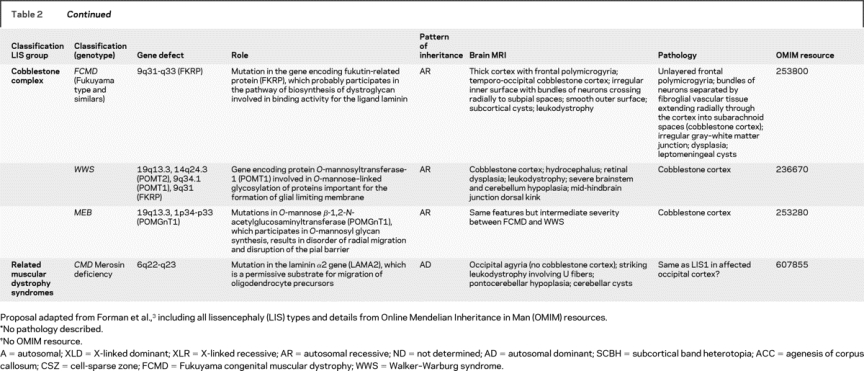
CBSC was formerly called type 2 lissencephaly.12 It is the main CNS feature of CMDs with O-glycosylation defects of α-dystroglycan, even though it is difficult to differentiate the genetic causes on the basis of brain MRI because of overlapping phenotypes.19–23 Cerebral, cerebellar, and brainstem abnormalities have been well documented in the major phenotypes: FCMD, WWS, and MEB. WWS displays severe cerebral cortical dysplasia, hypomyelination, dysplastic tectum, pontomedullary kink, severe hypoplasia of the vermis, and dysplastic cerebellar hemispheres.1,10 A ventral midline cleft of the ventral pons is often associated with MEB and WWS and could also be considered among their typical MR features. The midline cleft is probably due to reduced number of crossing pontine axons, possibly resulting from impaired tangential migration of pontine nuclei, as demonstrated in mice with mutation of Large.24,25
We found that callosal agenesis is not uncommon among patients with lissencephaly. ACC is among the characteristic features of ARX-associated lissencephaly.26,27 The frequent association of ACC with vLIS-ND patients in our series suggests considering this feature as a supplementary criteria to differentiate cLIS from vLIS. It is important to note, however, that ACC may also be found in other types of lissencephalies.28 Interestingly, the cerebellar vermis was dysplastic or severely hypoplastic in 13 of 14 patients with ACC; it will be interesting to see whether this association holds up in other series. A recent study of callosal agenesis associated with DCX found that the doublecortin protein might be involved in the early stages of corpus callosum formation29; however, none of the DCX or DCX-P patients in this study had ACC.
This study is limited by the small number of cases with established genetic diagnoses. An attempt was made to increase this number by assigning a presumed genetic classification based on established cerebral MRI features in different lissencephaly groups and correlation of imaging features with known pathologic features such as cell-sparse zones, associated callosal anomalies, and cortical thickness. This is obviously not scientifically rigorous and may be considered an artificial classification. However, we observed relative homogeneity of MHB involvement both within groups with presumed classification (LIS1-P) and between groups with known and presumed mutations of the same gene (DCX, RELN, and VLDLR). This homogeneity and our statistical results emboldened us to group together those with presumed and proved mutations for purposes of correlation. Results of these groupings are shown in the two first columns of table 2. For each structure analyzed, a gradient of involvement was found to be associated with each presumed genotype (mutation phenotype) (table e-2). We suggest that these presumed groupings might be useful to determine which tests for specific gene mutations should be performed in such patients.
Based on the data in this project and that in the literature, we propose a classification similar to that recently proposed,3 separating cLIS (LIS1 and DCX) from vLIS (RELN and ARX) (table 2). We included VLDLR and some ND cases with markedly atypical MR features, namely ND1, ND2, and ND3, within the vLIS group. ND3 refers to a case with microcephaly, paracentral pachygyria, and ACC resembling Baraitser–Winter syndrome, but other phenotypes may be associated with this syndrome.30 ND2 refers to two cases with undulating band heterotopia (a configuration that must be extremely rare, because we found no previous reports) and ACC. ND1 refers to two cases with complete agyria, SELH, ACC, and severe MHB involvement. Further investigations or more cases are needed to determine whether the ND groups should be classified with vLIS or in still another category.
As noted in the previous paragraph, it was difficult to determine which entities to include in the vLIS category and how to classify them. More than 80% of vLIS patients did not have characteristic imaging findings (ND). The protein products of VLDLR and RELN act in the same pathway but are coded by different genes that have different patterns of inheritance, so they were included as separate malformations. Based on a previously cited work, we included two-layered lissencephaly as part of the vLIS category.3
Another weakness of this study is a lack of clinical data that could help clinicians to better identify patients with these malformations. It would also have helped to better classify those patients who were ultimately assigned to the ND category. Despite this obvious deficiency, important information has emerged from this study for both radiologists and other physicians interested in brain development. This study expands the categories of lissencephalies and adds to the knowledge of associated malformations. This information will, hopefully, lead to further studies of the molecular disorders involved in the many brain anomalies associated with agyria and pachygyria and, ultimately, a better understanding of the mechanisms by which the brain is formed.
AUTHOR CONTRIBUTIONS
P.J.-T. conducted the statistical analysis.
ACKNOWLEDGMENT
The authors thank Drs. Chistopher Walsh, William Dobyns, Daniela Pilz, Sean Bryant, and Naci Koçer for the MRI scans that they kindly contributed to this work.
Supplementary Material
Address correspondence and reprint requests to Dr. Patrice Jissendi-Tchofo, Radiology Department, Erasme Hospital, Route de Lennik 808, B-1070 Brussels, Belgium jissendi@gmail.com
Supplemental data at www.neurology.org
Editorial, page 394
e-Pub ahead of print on November 19, 2008, at www.neurology.org.
Supported by R37 NS035129 from the National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD.
Disclosure: The authors report no disclosures.
Received March 7, 2008. Accepted in final form July 16, 2008.
REFERENCES
- 1.Barkovich AJ. Congenital malformations of the brain and skull. In: Pediatric Neuroimaging, 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005. [Google Scholar]
- 2.Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet 2003;12:R89–R96. [DOI] [PubMed] [Google Scholar]
- 3.Forman MS, Squier W, Dobyns WB, et al. Genotypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol 2005;64:847–857. [DOI] [PubMed] [Google Scholar]
- 4.Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology 1999;53:270–277. [DOI] [PubMed] [Google Scholar]
- 5.Dobyns WB, Reiner O, Carrozzo R, et al. Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 1993;270:2838–2842. [DOI] [PubMed] [Google Scholar]
- 6.Ross ME, Swanson K, Dobyns WB. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics 2001;32:256–263. [DOI] [PubMed] [Google Scholar]
- 7.van der Knaap MS, Smit LM, Barth PG, et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol 1997;42:50–59. [DOI] [PubMed] [Google Scholar]
- 8.Aida N, Tamagawa K, Takada K, et al. Brain MR in Fukuyama congenital muscular dystrophy. AJNR Am J Neuroradiol 1996;17:605–613. [PMC free article] [PubMed] [Google Scholar]
- 9.Valanne L, Pihko H, Katevuo K, et al. MRI of the brain in muscle-eye-brain (MEB) disease. Neuroradiology 1994;36:473–476. [DOI] [PubMed] [Google Scholar]
- 10.Mercuri E, Topaloglu H, Brockington M, et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol 2006;63:251–257. [DOI] [PubMed] [Google Scholar]
- 11.Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis 2006;29:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barkovich AJ, Kuzniecky RI, Jackson GD, et al. A developmental and genetic classification for malformations of cortical development. Neurology 2005;65:1873–1887. [DOI] [PubMed] [Google Scholar]
- 13.Schurig V, Orman AV, Bowen P. Nonprogressive cerebellar disorder with mental retardation and autosomal recessive inheritance in Hutterites. Am J Med Genet 1981;9:43–53. [DOI] [PubMed] [Google Scholar]
- 14.Boycott KM, Flavelle S, Bureau A, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet 2005;77:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leite CC, Lucato LT, Martin MGM. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol 2005;35:572–579. [DOI] [PubMed] [Google Scholar]
- 16.Philpot J, Pennock J, Cowan F, et al. Brain magnetic resonance imaging abnormalities in merosin-positive congenital muscular dystrophy. Eur J Paediatr Neurol 2000;4:109–114. [DOI] [PubMed] [Google Scholar]
- 17.Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 2000;26:93–96. [DOI] [PubMed] [Google Scholar]
- 18.Glass HC, Boycott KM, Adams C, et al. Autosomal recessive cerebellar hypoplasia in the Hutterite population. Dev Med Child Neurol 2005;47:691–695. [DOI] [PubMed] [Google Scholar]
- 19.Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 2003;12:2853–2861. [DOI] [PubMed] [Google Scholar]
- 20.Beltran-Valero de Bernabé D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet 2004;41:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 2005;42:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Reeuwijk J, Maugenre S, van den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 2006;27:453–459. [DOI] [PubMed] [Google Scholar]
- 23.van Reeuwijk J, Grewal PK, Salih MA, et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 2007;121:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu Q, Crandall JE, Luo T, et al. Defects in tangential neuronal migration of pontine nuclei neurons in the Largemyd mouse are associated with stalled migration in the ventrolateral hindbrain. Eur J Neurosci 2006; 23:2877–2886. [DOI] [PubMed] [Google Scholar]
- 25.Barkovich AJ, Millen KJ, Dobyns WB. A developmental classification of malformations of the brainstem. Ann Neurol 2007;62:625–639. [DOI] [PubMed] [Google Scholar]
- 26.Dobyns WB, Berry-Kravis E, Havernick NJ, et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet 1999;86:331–337. [PubMed] [Google Scholar]
- 27.Paul LK, Brown WS, Adolphs R, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 2007;8:287–299. [DOI] [PubMed] [Google Scholar]
- 28.Dobyns WB. Absence makes the search grow longer. Am J Hum Genet 1996;58:7–16. [PMC free article] [PubMed] [Google Scholar]
- 29.Kappeler C, Dhenain M, Tuy FPD, et al. Magnetic resonance imaging and histological studies of corpus callosal and hippocampal abnormalities linked to doublecortin deficiency. J Comp Neurol 2007;500:239–254. [DOI] [PubMed] [Google Scholar]
- 30.Ganesh A, Al-Kindi A, Jain R, Raeburn S. The phenotypic spectrum of Baraitser-Winter Syndrome: a new case and review of literature. J AAPOS 2005;9:604–606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.