

Abstract
Objective:
To compare patterns of gray matter loss in subjects with mutations in the progranulin (PGRN) gene to subjects with mutations in the microtubule-associated protein tau (MAPT) gene.
Methods:
We identified all subjects seen at the Mayo Clinic, Rochester, MN, who had screened positive for mutations in PGRN or MAPT and had a head MRI. Twelve cases with mutations in the PGRN gene were matched by time from disease onset to scan to 12 subjects with mutations in the MAPT gene. Voxel-based morphometry was used to assess patterns of gray matter loss in the PGRN and MAPT groups compared to a control cohort, and compared to each other. MAPT subjects were younger than the PGRN subjects; therefore, each group was also compared to a specific age-matched control group.
Results:
Both PGRN and MAPT groups showed gray matter loss in frontal, temporal, and parietal lobes compared to controls, although loss was predominantly identified in posterior temporal and parietal lobes in PGRN and anteromedial temporal lobes in MAPT. The MAPT group had greater loss compared to healthy subjects of the same age than the PGRN subjects when compared to healthy subjects of the same age. The MAPT subjects showed greater gray matter loss in the medial temporal lobes, insula, and putamen than the PGRN subjects.
Conclusion:
These results increase understanding of the biology of these disorders and suggest that patterns of atrophy on MRI may be useful to aid in the differentiation of groups of PGRN and MAPT mutation carriers.
GLOSSARY
- AD
= Alzheimer disease;
- ADPR
= Alzheimer's Disease Patient Registry;
- ADRC
= Alzheimer's Disease Research Center;
- bvFTD
= behavioral variant frontotemporal dementia;
- bvFTD+P
= bvFTD with parkinsonism;
- CBS
= corticobasal syndrome;
- CDR-SB
= Clinical Dementia Rating scale sum of boxes;
- DRS
= Dementia Rating Scale;
- FTLD
= frontotemporal lobar degeneration;
- MAPT
= microtubule-associated protein tau;
- MCI
= mild cognitive impairment;
- MMSE
= Mini-Mental State Examination;
- PGRN
= progranulin;
- PPA
= primary progressive aphasia;
- STMS
= Short Test of Mental Status;
- VBM
= voxel-based morphometry.
Frontotemporal lobar degeneration (FTLD) is a heterogenous progressive disorder that consists of a number of different clinical and pathologic variants. Approximately 40% of subjects have a family history with an autosomal dominant pattern of inheritance.1,2 Two of the most commonly mutated genes are the microtubule-associated protein tau (MAPT) gene3,4 and the progranulin (PGRN) gene5,6 that are both located on chromosome 17q21. Mutations in these genes account in combination for approximately 10–20% of all FTLD subjects.7,8 Mutations in PGRN are associated with deposition of TAR DNA-binding protein 43 (TDP-43)- and ubiquitin-positive neuronal inclusions in the frontotemporal cortices and hippocampus (FTLD-U).9,10 In contrast, mutations in MAPT are associated with deposits of the hyperphosphorylated protein tau in the form of intraneuronal neurofibrillary tangles or Pick bodies in the frontal and temporal cortices of the brain.11,12 While both PGRN and MAPT mutations are associated with a clinical diagnosis of frontotemporal dementia, there is some suggestion that the specific clinical phenotypes associated with these mutations differ.8,13–15
Using an unbiased voxel-level technique called voxel-based morphometry (VBM), we previously demonstrated patterns of frontal, temporal, and parietal gray matter loss in subjects with mutations in the PGRN gene, and demonstrated that these patterns are more severe and widespread than those observed in sporadic PGRN-negative FTLD-U cases.16 At an individual level, patterns of atrophy in PGRN have also been shown to be highly asymmetric.13,14,17,18 Voxel-based morphometry has also been applied to subjects with mutations in MAPT, particularly those with a IVS10 + 16C>T mutation, and has revealed gray matter loss predominantly in the temporal lobes.19 Atrophy in MAPT carriers has been shown to be relatively symmetric.13,14 These studies therefore suggest anatomic differences between PGRN and MAPT carriers.
The aim of this study was to use VBM to compare the patterns of gray matter loss in MAPT and PGRN mutation carriers.
METHODS
Subject selection.
We identified all subjects seen at Mayo Clinic, Rochester, MN, who had screened positive for mutations in either PGRN or MAPT, were symptomatic and had a volumetric MRI scan. Twelve subjects, representing six families, were identified who had screened positive for mutations in PGRN. Sequencing analyses showed that five different PGRN mutations were present in our cohort: c.154delA (p.Thr52HisfsX2), c.910_911insTG (p.Trp304LeufsX58), c.1395_1396insC (p.Cys466LeufsX46), c.1145delC (p.Thr382SerfsX30), and c.138 + 1G>A (IVS1 + 1G>A). The demographic features for these subjects are shown in table 1 and the detailed clinical features and diagnoses are shown in table 2. Clinical diagnoses were made according to consensus criteria for mild cognitive impairment,20 Alzheimer disease (AD),21 behavioral variant frontotemporal dementia (bvFTD),22 primary progressive aphasia,23 and corticobasal syndrome (CBS).24 Eight of these subjects have come to postmortem and all were given a diagnosis of FTLD-U. Imaging results have previously been published from these eight subjects.16
Table 1 Subject demographics
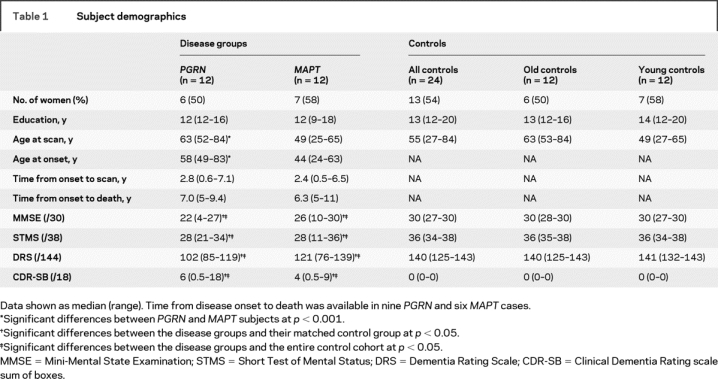
Table 2 Clinical features and diagnoses of each PGRN and MAPT subject
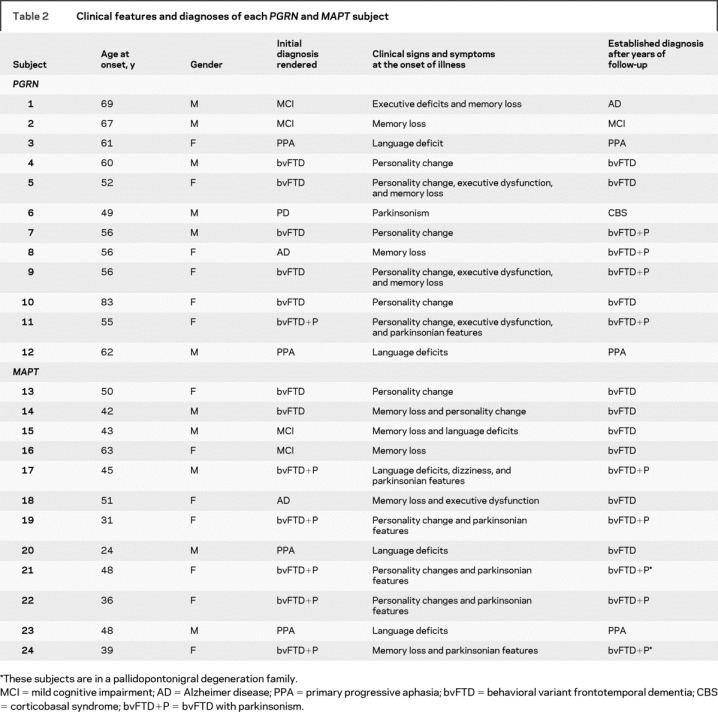
A total of 19 subjects were identified who had screened positive for mutations in MAPT. The 12 subjects with mutations in PGRN were frequency matched using time from disease onset to scan to 12 of the subjects with mutations in MAPT. In all subjects the first scan after the time of onset was used. This matching was performed to ensure that subjects in both groups had the disease for the same length of time. The 12 selected subjects represented seven families with six different MAPT mutations: c.1842T>G (p.Asn279Lys), c.1907C>T (p.Pro301L), c.1919G>A (p.Ser305Asn), c.1920 + 3G>A (IVS10 + 3G>A), c.1920 + 16C>T (IVS10 + 16C>T), and c.2170G>A (p.Gly389Arg). Subject demographics are shown in table 1 with clinical features and diagnoses shown in table 2. Four of these subjects underwent autopsy with pathologic findings of widespread tau deposition in neurons and glia.
The majority of all these cases had been prospectively studied in our Alzheimer's Disease Research Center (ADRC) or Alzheimer's Disease Patient Registry (ADPR). The historical records of all cases were reviewed by an expert in neurodegenerative diseases (K.A.J.) for the abstraction of data, including gender, age at onset, illness duration, Mini-Mental State Examination (MMSE),25 Short Test of Mental Status (STMS),26 Dementia Rating Scale (DRS),27 and the Clinical Dementia Rating sum of boxes score (CDR-SB)28 (table 1). Informed consent was obtained for participation in the studies, which were approved by the Mayo Institutional Review Board.
Each of the 24 subjects was then also matched by age and gender to a cognitively normal control subject. All control subjects were prospectively recruited into the ADRC or the ADPR and were identified from the ADRC/ADPR database. Control subjects were cognitively normal individuals that had been seen in internal medicine for routine physical examinations and asked to enroll in the ADRC or ADPR. All subjects were then evaluated by a neurologist to verify the normal diagnosis. Controls were identified as individuals who 1) were independently functioning community dwellers, 2) did not have active neurologic or psychiatric conditions, 3) had no cognitive complaints, 4) had a normal neurologic and neurocognitive examination, and 5) were not taking any psychoactive medications in doses that would affect cognition.
Genetic analysis.
Exons 0–13 and the 3′ untranslated region of the PGRN gene were amplified by PCR using our previously published primers and protocol.5,7 Analysis of MAPT exons 1, 7, and 9–13 was also performed using primers and conditions that were previously described.3 For both genes, the PCR amplicons were purified using the Multiscreen system (Millipore, Billerica, MA) and then sequenced in both directions using Big Dye chemistry following manufacturer's protocol (Applied Biosystems, Foster City, CA). Sequence products were purified using the Montage system (Millipore) before being run on an ABI 3730 DNA Analyzer. Sequence data were analyzed using either SeqScape or Sequenom software.
Voxel-based morphometry.
A standardized imaging protocol was performed on all subjects that included a coronal T1-weighted three-dimensional volumetric sequence with 124 contiguous partitions and 1.6 mm slice thickness (22 × 16.5 cm field of view, 25° flip angle). Patterns of cerebral atrophy were assessed using the automated and unbiased technique of VBM.29 An optimized method of VBM was applied using both customized templates and prior probability maps,30 implemented using SPM2 (http://www.fil.ion.ucl.ac.uk/spm). The processing steps were performed as previously described.16 Briefly, all images were normalized to a customized template created from all subjects in the study. The spatial normalization was optimized by normalizing the gray matter images to the customized gray matter template. Images were segmented using customized prior probability maps, modulated, and smoothed with an 8 mm full-width at half-maximum smoothing kernel.
Three types of VBM comparisons were performed. First, statistical comparisons were performed between the MAPT mutation carriers and the entire control group and between PGRN mutation carriers and the entire control group. Adjustment for the potential confounders of age and gender was performed by including them as nuisance variables in the statistical model. This analysis was also repeated assessing only the PGRN and MAPT subjects with a clinical diagnosis of bvFTD (n = 7 PGRN and n = 11 MAPT) in order to account for potential clinical confounders. However, since the MAPT subjects were, as a group, approximately 14 years younger than the PGRN mutation carriers, we also divided the control population into two groups: 1) those subjects who were individually age-matched to the MAPT subjects (i.e., young control group), and 2) those subjects who were individually age-matched to the PGRN subjects (i.e., old control group). We therefore performed a second type of statistical comparison between the PGRN group and the old controls, and also the MAPT group and the young controls. Once again age and gender were included in the statistical models as nuisance variables. This analysis was important to determine how much of the atrophy in the PGRN group was due to age rather than the disease itself. The characteristics of the PGRN and MAPT mutation carriers and the control cohorts are shown in table 1. These results were assessed after correction for multiple comparisons using the false discovery rate at p < 0.01. Finally, direct comparisons were also performed between the MAPT carriers and the PGRN carriers, including age and gender as nuisance variables. The results of this analysis were assessed at a more lenient statistical threshold of p < 0.001 uncorrected for multiple comparisons due to the hypothesis-driven nature of this comparison.
Statistics.
Statistical analyses were performed utilizing JMP computer software (JMP Software, version 6.0.0; SAS Institute Inc., Cary, NC) with α set at 0.05. Gender ratios were compared across groups with χ2 test. Kruskal-Wallis test was used to compare continuous data across the three groups.
RESULTS
The subject demographics are shown in table 1. The subjects with a mutation in MAPT were significantly younger at disease onset and scan than the subjects with a mutation in PGRN, although there was no difference between the groups in time from disease onset to scan, disease duration (time from onset to death), gender ratio, or education. The cognitive test scores were significantly different between the disease groups and the control groups, but there were no significant differences between the PGRN and MAPT groups.
Figure 1 and table e-1 (on the Neurology® Web site at www.neurology.org) show the patterns of gray matter loss in the PGRN and MAPT groups when compared to the entire control cohort. Both disease groups showed widespread patterns of gray matter loss involving frontal, temporal, and parietal lobes, compared to controls. However, the distribution of gray matter loss differed across the two disease groups. In the PGRN group, gray matter loss was identified predominantly in posterior temporal regions, with severe involvement of parietal lobes and additional involvement of the posterior cingulate gyrus and precuneus. In the MAPT group, the gray matter loss was focused predominantly in the temporal lobes, particularly anterior and medial temporal lobes, with less involvement of the parietal lobes and no involvement of cingulate gyrus or precuneus. The cerebellum was also involved in the MAPT group. Very similar patterns of gray matter loss were observed in both the PGRN and MAPT groups when only subjects with bvFTD were assessed (figure e-1).

Figure 1 Patterns of gray matter loss in subjects with mutations in PGRN and subjects with mutations in MAPT compared to the entire control cohort
Results are shown on three-dimensional renders of the brain and coronal slices through the frontal, temporal, and parietal lobes.
Figure 2 shows the patterns of gray matter loss in the PGRN and MAPT groups when each is compared to a specific age-matched control group. It is notable from this figure that the PGRN group shows a less severe and widespread pattern of gray matter loss compared to the age-matched old controls than the MAPT group compared to the age-matched young controls. The PGRN group shows gray matter loss mainly restricted to the posterior temporal lobe and parietal lobe compared to the old controls. The patterns of loss observed in the MAPT group compared to the young controls are similar to those identified in figure 1, with widespread involvement of the frontal, temporal, and parietal lobes, with particular focus on the anterior and medial temporal lobes, and some involvement of the cerebellum.

Figure 2 Patterns of gray matter loss in subjects with mutations in PGRN compared to age-matched old controls, and subjects with mutations in MAPT compared to age-matched young controls
Results are shown on three-dimensional renders of the brain and coronal slices through the frontal, temporal, and parietal lobes.
Direct comparisons were performed between the two groups. The subjects with mutations in MAPT showed greater gray matter loss predominantly in the temporal lobes than the subjects with mutations in PGRN (figure 3). Regions of loss were identified in the bilateral amygdala, hippocampus and parahippocampal gyrus, and left inferior temporal lobe, as well as the insula and left putamen. The reverse comparison showed that no regions of greater gray matter loss were identified in subjects with mutations in PGRN compared to subjects with mutations in MAPT.

Figure 3 Regions that show greater gray matter loss in subjects with mutations in MAPT compared to subjects with mutations in PGRN
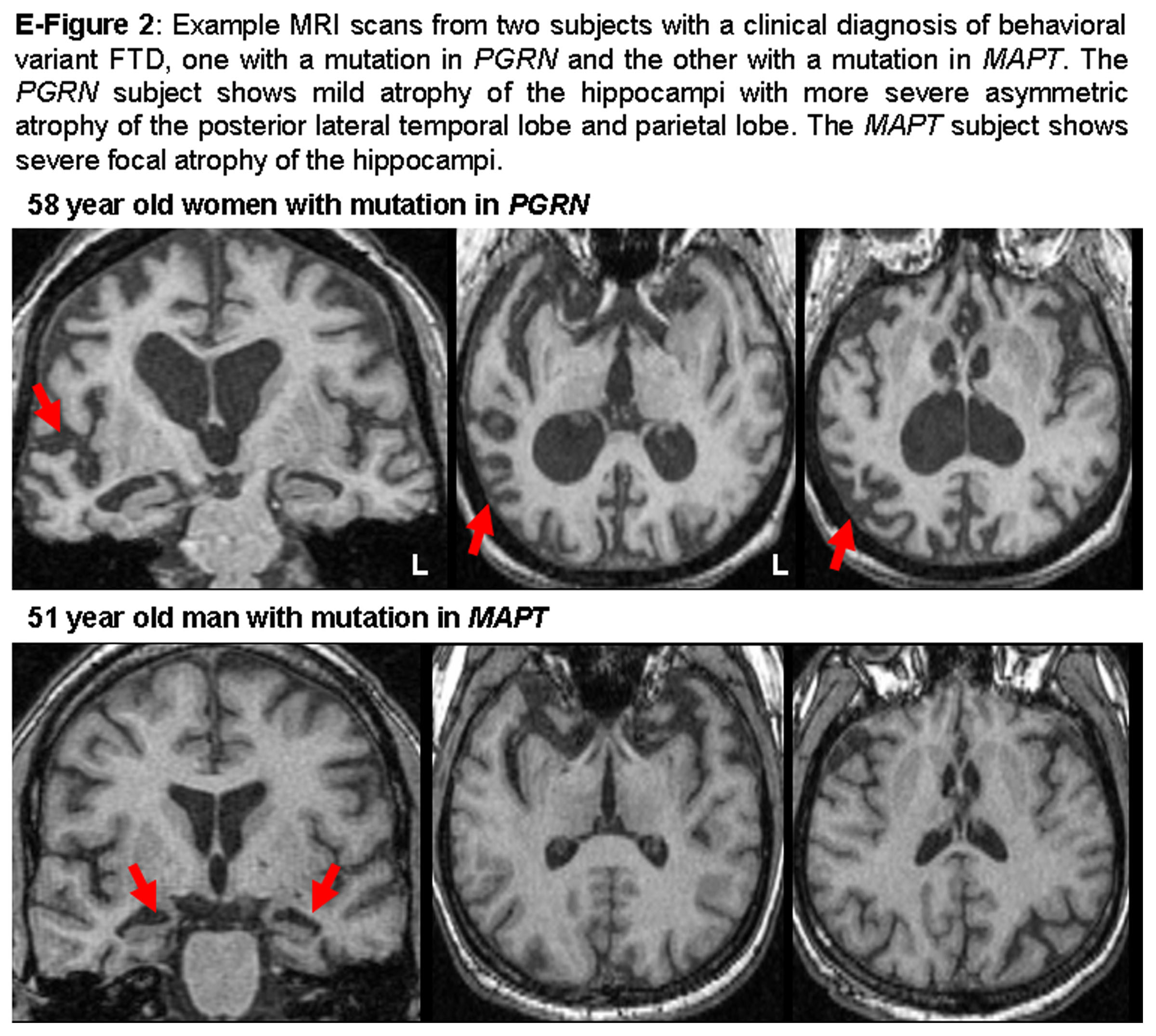
Example MRI scans from individual cases with mutations in PGRN or MAPT illustrating these observed differences in patterns of atrophy are shown in figure e-2.
DISCUSSION
In this study, we compared regional patterns of gray matter loss across subjects with mutations in MAPT and PGRN using automated and unbiased methodology. The subjects with mutations in the PGRN gene showed patterns of gray matter loss predominantly affecting the posterior temporal and parietal lobes, with additional involvement of the frontal lobes. Patterns of posterior temporal gray matter loss have previously been reported in subjects with a pathologic diagnosis of FTLD-U.19,31 Atrophy of the parietal lobes13,16–18,32 and parietal lobe dysfunction32 have also been reported to be common in subjects with mutations in PGRN. Furthermore, parietal lobe atrophy was shown in a pathologic study.9 Gray matter loss of the posterior cingulate and precuneus was also present in the subjects with PGRN mutations compared to controls, suggesting that PGRN is associated with both medial and lateral parietal lobe damage. We have previously shown that subjects with PGRN mutations show a greater degree of parietal lobe involvement than subjects with FTLD-U without a mutation in PGRN.16 The current study, however, shows that the parietal lobe is also affected in subjects with mutations in the MAPT gene and we found no differences in the parietal lobe between the PGRN and MAPT subjects on direct comparison. The involvement of the parietal lobe in PGRN concurs with the fact that subjects with PGRN can have a clinical presentation and diagnosis of AD or CBS.8,13,17,18 However, our results show that the parietal gray matter loss is not driven solely by the presence of AD and CBS phenotypes since similar patterns of loss were observed in the bvFTD subjects with mutations in PGRN. Although the parietal lobe was affected in subjects with mutations in MAPT it was involved to a lesser degree than the frontal and temporal lobes, and therefore appears to be a feature of widespread disease rather than being the focus of loss as seen in PGRN.
A widespread pattern of frontotemporal gray matter loss was observed in the subjects with mutations in the MAPT gene, although, in contrast to the PGRN subjects, the most severe regions of loss were identified in the anteromedial temporal lobes. In fact, subjects with mutations in MAPT showed greater temporal lobe atrophy, particularly involving the medial temporal lobe, than the subjects with mutations in PGRN on direct comparison. Patterns of severe temporal lobe atrophy have previously been demonstrated in studies that have investigated subjects with tau exon mutations IVS10 + 16C>T,19,33 IVS10 + 3G>A,34 and p.Asn279Lys,35,36 suggesting that MAPT mutations may predispose to temporal lobe atrophy. These findings fit with previous studies that have shown that while the majority of MAPT cases have a diagnosis of behavioral variant FTD,8,13,15,37 a high proportion show language deficits.8,15,17 Greater gray matter loss in MAPT subjects was also identified in the putamen. Reduced uptake of fluoro-l-dopa has previously been reported using PET in the striatum of subjects with pallidopontonigral degeneration and p.Asn279Lys mutations.38 While in the comparisons with controls the MAPT group appeared to show greater involvement of the cerebellum than the PGRN group, this finding did not appear on direct comparison, suggesting that it is unlikely to be an important disease-specific finding.
The MAPT subjects were however approximately 14 years younger at disease onset and scan than the subjects with PGRN mutations. This trend for younger age at onset in subjects with mutations in MAPT has been previously noted.8,13 Age was included in the main analysis as a confounding variable; however, in order to more accurately correct for this age difference, we compared the disease groups to different and specifically age-matched control subjects. This analysis showed that the MAPT subjects have greater gray matter loss compared to healthy subjects of the same age than the amount present in the PGRN subjects when compared to healthy subjects of the same age. The time from onset to scan was similar in the PGRN and MAPT groups and therefore one could infer from these cross-sectional results that the rate of gray matter atrophy may be higher in the MAPT subjects, since they have lost a greater degree of gray matter from normal in the same time. A trend for more severe atrophy in young-onset subjects has previously been demonstrated in subjects with AD.39 These results suggest the same may be true for subjects with frontotemporal dementia. However, it has also been suggested that subjects with mutations in MAPT have a strong neurodevelopmental component with lower baseline functioning,40 perhaps indicating that they also have a lower premorbid brain reserve. Longitudinal studies will be needed to solve this issue. The fact that less gray matter loss was observed when the PGRN group was compared to the old controls than when they were compared to the on average younger entire control cohort also suggests that significant age-related gray matter loss was present in the PGRN results shown in figure 1. In fact, the most striking difference in the PGRN maps between figure 1 and figure 2 was the lack of frontal lobe signal when PGRN subjects are compared with age-matched controls, suggesting that greatest gray matter loss in the old controls occurred in the frontal lobes.
The assessment of patterns and severity of gray matter loss could however be limited by the fact that subjects with mutations in PGRN typically show very asymmetric patterns of atrophy, affecting either the left or right hemisphere.13,14,17,18 In contrast, subjects with MAPT mutations typically show more symmetric patterns of atrophy.13,14 The asymmetry present in the PGRN subjects would not necessarily be evident from a VBM analysis, which averages the patterns of loss across groups of subjects. Variability in hemispheric atrophy could reduce the signal identified by the VBM analysis. The patterns of loss in each hemisphere are also more likely to be driven by fewer subjects in the PGRN group. Therefore, it will be important for these patterns of hemispheric atrophy to be confirmed in larger cohorts of subjects with mutations in PGRN.
While this is a group study and individual patterns of atrophy, and hence clinical phenotypes, may vary across these genetic types,17 these results show trends for differences in patterns of atrophy on MRI across PGRN and MAPT mutation carriers. Both groups show involvement of the frontal, temporal, and parietal lobes, but there was a trend for subjects with mutations in MAPT to show a predominance of anteromedial temporal lobe atrophy while subjects with mutations in PGRN show heavy involvement of the posterior temporal and parietal lobes. These imaging findings, in conjunction with clinical measures, may aid in the differentiation of PGRN and MAPT mutation carriers. It will be important to determine whether these patterns can be identified early in the disease course, especially in asymptomatic family members, and therefore allow the early prediction of genetic status and hence resultant pathologic outcome.
ACKNOWLEDGMENT
The authors thank Dr. Dennis W. Dickson and Dr. Joseph E. Parisi for conducting pathologic analyses.
Supplementary Material
Address correspondence and reprint requests to Dr. Keith A. Josephs, Department of Neurology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905 josephs.keith@mayo.edu.
Supplemental data at www.neurology.org
Supported by the NIH Roadmap Multidisciplinary Clinical Research Career Development Award Grant (K12/NICHD)-HD49078, NIH grants P50-AG16574, U01-AG06786, R01-AG11378, as well as the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation, the NIH Construction Grant (NIH C06 RR018898), and the Pacific Alzheimer's Research Foundation (PARF) grant C06-01. K.A.J. and Z.K.W. are also supported by Morris K. Udall PD Research Center of Excellence (NIH/National Institute of Neurological Disorders and Stroke P50 #NS40256).
Disclosure: D.S.K. has been a consultant to GE Healthcare, GlaxoSmithKline, and Myriad Pharmaceuticals, has served on a Data Safety Monitoring Board for Neurochem Pharmaceuticals, and is an investigator in a clinical trial sponsored by Elan Pharmaceuticals. B.F.B. has been a consultant to GE Healthcare and was an investigator in a clinical trial sponsored by Myriad Pharmaceuticals. R.C.P. has been a consultant to GE Healthcare and has served on a data safety monitoring board in a clinical trial sponsored by Elan Pharmaceuticals. C.R.J. receives research support from Pfizer in the form of research grants.
Received September 15, 2008. Accepted in final form November 24, 2008.
REFERENCES
- 1.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol 1999;56:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 1999;64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 4.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 2004;24:277–295. [DOI] [PubMed] [Google Scholar]
- 5.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 6.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006;442:920–924. [DOI] [PubMed] [Google Scholar]
- 7.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Molec Genet 2006;15:2988–3001. [DOI] [PubMed] [Google Scholar]
- 8.Pickering-Brown SM, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–731. [DOI] [PubMed] [Google Scholar]
- 9.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 2007;66:142–151. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie IR, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 2006;129:3081–3090. [DOI] [PubMed] [Google Scholar]
- 11.Hutton M. Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology 2001;56:S21–25. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi S, McDonagh AM, Pickering-Brown SM, et al. The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 2004;30:1–18. [DOI] [PubMed] [Google Scholar]
- 13.Beck J, Rohrer JD, Campbell T, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008;131:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghetti B, Spina S, Murrell JR, et al. In vivo and postmortem clinicoanatomical correlations in frontotemporal dementia and parkinsonism linked to chromosome 17. Neurodegenerative Dis 2008;5:215–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snowden JS, Pickering-Brown SM, Mackenzie IR, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 2006;129:3091–3102. [DOI] [PubMed] [Google Scholar]
- 16.Whitwell JL, Jack CR Jr, Baker M, et al. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol 2007;64:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in progranulin. Neurobiol Aging Epub 2007 Oct 18. [DOI] [PMC free article] [PubMed]
- 18.Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131:732–746. [DOI] [PubMed] [Google Scholar]
- 19.Whitwell JL, Josephs KA, Rossor MN, et al. Magnetic resonance imaging signatures of tissue pathology in frontotemporal dementia. Arch Neurol 2005;62:1402–1408. [DOI] [PubMed] [Google Scholar]
- 20.Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999;56:303–308. [DOI] [PubMed] [Google Scholar]
- 21.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 22.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 23.Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol 1982;11:592–598. [DOI] [PubMed] [Google Scholar]
- 24.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54 suppl 5:S15–19. [DOI] [PubMed] [Google Scholar]
- 25.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 26.Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc 1987;62:281–288. [DOI] [PubMed] [Google Scholar]
- 27.Mattis S. Mental status examination for organic mental syndrome in the elderly patient. In: Bellak L, Karasu T, eds. Geriatric Psychiatry. New York: Grune and Stratton; 1976: 77–121. [Google Scholar]
- 28.Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 30.Senjem ML, Gunter JL, Shiung MM, et al. Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage 2005;26:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitwell JL, Jack CR Jr, Senjem ML, Josephs KA. Patterns of atrophy in pathologically confirmed FTLD with and without motor neuron degeneration. Neurology 2006;66:102–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohrer JD, Warren JD, Omar R, et al. Parietal lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol 2008;65:506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lantos PL, Cairns NJ, Khan MN, et al. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 2002;58:1169–1175. [DOI] [PubMed] [Google Scholar]
- 34.Spina S, Farlow MR, Unverzagt FW, et al. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain 2008;131:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arvanitakis Z, Witte RJ, Dickson DW, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Relat Disord 2007;13:230–239. [DOI] [PubMed] [Google Scholar]
- 36.Frank AR, Wszolek ZK, Jack CR Jr, Boeve BF. Distinctive MRI findings in pallidopontonigral degeneration (PPND). Neurology 2007;68:620–621. [DOI] [PubMed] [Google Scholar]
- 37.Van Deerlin VM, Wood EM, Moore P, et al. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol 2007;64:1148–1153. [DOI] [PubMed] [Google Scholar]
- 38.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 39.Josephs KA, Whitwell JL, Ahmed Z, et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol 2008;63:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geschwind DH, Robidoux J, Alarcon M, et al. Dementia and neurodevelopmental predisposition: cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Ann Neurol 2001;50:741–746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}