

Summary
The basis for selective death of specific neuronal populations in neurodegenerative diseases remains unclear. Parkinson's disease (PD) is a synucleinopathy characterized by a preferential loss of dopaminergic neurons in the substantia nigra (SN), whereas neurons of the ventral tegmental area (VTA) are spared. Using intracellular patch electrochemistry to directly measure cytosolic dopamine (DAcyt) in cultured midbrain neurons, we confirm that elevated DAcyt and its metabolites are neurotoxic and that genetic and pharmacological interventions that decrease DAcyt provide neuroprotection. L-DOPA increased DAcyt in SN neurons to levels 2-3-fold higher than in VTA neurons, a response dependent on dihydropyridine-sensitive Ca2+ channels, resulting in greater susceptibility of SN neurons to L-DOPA-induced neurotoxicity. DAcyt was not altered by α-synuclein deletion, although dopaminergic neurons lacking α-synuclein were resistant to L-DOPA-induced cell death. Thus, an interaction between Ca2+, DAcyt and α-synuclein may underlie the susceptibility of SN neurons in PD, suggesting multiple therapeutic targets.
Introduction
Parkinson's disease (PD) is characterized by aggregation of alpha-synuclein (α-syn) into “Lewy bodies” and “Lewy neurites”, and a progressive loss of specific neuronal populations. In particular, ventral midbrain (VM) dopamine (DA) neurons of the substantia nigra (SN) preferentially degenerate in PD, while neighboring ventral tegmental area (VTA) DA neurons are relatively spared (Dauer and Przedborski, 2003). A role of α-syn in PD pathogenesis is demonstrated by cases of familial PD that result from mutations or overexpression of α-syn, as well as by the observation that SN neurons in mice with α-syn deletion are protected against the parkinsonian neurotoxins MPTP and 6-OHDA (Alvarez-Fischer et al., 2008; Dauer et al., 2002). Several hypothesis may explain α-syn-mediated cytotoxicity, including the formation of toxic aggregates that disrupt membrane (Conway et al., 2001), a blockade of lysosomal protein degradation (Martinez-Vicente et al., 2008), and mitochondrial dysfunction (Ved et al., 2005). It has been also recently suggested that high Ca2+ levels due to Cav1.3 channel-dependent pacemaking activity (Nedergaard et al., 1993) may contribute to SN susceptibility, as VTA neurons use HCN/Na+ channels for pacemaking (Chan et al., 2007). Accordingly, Cav1.3 antagonists block SN death from MPTP and other neurotoxin models (Chan et al., 2007). The reason for the preferential death of SN DA neurons is, however, unclear as both α-syn and Cav1.3 channels are expressed throughout the CNS in neurons that are not lost in PD (Clayton and George, 1998; Rajadhyaksha et al., 2004; Striessnig et al., 2006).
A long-standing hypothesis of neuronal neurodegeneration in PD postulates that the buildup of cytosolic DA (DAcyt) with associated oxyradical stress and its possible interaction with α-syn and other PD-related proteins underlie neurotoxicity (Caudle et al., 2008; Chen et al., 2008; Edwards, 1993; Pardo et al., 1995; Sulzer and Zecca, 2000). A role for DAcyt in the selectivity of cell death in PD, however, has never been directly studied due to a lack of means to measure DAcyt. Here we use a new electrochemical approach to measure DAcyt in neurons following various pharmacological and genetic interventions. We report that “multiple hits” consisting of high cytoplasmic Ca2+, elevated DAcyt and α-syn expression are required to evoke selective death of dopaminergic neurons from SN and show that interference with any of these three factors rescues the neurons.
Results
We previously introduced intracellular patch electrochemistry (IPE) to study the regulation of cytosolic catecholamine homeostasis in cultured primary murine adrenal chromaffin cells (Mosharov et al., 2003) and the PC12 cell line (Mosharov et al., 2006). To extend IPE measurements to DA neurons, we employed ventral midbrain (VM) cultures from mice that express green fluorescent protein under the control of the tyrosine hydroxylase (TH) promoter (TH-GFP, Figure S1) (Sawamoto et al., 2001). Immunolabeling of fixed two-week-old cultures of VM neurons for TH showed that approximately 97% of GFP+ cells were TH+ (185 of 191 cells).
Dependence of DAcyt on extracellular L-DOPA
IPE measurements in a cyclic voltammetric mode that detects DA preferentially over other intracellular metabolites (including L-DOPA and DOPAC) revealed that DAcyt in untreated GFP+ neurons was below the detection limits of the technique (< 0.1 μM). This was similar to DAcyt levels in PC12 cells (Mosharov et al., 2006), but differed from the 10-20 μM cytosolic catecholamine concentrations found in untreated chromaffin cells (Mosharov et al., 2003). We previously demonstrated that 1 h pre-treatment with 100 μM L-DOPA produces a 2-3-fold increase of cytosolic catecholamine concentration in chromaffin cells (Mosharov et al., 2003). The same dose of L-DOPA increased DAcyt in GFP+ neurons to 17.4 ± 1.7 μM (mean ± SEM; n = 74 cells).
To determine the kinetics of DAcyt changes after L-DOPA treatment, we performed IPE at 1, 8, 15 and 24 h after L-DOPA addition to the media. After 1 h of 100 μM L-DOPA exposure, DA in the cytosol reached a steady state level that was maintained for ∼8 h, followed by a decline to control levels over the succeeding 24 h of drug treatment (Figure 1A). Interestingly, 500 μM L-DOPA increased DAcyt to the same maximum level, but the elevated steady state was maintained longer, and 24 h after L-DOPA treatment, DAcyt was still higher than in untreated cells. To study the dependence of DAcyt on extracellular L-DOPA, neuronal cultures were treated with a range of L-DOPA concentrations for 1 h at 37°C, followed by IPE measurements in the presence of the same L-DOPA concentrations in the bath and in the pipette solutions within the following 30 min at room temperature. The drug response curve was hyperbolic (Figure 1B) with an apparent K0.5 of ∼10 μM L-DOPA.
Figure 1. L-DOPA metabolic consumption by VM neurons.

(A) Time dependence of DAcyt following cell treatment with two L-DOPA concentrations. * - p<0.05 vs. 1h time-point by one-way ANOVA (n = 9-52 cells in different groups).
(B) Dependence of DAcyt on extracellular L-DOPA in mouse neurons treated with the drug for 1h (n = 10-24 cells). Solid line indicates the fit of the data-points using the equation: [DAcyt] = DAmax * [L-DOPA] / (K0.5 + [L-DOPA]).
(C) HPLC-EC measurements of the total intracellular DA and DOPAC in rat VM (n = 9 dishes) and cortical (n = 3) neuronal cultures exposed to 100 or 500 μM L-DOPA for 1h; N.D. is ‘not detected’. * - p<0.05 vs. untreated cultures.
(D) HPLC measurements of intracellular L-DOPA contents in rat VM cultures pretreated with AADC inhibitor benserazide (2 μM; 1 h) and then treated with 50, 100 or 500 μM L-DOPA for 1h (n = 3-8 dishes). Solid line indicates the fit of the data-points using the equation: [L-DOPAintracell] = DOPAmax * [L-DOPAextracell] / (K0.5 + [L-DOPAextracell]).
(E) Dependence of the rate of L-DOPA auto-oxidation on the initial L-DOPA concentration determined by HPLC-EC in cell-free system. The half-life was calculated from the incline (k) of the linear fit of the data-points as t1/2 = Ln(2)/k.
(F) Time dependence of extracellular L-DOPA concentration modeled using kinetic constants from (E). Dashed line indicates K0.5 from (B).
Data on panels A-D are means ± SEM.
As the steady state DA concentration in neuronal cytosol is regulated by multiple enzymes and transporters, we attempted to determine which metabolic step limited DAcyt accumulation in L-DOPA-treated cells. HPLC-EC measurements of the total intracellular DA (the majority of which represents vesicular DA (Chien et al., 1990; Mosharov et al., 2003)) and DOPAC contents showed that these metabolites were elevated to the same degree in neurons exposed to 100 and 500 μM L-DOPA (Figure 1C), indicating that there was no difference in the rates of DA oxidation by MAO and vesicular uptake by VMAT2. To establish whether L-DOPA transport into the cell or its conversion to DA by AADC limited the L-DOPA metabolic consumption, we measured total intracellular L-DOPA levels in VM cultures after 1 h treatment with 100 and 500 μM L-DOPA concentrations in the presence of AADC inhibitor benserazide to block DA synthesis (Figure 1D). The initial rate of L-DOPA accumulation into the cells was not saturated under these conditions, consistent with previously published data (Sampaio-Maia et al., 2001) on the kinetics of L-DOPA uptake by cell lines derived from astrocytes and neurons (Km = 50 - 100 μM). Overall, these data suggest that the activity of AADC limits the steady state DAcyt concentration following L-DOPA treatment.
The data in Figure 1C also provide information about the rate of L-DOPA turnover by the cells. The combined rate of DA and DOPAC synthesis during 1 h of L-DOPA exposure was ∼40 fmol/μg of total protein. As each culture dish typically contained 50 - 100 μg of total protein, the rate of L-DOPA consumption during 10 h of incubation was 20-40 pmol per culture. This rate, however, is too low to account for the decline in the DAcyt observed after 10 h of cell incubation with L-DOPA (Figure 1A), as the total amount of available L-DOPA was >3 orders of magnitude higher (200 nmol in 2 ml of 100 μM solution). We therefore examined the availability of extracellular L-DOPA, which is known to auto-oxidize to DOPA-semiquinone and DOPA-quinone derivatives (Sulzer and Zecca, 2000). HPLC-EC measurements of L-DOPA concentration in cell-free media showed that the drug disappeared with first order kinetics and a half-life of 4.7 h (Figure 1E). This suggests that after 100 or 500 μM L-DOPA initial doses, its concentration in the media would reach the K0.5 levels in 16 h or 26 h, respectively (Figure 1F), which is in close agreement with the kinetics of DAcyt changes, as a significant decrease in its initial steady state was observed at approximately these time points (Figure 1A). Together, these data allow us to predict the time dependence of DAcyt changes based on the concentration of L-DOPA added to the media and the initial increase in DAcyt determined by IPE (see Discussion).
L-DOPA-induced neurotoxicity
We next investigated whether exposure to various L-DOPA concentrations, and thereby different durations of sustained elevated DAcyt, correlated with neurotoxicity. We identified an exponential dependence of neurotoxicity on the extracellular L-DOPA concentration (Figure 2A), whereas the time dependence for cell survival in cultures treated with 250 μM L-DOPA demonstrated that the number of TH+ neurons declined linearly after drug exposure, reaching ∼50% of the control levels after 4 days, with no subsequent neuronal loss (Figure 2B). As L-DOPA at this concentration is cleared within ∼22 h, the data indicate a lag between the end of L-DOPA/DA-mediated stress and the completed course of the catechol-induced toxicity. For further experiments we exposed cultures to 250 μM L-DOPA, which consistently produced ∼50% loss of VM dopaminergic neurons 4 days following drug addition.
Figure 2. L-DOPA-induced neurotoxicity in DA and non-DA neurons.

(A) The loss of TH+ neurons in mouse VM cultures treated with different L-DOPA concentrations for 4 days. Similar decline in the number of VMAT2-positive neurons was observed (data not shown). The average number and the density of TH+ neurons in untreated cultures were 200-250 neurons/culture and 10-15 neurons/mm2, correspondingly. Solid line is the fit of the data with an exponential decay function. * - p<0.001 vs. age-matched controls by one-way ANOVA.
(B) Time-dependence of DA neurons survival in cultures treated with 250 μM L-DOPA. ** - p<0.001 vs. untreated cells by two-way ANOVA (n = 3-4 dishes).
(C) DAcyt in mouse cortical (n = 59) and striatal (n = 21) neurons incubated with 100 μM L-DOPA for 1h.
(D) L-DOPA-induced toxicity in cortical and striatal neurons exposed to 250 μM for 4 days and immunostained for MAP2. * - p<0.01 vs. untreated cells by t-test (n = 3-5 dishes).
Dotted line and shadowed box on (A) and (D) represent mean ± SEM in untreated cells.
Extracellular DA released by neuronal activity did not seem to play a significant role in the L-DOPA-induced cell death, as neuronal viability was not affected by Na+ channel blocker tetrodotoxin (TTX, 1 μM), which inhibits stimulation-dependent transmitter release (data not shown). To further determine which pool of DA was responsible for the observed cell damage, we employed postnatal cultures of cortical and striatal neurons, which do not express TH, DAT or VMAT. Following L-DOPA treatment of cortical neurons, we detected small amounts of DA measured by HPLC-EC (Figure 1C) and a substantial elevation of DAcyt measured by IPE (Figure 2C). This was accompanied by >60% loss of cells immunoreactive to microtubule-associated protein 2 (MAP2), which was used as a neuronal marker (Figure 2D). L-DOPA neurotoxicity was NSD-1015-sensitive, confirming AADC-mediated DA synthesis in cortical neurons (data not shown). In contrast, almost no DAcyt was detected in L-DOPA-treated striatal neurons and they were spared following L-DOPA challenge (Figures 2C and 2D). Together, our data suggest DAcyt as the primary source of the L-DOPA-induced neurotoxicity.
Pharmacological manipulation of DAcyt and neurotoxicity
AADC, MAO and VMAT inhibitors
To study the contribution of individual metabolic pathways in the maintenance of DAcyt steady state and to determine if changes in DA level correlate with neurotoxicity, we performed metabolite measurements and toxicity studies on L-DOPA - treated neurons that were pre-incubated with specific inhibitors of AADC, MAO and VMAT. Inhibition of the DA synthesizing enzyme AADC with NSD-1015 (Figure 3A and 3B) or benserazide (not shown) blocked L-DOPA-induced elevation of whole-cell intracellular DA and DOPAC. Benserazide also inhibited the buildup of DAcyt following L-DOPA treatment (Figure 3C), consistent with the idea that the IPE oxidation signal comes from DA synthesized by AADC in the cytosol (note that NSD-1015 alters IPE sensitivity for DA, and therefore was not examined: see Methods). Moreover, the blockade of L-DOPA decarboxylation completely prevented drug-induced cell death (Figure 3D), indicating that an L-DOPA metabolite, but not L-DOPA itself, was responsible for the toxicity.
Figure 3. Effect of pharmacological inhibitors on DAcyt and neurotoxicity.

(A,B) HPLC-EC measurements of total intracellular DA (A) and DOPAC (B) in rat VM neuronal cultures pretreated for 1h with 500 μM NSD-1015, 2 μM reserpine (res) or 10 μM pargyline (PGL) and then treated with 100 μM L-DOPA for 1h (n = 6-10 dishes). N.D. - ‘not detected’.
(C) DAcyt in mouse VM neurons under the same treatments as above accepts 2 μM benserazide (Bsrz) to block AADC. n = 22-81 cells.
(D) The effect of NSD-1015, benserazide, reserpine and pargyline on the survival of mouse TH+ neurons treated with 250 μM L-DOPA for 4 days (n = 6-20 dishes).
(E) DAcyt concentration in TH-GFP neurons pre-treated with 10 μM cocaine for 15 min, then exposed to 100 μM L-DOPA for 1 h, and then treated with 50 μM METH for 15-30 min (n = 19-46 cells).
(F) Cell survival of mouse TH+ neurons pre-treated with METH and cocaine as in (E) and exposed to 250 μM L-DOPA for 4 days (n = 3-8 dishes).
* - p<0.05 vs. cells treated with L-DOPA only by one-way ANOVA. Dotted lines and shadowed boxes on (D) and (F) represent mean ± SEM in untreated cells.
The contribution of DA catabolism was investigated by treating cells with pargyline, an inhibitor of MAO that almost completely abolished DOPAC synthesis (Figure 3B). Pargyline also produced a several-fold increase in both the total amount of intracellular DA (Figure 3A) and DAcyt (Figure 3C) in L-DOPA-treated neurons, which further correlated with a significantly greater neuronal loss (Figure 3D).
The blockade of VMAT-mediated DA uptake into the vesicles depletes vesicular catecholamine storage, as observed by a reduction in the exocytotic quantal size (Colliver et al., 2000). Consistently, pre-treatment of VM neurons with reserpine decreased the amount of total intracellular DA synthesized from L-DOPA by ∼2-fold both with and without pargyline (Figure 3A). We observed, however, no effect of reserpine, or another VMAT inhibitor, tetrabenazine (10 μM; data not shown) on either DAcyt or the number of surviving dopaminergic neurons (Figure 3C and 3D).
Methamphetamine and cocaine
The discrepancy between the reduction of the total DA in reserpine-treated neurons and the lack of the effect of VMAT inhibition on DAcyt might be explained by the distribution of synaptic vesicles between neuronal cell bodies and the neurites. If the majority of vesicles filled with DA are located in synaptic terminals far from the cell bodies, any transmitter redistributed from them would be invisible to IPE (see Methods).
To examine the effects of other DA-releasing drugs on somatic DA concentration, we exposed L-DOPA-treated neurons to methamphetamine (METH), which disrupts the vesicular proton gradient and redistributes DA from synaptic vesicles to the cytosol (Sulzer et al., 2005). In contrast to the increased cytosolic catecholamine levels observed in chromaffin cells acutely treated with METH (Mosharov et al., 2003), neurons exposed to 50 μM METH showed decreased DAcyt (Figure 3E); 5 μM METH produced the same decrease of DAcyt (38 % of untreated cells; p<0.005 by t-test). METH-mediated reduction of DAcyt was blocked by the DA uptake transporter (DAT) blocker cocaine, suggesting that the effect was due to reverse transport through DAT (Sulzer et al., 2005). METH-mediated decrease of DAcyt and its blockade by cocaine were also observed in L-DOPA-treated cells in the presence of PGL (data not shown). Note that DAcyt in L-DOPA-treated neurons was unaffected by DAT inhibitors cocaine (Figure 3E) or nomifensine (5 μM; data not shown), supporting the idea that reverse transport is not induced by the elevated DAcyt alone, but requires additional METH-mediated effects on DAT (Kahlig et al., 2005).
Consistent with the IPE results, METH protected neuronal cell bodies from L-DOPA toxicity (Figure 3F), despite the considerable neurite loss that occurs in METH-treated cultured DA neurons (Cubells et al., 1994; Larsen et al., 2002). When applied for 4 days without L-DOPA, neither METH or cocaine nor the combination of the two drugs had a significant effect on the number of surviving neurons (data not shown).
Genetic manipulation of DAcyt and neurotoxicity
VMAT2 overexpression
We previously developed a recombinant adenovirus that overexpresses VMAT2 (rVMAT2), resulting in reduced neuronal neuromelanin content, enhanced quantal size and increased the number of evoked quantal neurotransmitter release events from the terminals of cultured VM dopaminergic neurons (Pothos et al., 2000; Sulzer and Pothos, 2000). To investigate whether enhanced vesicular uptake may decrease DAcyt and rescue neurons from L-DOPA-induced toxicity, we employed an adenoviral construct that resulted in overexpression of the recombinant VMAT2 in 80-90% of both dopaminergic and non-dopaminergic cells (Figure S2).
VMAT2 overexpression significantly decreased DAcyt in L-DOPA-treated GFP+ VM neurons (from 17.4 ± 1.7 μM, n = 74 neurons to 3.1 ± 0.8 μM, n = 28; Figure 4A), but had no effects on GFP- neurons from the same culture (2.4 ± 0.8 μM, n = 33 vs. 2.3 ± 1.9 μM, n = 7). GFP+ neurons in cultures treated with a virus that did not contain rVMAT2 displayed the same DAcyt levels after L-DOPA treatment as control cells (data not shown). Consistent with the kinetics of L-DOPA metabolic consumption (Figure 1), rVMAT2 lowered DAcyt to the same extent in cells treated with 100 μM and 500 μM L-DOPA (Figure 4A). Moreover, infection with rVMAT2 effectively protected TH+ neurons from the L-DOPA-mediated neurotoxicity (Figure 4B). It should be noted, however, that these data also suggest that somatic vesicles or other organelles that do not normally sequester substantial levels of DA can do so after transporter overexpression, as has been demonstrated for non-catecholaminergic AtT-20 cells (Pothos et al., 2000) and hippocampal neurons (Li et al., 2005).
Figure 4. Effect of VMAT2 overexpression and α-syn knock-out on DAcyt and toxicity.

(A) DAcyt in sister cultures of mouse TH+ neurons treated with L-DOPA for 1h; cells were either untransfected or were transfected with rVMAT2 or empty vector (not shown) one day before the application of L-DOPA. * - p<0.01 vs. cells treated with L-DOPA only by one-way ANOVA (N=15-28 cells).
(B) Neuroprotection of TH+ neuron in rat VM cultures infected with rVMAT2 or empty vector and exposed to varying concentrations of L-DOPA on day one and accessed for survival on day 7. * - p<0.05 vs. by two-way ANOVA.
(C) Reduced sensitivity of TH+ neurons from α-syn deficient mice to L-DOPA-induced neurotoxicity. Neurons from α-syn wild-type, heterozygous and knock-out littermate mice were treated with indicated L-DOPA concentrations for 4 days. * - p<0.05 and ** - p<0.001 vs. the WT by two-way ANOVA with Bonferroni post-hoc test (n = 10-29 dishes).
(D) Comparison of DAcyt levels in neurons from α syn knock-out, heterozygous and wild-type mice treated with 100 μM L-DOPA treated for 1 h (n = 20-49 cells).
Alpha-synuclein knock-out
To investigate the role of α-syn in mediating DAcyt neurotoxicity, we generated α-syn deficient mice that express eGFP in dopaminergic neurons (see Methods). While VM neurons from α-syn-/- and α-syn+/- mice were more resistant to L-DOPA-induced stress than neurons from their wild-type littermates (Figure 4C), IPE measurements indicated no difference in the DAcyt concentration between the three groups (Figure 4D). These data suggest that the pathogenic effect of α-syn is downstream of DA synthesis (see Discussion), and that the presence of both elevated DAcyt and α-syn is required for L-DOPA-induced neurotoxicity.
DAcyt in SN and VTA neurons
To address the question of differential susceptibility of neurons from different brain regions, we prepared cultures that were enriched with cells from either SN or VTA, as previously published (Burke et al., 1998). Immunolabel for calbindin, a protein that is expressed at higher levels in VTA than in SN (Thompson et al., 2005), showed that in mixed VM cultures ∼50% of TH+ neurons were also calbindin+, while the proportion of TH+/calbindin+ cells was ∼85% in VTA cultures and ∼25% in SN cultures (Figure 5A and 5B).
Figure 5. Sensitivity of SN and VTA DA neurons to L-DOPA challenge.
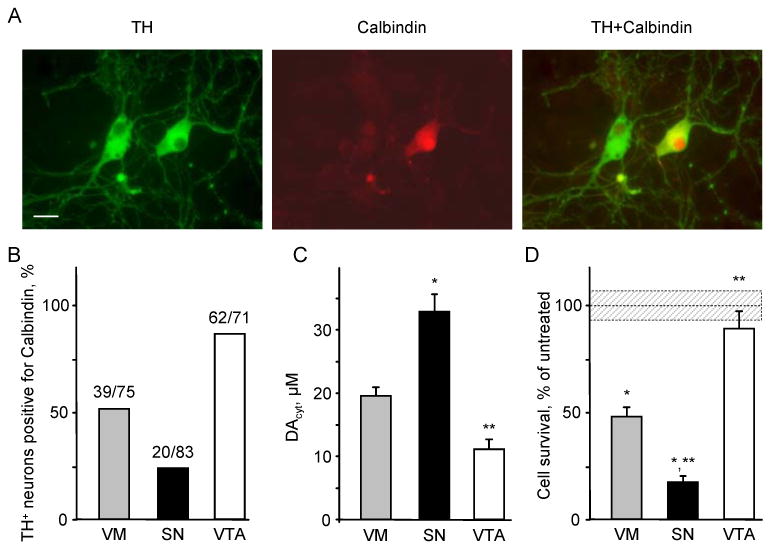
(A) Representative images of VM neuronal cultures double stained for TH and calbindin. Scale bar is 20 μm.
(B) Relative number of calbindin+ cells within the population of TH+ neurons.
(C) Cytosolic DA concentrations in VM, SN and VTA TH+ neurons treated with 100 μM L-DOPA for 1h. A representative experiment shown was repeated 5 times with 9-16 cells in each group in each experiment. p < 0.01 vs. VM (*) or vs. VM and SN (**) by one-way ANOVA.
(D) L-DOPA-induced neurotoxicity in cultures treated with 250 μM L-DOPA for 4 days. Dotted line and shadowed box represents mean ± SEM in untreated cells. p < 0.01 vs. untreated neurons of the same group (*) or vs. L-DOPA-treated neurons of all other groups (**) by one-way ANOVA (n = 10-15 dishes).
IPE measurements showed that L-DOPA treated SN neurons displayed 2-3-fold higher DAcyt than VTA neurons (Figure 5C). This difference in DAcyt translated into a significantly higher susceptibility of SN neurons to L-DOPA-induced toxicity, while VTA neurons were almost completely protected (Figure 5D). The resistance of VTA neurons to L-DOPA challenge was also seen in mixed VM cultures, where after 4 days of 250 μM L-DOPA the percentage of TH+/calbindin+ neurons increased from 52% to 88% (p<0.05 by χ2 test).
Relationship between cytoplasmic Ca2+ and DAcyt
The regulation of intracellular Ca2+ differs significantly between SN and VTA neurons (Gerfen et al., 1985; Surmeier, 2007; Thompson et al., 2005). Thus, to search for the mechanism that governs the difference in DAcyt between the two neuronal populations, we investigated whether intracellular Ca2+ had an effect on L-DOPA-induced DAcyt accumulation.
Cell pretreatment with the voltage-gated calcium channel blocker CdCl2 or buffering cytoplasmic Ca2+ with membrane-permeable chelator BAPTA-AM, both significantly decreased DAcyt in dopaminergic neurons from SN and VTA (Figure 6A). This suggests that the regulatory mechanism that links cytoplasmic Ca2+ and DAcyt exists in both cell populations, and that the difference between SN and VTA neurons might be in the steady-state Ca2+ level. This would be consistent with SN neurons relying on dihydropyridine-sensitive L-type Cav1.3 channels for autonomous pacemaking, in contrast to VTA neurons that use HCN/Na+ channels for pacemaking. Immunostaining of VM cultures for the α1D subunit of the L-type Ca2+ channels, which is specifically present in the Cav1.3 channels, showed that its expressed in both calbindin+ and calbindin- DA neurons (Figure S3), consistent with previous reports (Rajadhyaksha et al., 2004; Striessnig et al., 2006). The Cav1.2/Cav1.3 blockers nimodipine (Figure 6A) and nitrendipine (not shown), however, had no effect on DAcyt in L-DOPA exposed VTA neurons, but decreased DAcyt in SN neurons to the levels found in VTA neurons (Figure S4). VM cultures treated with nimodipine were far more resistant to L-DOPA-induced neurodegeneration (Figure 6B). Pre-treatment of SN neurons for 1h with HCN channel antagonist ZD7288 (50 μM) or TTX (1 μM) had no significant effect on DAcyt reached after L-DOPA treatment (data not shown).
Figure 6. Regulation of DAcyt by cytoplasmic Ca2+.

(A) DAcyt in SN and VTA neurons pretreated with 30 μM CdCl2, 10 or 100 μM BAPTA-AM, or 10 μM nimodipine (Nmdp) for 1h, and then exposed to 100 μM L-DOPA for 1h. Data are presented as relative changes compared to SN and VTA neurons treated with L-DOPA only. * - p<0.05 vs. L-DOPA only by one-way ANOVA (n = 11-52 cells).
(B) Protection of SN neurons from L-DOPA-induced neurotoxicity by L-type Ca2+ channel blocker nimodipine (10 μM; 1h pretreatment). * - p<0.05 vs. L-DOPA only by one-way ANOVA (n = 3-23 dishes).
(C) DAcyt levels in SN and VTA neurons pretreated with 2 μM reserpine and 10 μM pargyline for 1h and then exposed to 100 μM L-DOPA for 1h. * - p<0.05 vs. corresponding L-DOPA only group by one-way ANOVA (n = 18-27 cells).
(D) Whole-cell L-DOPA concentration in VM cultures pretreated for 1h with 2 μM benserazide with and without 10 μM BAPTA-AM and then exposed to 100 μM L-DOPA for 1h (n = 4 dishes).
(E) Total cytosolic catechol concentrations (IPE in amperometric mode) in VM neurons either untreated or treated as in (D). n = 10 cells.
To determine which enzyme or transporter is responsible for Ca2+-induced upregulation of DA homeostasis, we first compared DAcyt in SN and VTA neurons treated with pargyline and reserpine. DAcyt was increased by the same percentile in both types of neurons (Figure 6C), suggesting that MAO and VMAT2 activities did not underlie the difference between DAcyt handling by SN and VTA neurons and were not the regulatory targets. Next, DAcyt in SN and VTA neuronal cultures was unaffected by the presence of DAT blocker cocaine (data not shown). Finally, the uptake of L-DOPA in VM cultures pre-treated with BAPTA-AM and benserazide was monitored by HPLC-EC and IPE in the amperometric detection mode, which in contrast to CV produces similar oxidative currents for DA and L-DOPA (Mosharov et al., 2003). Measurements by both methods demonstrated that L-DOPA uptake was unaffected by Ca2+ buffering (Figure 6D and 6E). Overall, our data suggest that MAO, VMAT, DAT and the L-amino acid plasma transporter were not responsible for the difference in DAcyt observed between SN and VTA neurons. Thus, it appears that the activity of AADC may provide the Ca2+-sensitive metabolic step that leads to higher DAcyt levels in SN neurons (see Discussion).
Discussion
Efforts over the past decade have identified genes that cause familial instances of several neurodegenerative disorders, including Parkinson's disease. The steps underlying the degeneration of the specific neuronal populations associated with these diseases, however, have proven elusive. Here we show that selective death of SN dopaminergic neurons responsible for the definitive motor deficits in PD can result from a combination of “multiple hits” resulting from the activity of the L-type Ca2+ channels that create high cytoplasmic Ca2+ levels, an upregulation of DAcyt synthesis by Ca2+, and the presence of α-synuclein.
Dependence of neurotoxicity on DAcyt
A series of studies have suggested that dysregulation of DAcyt maintenance underlies neuropathology in PD, but characterization of DAcyt and its relationship to neurotoxicity has not been possible due to a lack of means to measure DAcyt. We have adapted the recently introduced technique of IPE to provide the first detailed characterization of DA homeostasis in neuronal cytosol.
Our results confirm a relationship between high levels of DAcyt and neurotoxicity. A variety of pharmacological and genetic means to influence DA homeostasis indicate that neurotoxicity was mediated by the cytosolic pool of DA, while other potential sources of cellular stress, including extracellular DA and L-DOPA, played limited roles under our experimental conditions. To compare the data from different experiments, we examined the dependence of cell survival on the integral of cell exposure to elevated DA levels (DAcyt dose; Figure 7A and S4). The relationship was close to linear, with doses above 100-200 μM·h causing significant neurotoxicity. This finding was consistent over a wide variety of interventions that altered DAcyt in different manners, including inhibition of L-DOPA conversion to DA, VMAT2 blockade or overexpression, and treatment with METH and cocaine. Cortical neurons were less resistant to L-DOPA challenge than midbrain DA neurons, although there are no reports of cell loss in the cortex of PD patients who received L-DOPA for many years. Conversely, VM neurons treated with the MAO inhibitor pargyline were significantly more resistant to DAcyt-mediated toxicity (Figure 7A; asterisk). One possible explanation for the lower than expected levels of cell death observed in the presence of MAO blockade is a contribution of DA catabolic products that arise via MAO activity, i.e., H2O2 and DOPAL, as has been recently suggested (Burke et al., 2004; Eisenhofer et al., 2004). Although additional studies are required to characterize other factors that modulate susceptibility of neurons to stress, our data support the exploration of central MAO inhibitors as treatments for PD either as monotherapy at early stages of the disease or in combination with L-DOPA in the advanced disease stages (Chen et al., 2007; Hauser et al., 2008); in both cases MAO blockers would increase the availability of DAcyt for vesicular sequestration while preventing the buildup of toxic DA metabolites.
Figure 7. DAcyt dose and neurotoxicity.

(A) Dependence of cell survival under L-DOPA-induced stress on the DAcyt dose in mouse neurons. DAcyt dose was estimated as [DAcyt] · TExposure = [DAcyt] · Ln ([L-DOPA] / K0.5) / k, where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (>50 μM) of L-DOPA for 1h, [L-DOPA] is the initial drug concentration, and K0.5 = 9.7 μM and k = 0.15 h-1 are the kinetic constants derived from Figure 1B and 1E, correspondingly. TExposure therefore approximates the time during which extracellular L-DOPA remained higher than K0.5 (Figure 1F). The datapoints are (from left to right): (filled circles) VM cultures treated with 25, 100, 250, 500 and 1000 μM L-DOPA alone; (open circles) VM neurons treated with 250 μM L-DOPA in the presence of benserazide, METH, reserpine, PGL and PGL+reserpine; (diamonds) VTA and SN neurons, and (triangles) striatal and cortical neurons treated with 250 μM L-DOPA. Dotted line and shadowed box represent mean ± SEM in untreated cells. Solid line is the linear fit of all datapoints, excluding striatal and cortical neurons, and two datapoints indicated by asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; data points to the left of the line are more susceptible to DAcyt stress.
(B) Multi-hit model of PD pathogenesis. Neurotoxicity is a result of multiple factors, including the presence of α-syn, elevation of cytoplasmic Ca2+ and the buildup of DAcyt and its metabolites. Non-exclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Decreasing the levels of any of the three players provides neuroprotection.
DAcyt maintenance in neuronal somas and synaptic terminals
Perhaps surprisingly, reserpine and METH, drugs that disrupt vesicular DA sequestration, did not affect DAcyt, despite the depletion of vesicular DA stores evident from HPLC-EC measurements of the total intracellular DA (Figure 3; (Larsen et al., 2002)). These results are however consistent with diffusion calculations indicating that IPE measures only DA in the cell body (see Methods), i.e., the number of DA storage vesicles in the soma might be insufficient to detectably increase DAcyt following reserpine or METH. Although some DA secretory organelles reside in or close to neuronal cell bodies, as evident from somatodendritic vesicular DA release from midbrain neurons (Jaffe et al., 1998; Rice et al., 1997), emptying one synaptic vesicle containing 10,000 DA molecules (Pothos et al., 1998) would increase DAcyt by only 0.01 μM (see Methods), and 150-200 such vesicles would need to be emptied to produce a 10% increase over the 15-20 μM DAcyt observed in L-DOPA-treated cells. In comparison, adrenal chromaffin cells, which lack neurites and are filled with secretory vesicles that each contain ∼1,000,000 transmitter molecules (Wightman et al., 1991), show increased cytosolic catechol concentrations following reserpine or METH (Mosharov et al., 2003). While IPE cannot measure DAcyt in axonal terminals, it is possible that a similarly large increase in DAcyt occurs in METH-treated neuronal terminals and, if so, this may explain the unusual pattern of METH induced neurodegeneration in which neurites but not cell bodies are damaged in vitro (Cubells et al., 1994; Larsen et al., 2002), in animal models in vivo (Ricaurte et al., 1982), and in imaging studies of METH abusers after prolonged abstinence (Chang et al., 2007). Post-mortem examination of chronic METH users reveals a more severe DA axonal loss in the caudate than in the putamen, presumably explaining the lack of parkinsonian symptoms in METH abusers (Moszczynska et al., 2004).
Upregulation of DA homeostasis by Ca2+ explains higher vulnerability of SN neurons to L-DOPA-induced stress
We found that DAcyt reached in L-DOPA-treated dopaminergic neurons directly correlated with intracellular Ca2+, as lower DAcyt levels were found in SN and VTA neurons following voltage-gated calcium channels blockade by CdCl2 or cytoplasmic Ca2+ buffering by BAPTA-AM. Conversely, significantly higher DAcyt was reached in SN than in VTA neurons, consistent with the pacemaking activity of dihyrdropyridine-sensitive Cav1.3 channels in SN but not VTA neurons. Notably, while dihydropyridines nimodipine and nitrendipine had no effect on DAcyt concentration in VTA neurons, they decreased DAcyt in SN neurons to the levels in the VTA, and provided neuroprotection from L-DOPA-induced cell death.
The target of Ca2+-dependent upregulation of DA homeostasis appears to be the conversion of L-DOPA to DA by AADC. Although regulation of AADC activity via protein kinases A and C - mediated phosphorylation has been reported (Duchemin et al., 2000; Young et al., 1998), these mechanisms require further characterization, as Cav1.3 channels are also regulated by these second messenger pathways (Baroudi et al., 2006; Rajadhyaksha and Kosofsky, 2005). It should also be noted that Ca2+-dependant phosphorylation of TH (Dunkley et al., 2004) is likely to play a role in the absence of exogenously added L-DOPA.
A “multiple hit” hypothesis of SN neurodegeneration in PD
Multiple genes have been identified to cause familial PD, but the disease may require multiple steps, such as an initial insult together with an insufficient stress response (Abou-Sleiman et al., 2006; Conway et al., 2001; LaVoie et al., 2005; Martinez-Vicente et al., 2008; Moore et al., 2005; Sang et al., 2007; Sulzer, 2007), which may explain why known genetic and environmental factors do not straightforwardly account for idiopathic PD. The interaction between high DAcyt and α-syn may provide one of the “multiple hit” mechanism. Due to the presence of α-syn in Lewy bodies and the role of α-syn mutations or gene multiplication in some forms of familial PD, it is widely believed that this protein plays a central role in the degeneration of VM DA neurons. Consistent with previous reports that dopaminergic neurons in α-syn deficient mice show reduced susceptibility to neurotoxins (Alvarez-Fischer et al., 2008; Dauer et al., 2002), we found that α-syn deletion makes VM neurons resistant to L-DOPA-induced toxicity. On the other hand, neuronal DAcyt was unaffected by the absence of α-syn (Figure 4D), similar to cytosolic catecholamine levels in chromaffin cells from α-syn null animals (Mosharov et al., 2006). These data are consistent with a toxic mechanism downstream of DA synthesis that may involve stabilization of toxic a-syn protofibrils by DA, inhibition of chaperone-mediated autophagy by DA-modified α-syn, or a cumulative stress from various sources, which in addition to DAcyt, Ca2+ and α-syn may include mitochondrial and proteasomal dysfunction and inflammatory responses (Figure 7B) (Burke et al., 2008; Conway et al., 2001; Dauer and Przedborski, 2003; Gao et al., 2008; Martinez-Vicente et al., 2008; Ved et al., 2005). Additionally, although normal expression levels of mouse wild-type α-syn do not seem to affect DAcyt concentration, α-syn mutation or overexpression may lead to elevated DAcyt (Lotharius et al., 2002; Mosharov et al., 2006) that would further exacerbate neurotoxicity.
It is striking that L-DOPA doses that produced neurotoxicity in this study also induce the synthesis of neuromelanin (Sulzer et al., 2000) - an autophagosome containing oxidized dopamine products (Sulzer et al., 2008). Moreover, VMAT2 overexpression, which as we show here decreased DAcyt and abolished L-DOPA-induced neuronal death, has also been demonstrated to inhibit neuromelanin synthesis (Sulzer et al., 2000). Together, these data support the idea that neuromelanin synthesis is a stress response and that the presence of neuromelanin in human SN and its accumulation over time are indicative of an ongoing DAcyt damage even in normal non-PD human brain. Conversely, dopaminergic neurons in the VTA, which are spared in PD, produce minimal neuromelanin over a lifetime (Hirsch et al., 1988; Liang et al., 2004; Zecca et al., 2001).
Whereas DA neurons are comparatively resistant to DAcyt toxicity, it remains unclear whether neurotoxic DAcyt levels are reached in humans, particularly during treatment of PD patients with L-DOPA. Although L-DOPA therapy is linked to increased levels of the products of peroxynitrite-mediated oxidative stress in cerebrospinal fluid (CSF) (Isobe et al., 2006) and a more rapid decline in striatal dopamine transporter over the course of L-DOPA therapy, there is no increase in the rate of symptomatic progression in PD patients with L-DOPA (Fahn, 2005). Moreover, while recent reports demonstrate L-DOPA-induced neurotoxicity in rodents (Chen et al., 2008; Jeon et al., 2007), other in vivo studies found L-DOPA to be benign or even neuroprotective (reviewed by (Olanow et al., 2004)). Important factors may include the ability of L-DOPA-induced mild oxidative stress to activate glutathione synthesis (Han et al., 1996; Mena et al., 1997) and induce macroautophagy (Sulzer et al., 2008), and a likely protective action of L-DOPA therapy on the striatopallidal pathway (Day et al., 2006). Normal L-DOPA concentration in the CSF is only ∼5 nM (Tohgi et al., 1997) and increases to ∼300 nM in L-DOPA treated patients (Tohgi et al., 1995), which is likely within a range that cellular stress response pathways can handle. It should be emphasized that the meaning of our findings is not that L-DOPA is neurotoxic to humans, as it is artificially used as a tool to enhance DA: genuine PD does not result from L-DOPA exposure, but may result following chronically elevated DAcyt and the presence of other genetic and environmental “hits”.
The newly identified relationship between high Ca2+ and DAcyt may underlie the selective vulnerability of dopaminergic neurons in SN and locus coeruleus, a group of noradrenergic neurons that also rely on Ca2+ channel pacemaking (Williams et al., 1984). If so, various strategies could be employed to prevent neuronal death in PD, including an inhibition of Cav1.3 channel activity (Surmeier, 2007) or blocking the deleterious effects of DA-protein interactions (Martinez-Vicente et al., 2008). On the other hand, symptomatic treatment of PD relies on therapies that enhance synaptic vesicle sequestration of DA, thereby reconstituting the dopaminergic tone in the striatum. As discussed above, our data may indicate advantages to using central MAO inhibitors as PD drugs. Likewise, as we show here, VMAT2 overexpression enhances exocytotic DA release while decreasing DAcyt. Since VMAT2 overexpression not only promotes DA neurotransmission in a manner similar to L-DOPA but also confers neuroprotection, controlled VMAT2 overexpression may provide a therapeutic intervention for PD, either alone or in combination with L-DOPA or MAO blockers.
Experimental Procedures
Mouse strains
For neurotoxicity and IPE experiments, neurons from C57/B6 mice or from transgenic mice that express green fluorescent protein (TH-GFP+/-) under the control of the rat tyrosine hydroxylase (TH) promoter (Sawamoto et al., 2001) were used. α -Synuclein null mice that express eGFP under the control of TH promoter were generated in two steps. TH-GFP+/- mice were crossed with α-syn-/- mice (strain B6;129X1-Sncatm1Rosl/J from Jacksons Labs; originally from (Abeliovich et al., 2000)). Next, the resulting TH-GFP+/- / α-syn+/- mice were crossed with each other and genotyped for the presence of both eGFP and α-syn to obtain TH-GFP+/- / α-syn+/-, TH-GFP+/- / α-syn-/- and TH-GFP+/- / α-syn+/+ littermates. For HPLC measurements, cells from Sprague-Dawley rats were used. Animals were used in accordance with the National Institutes of Health guidelines for the use of live animals and the animal protocol was approved by the Institutional Animal Care and Use Committee of Columbia University.
Primary neuronal cultures
Ventral midbrain (VM), cortical or striatal neurons from postnatal day 0-2 mice or rats were dissected, dissociated and plated on a monolayer of cortical astrocytes at the plating density of ∼100,000 cells/cm2, as previously described (Burke et al., 1998; Rayport et al., 1992). Mouse neurons were cultured on glass poly-D-lysine-coated coverslips attached to ∼0.8 cm2 wells cut into 50 mm dishes. Rat neurons were cultured on 35 mm tissue culture dishes (∼9.6 cm2).
We previously reported (Mena et al., 1997) that lower levels of L-DOPA provided a short-lasting enhancement of dopaminergic neuron survival due to neuroprotective antioxidant responses to L-DOPA-induced oxidative stress. We have since demonstrated that glial-derived neurotrophic factor (GDNF) is selectively neuroprotective for postnatally-derived dopaminergic neurons, providing markedly enhanced survival (Burke et al., 1998) and now routinely include this component in the culture medium. As shown in the present study, L-DOPA is not neuroprotective in the presence of GDNF (Figure 2A), presumably because the neuroprotective response has been occluded, allowing the toxic response to be revealed.
Experiments were conducted 6-10 days post-plating; all treatments were performed at 37°C. Pseudo-3D images of cultured TH-GFP neurons (Figure S1) were obtained with a two-photon microscope (Zeiss Axioskop2 FS MOT upright microscope, LSM 510 Coherent 2-photon setup), using 850 nm excitation and 525 nm emission wavelengths. Z sections were taken at 1 μm increments. Other images were acquired on a conventional fluorescence microscopy setup (Axiovert 100 microscope; Carl Zeiss MicroImaging, Inc. Thornwood, NY) equipped with Zeiss AxioCam MRm camera and FITC and rhodamine filter sets (Chroma, Rockingham, VT).
Adenoviral vector construction and transfection of neurons with rVMAT2
A hemagglutinin (HA) - tagged VMAT2 cDNA was first subcloned into the shuttle vector pTet-EF, then co-infected with donor virus DNA (φ5) into HEK293 cells expressing Cre recombinase, and the resultant VMAT2-HA harboring adenovirus was purified and stored as previously described at ∼8000 pfu/μl (Krantz et al., 1997). Primary rat VM cultures were incubated with 1 μl of rVMAT2 diluted in 100 μl media for 5 h and then 2 ml of media was added in each dish; φ5 alone was used as a negative (empty virus) control. Transfection efficiency was 80-90% for both dopaminergic and GABAergic neuronal types (Figure S2).
Immunocytochemistry and western blots
Immunostaining of 4% paraformaldehyde-fixed cultures was performed using mouse or rabbit anti-TH (1:1000; Chemicon, Temecula, CA), rabbit anti-MAP2 (1:500; Chemicon), and mouse anti-calbindin D-28K (1:200; Sigma, St. Louis, MO) antibodies, followed by secondary antibodies conjugated with Alexa 488 or Alexa 594 (1:200; Molecular Probes, Eugene, OR). Polyclonal rabbit anti-VMAT2 antibody (1:200; Pel-Freez Biologicals, Rogers, AR) was used to detect endogenous and exogenous VMAT2 protein. For immunodetection of HA-tagged rVMAT2, cultures were stained using monoclonal anti-HA antibody (1:200; BabCO, Richmond, CA) (Sulzer and Pothos, 2000). Western immunoanalysis of VMAT2 protein was performed as described (Krantz et al., 1997) with VMAT2 antibody (1:500; Chemicon), horseradish peroxidase conjugated secondary antibody and visualization by enhanced chemiluminescence (Pierce Biotech, Rockford, IL).
Neurotoxicity
Cells were pre-incubated with rVMAT2 for one day and with various DA metabolism inhibitors for 1h before the application of L-DOPA, unless stated otherwise. Following 1-7 days incubation, neuronal cultures were fixed and stained for TH (VM neurons) or MAP2 (cortical and striatal neurons). The total numbers of immunoreactive neurons were then tallied and analyzed as previously described (Larsen et al., 2002). Alternatively, neuronal density was assessed by counting the number of immunoreactive cells in 24 fields of view at 200× magnification (Plan-Neofluar 20× objective; ∼0.8 mm2 viewing field) and taking the average as a representative for each dish. Although both methods yielded similar results, the second approach was more robust, producing lower deviation within experimental groups. The counts were performed by an observer blind to the experimental treatments.
High performance liquid chromatography with electrochemical detection (HPLC-EC)
Whole-cell DA, dihydroxyphenylacetic acid (DOPAC) and L-DOPA levels were determined by HPLC-EC as previously described (Feigin et al., 2001; Larsen et al., 2002). DA, DOPAC and L-DOPA amounts were calculated from areas under HPLC peaks using calibration curves and normalized to protein concentrations in each sample.
Intracellular patch electrochemistry (IPE)
Measurements of cytosolic DA (DAcyt) concentrations in neurons were based on protocols for PC12 cells and chromaffin cells (Mosharov et al., 2003; Mosharov et al., 2006). A polyethylene-coated 5 micron carbon fiber electrode (CFE) was placed inside a glass patch pipette and used in cyclic voltammetric (CV) mode of detection where voltage ramps from -450 mV holding potential to +800 mV and back to -450 mV over 10 ms (scan rate of 250 mV/ms) were applied at 100 ms intervals. After achieving a seal between the cell and the patch pipette, the plasma membrane was ruptured by suction and substances diffusing from the cytosol into the pipette were observed as a slow wave of oxidation current. As no difference was found between IPE signal using intracellular and extracellular ionic compositions as the patch pipette saline (Mosharov et al., 2003), the same saline was employed as the bath and the pipette solutions in the present study. The saline contained (in mM): 118 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1 NaH2PO4, 10 glucose, 25 HEPES-NaOH (pH 7.4); the same concentrations of all drugs, including L-DOPA were present in the bath and in the patch pipette. After background current subtraction, the concentration of DA at the pipette tip was calculated as previously described (Mosharov et al., 2003). IPE sensitivity, estimated as the current 3-fold higher than the RMS noise on the subtraction voltammogram, was ∼100 nM DA; the calibration curves were log-linear for up to at least 1 mM DA concentration (Mosharov et al., 2003). The initial concentration of DAcyt was calculated using neuronal cell body volume and the volume of the pipette tip estimated from photographs taken before each recording (Figure S1).
To investigate if the electrode sensitivity for DA was affected by the presence of compounds present in the recording media, we performed a series of calibration measurements of 5 μM DA with and without the drugs (Supplementary Table 1). Based on these data, NSD-1015 cannot be used for IPE experiments because it abolished the oxidation signal of DA.
Although CV voltammograms cannot differentiate between DA, L-DOPA, and the product of DA catabolism, DOPAC, there are several reasons to suspect that DA represented the majority of the cytosolic oxidation signal estimated by IPE. First, the sensitivity of CV is 15-20-fold higher for DA molecules than for other catechols (Mosharov et al., 2003). Second, there were identical concentrations of L-DOPA present in the patch pipette and extracellular saline during the recordings; thus any change in the oxidation signal after achieving the whole-cell configuration could not be due to the influx of L-DOPA into the patch pipette per se. Third, L-DOPA is rapidly converted to DA once in the cytosol, and enhanced DA can be measured in these cultures within 90 seconds (Pothos et al., 1998), while, as shown in Results, inhibition of AADC almost completely abolished the oxidation signal from L-DOPA-treated cells. Finally, neither L-DOPA nor DOPAC are VMAT2 substrates, arguing against a possibility that the observed decrease in the cytosolic signal in neurons overexpressing VMAT2 was due to nonspecific catechol uptake into the vesicles.
To estimate the contribution of neurotransmitter from neurites to the CV oxidation signal, we calculated the time required for DA to diffuse into the cell body and then into the patch pipette. The average neuronal soma diameter and the distance from the cell to the CFE measured from photographs acquired before the recordings were 17.3 ± 3.9 μm and 16.6 ± 3.9 μm, respectively. Diffusion time (t) was calculated as t = x2/2D, where x is the distance and D is DA diffusion coefficient, 2.7*10-6 cm2/s (Nicholson, 1999). It follows that DA from the middle of the cell body (x = 25.3 μm) would reach the CFE in 1.2 seconds, which was close to the average time to oxidation peak maximum experimentally observed in IPE recordings (1.4 ± 0.8 s, mean ± SEM, n = 34 neurons; Figure S1). In contrast, it would take ∼10-times longer for DA to diffuse from a varicosity positioned 3 cell body diameters away (x = 77.1 μ); this delay would be longer still if the shape and diffusional barriers within the neurite were taken into account. We conclude that the majority of DA measured by IPE represents neurotransmitter that resided within the neuronal soma.
Possible influence of vesicular DA cargo on the DAcyt measurements
If one synaptic vesicle releases all its content inside the neuronal soma, the change in the DAcyt concentration will equal QN / NA / Vcell, where QN is vesicular quantal size (∼10,000 molecules in L-DOPA-treated neuronal synaptic vesicles (Pothos et al., 1998)), NA is Avogadro's Constant (6.023*1023 molecules/mole), and Vcell is the cell soma volume (∼1.77 pL for a cell with a diameter of 15 μm). It follows that emptying one vesicle would increase DAcyt by 10 nM, although this value might be underestimated since the effective cytosolic volume is smaller due to the presence of other organelles. In a similar scenario inside a synaptic terminal (∼1 μm in diameter), DAcyt will be increased by >30 μM.
Statistical analyses
Statistical analysis was performed in Prism 4 (GraphPad Software, La Jolla, CA), using one-way analysis of variance (ANOVA) followed by a Tukey's post-hoc test for comparisons across multiple groups or two-way ANOVA with Bonferroni post-test for the paired data.
Supplementary Material
Figure S1. IPE measurements from TH-GFP neurons.
A. Pseudo-3D image of a live ventral midbrain GFP-positive neuron from TH-GFP mice. Insets on the top and right show vertical sections of the neuron at planes indicated by the blue and red lines, respectively. An approximation of neuronal soma is shown as yellow oval.
B. Representative cyclic voltammetry trace from GFP-positive neuron treated with 100 μM L-DOPA for 1h; the current was sampled at 450 mV. Cell membrane rupture is indicated by the dotted line. The inset shows a typical catecholamine voltammogram taken at the maximum of DA oxidation wave.
Figure S2. Overexpression of VMAT2 in ventral midbrain neurons.
Representative images of mouse VM neurons (7-day post-plating) infected with rVMAT and double-stained for TH (green) and VMAT2 (red). Overlaid fluorescent images show complete colocalization of the two fluorescent markers in untreated neurons. Cultures exposed to rVMAT2 show increased VMAT2 immunoflourescence in the cell bodies of TH+ neurons, as well as an expression of the protein in non-dopaminergic cells.
As previously published1, expression of rVMAT2 was confirmed by immunostaining for both VMAT2 and the HA epitope placed in the lumenal loop between TMDs 1 and 2 that does not interfere with transporter activity or its subcellular localization. Overexpression of VMAT2 in VM neurons increased the overall transporter levels measured by western blot, and enhanced the depolarization-evoked release of DA and the total intracellular DA by ∼2-fold relative to control cells as assessed by HPLC-EC2. Transfection efficiency reached 80-90% with expression of VMAT2 protein observed in both dopaminergic and non-dopaminergic neurons.
1. Krantz, D. E., Peter, D., Liu, Y. & Edwards, R. H. Phosphorylation of a vesicular monoamine transporter by casein kinase II. J Biol Chem 272, 6752-6759 (1997).
2. Pothos, E. N. et al. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 20, 7297-306. (2000).
Figure S3. Immunocytochemistry of Cav1.3 channel in SN and VTA neurons.
Mouse VM neurons were fixed and stained for TH, calbindin and the α1D subunit of the L-type Ca2+ channels, which is uniquely present in the Cav1.3 channel. No difference in the Cav1.3 staining was observed between SN (calbindin-) and VTA (calbindin+) DA neurons. Scale bar is 20 microns.
Figure S4. Comparison of DAcyt in SN and VTA neurons before and after nimodipine treatment.
SN and VTA neurons were pre-treated with 10 μM nimodipine for 1h, and then exposed to 100 μM L-DOPA for 1h. * -p<0.05 vs. SN with L-DOPA only by one-way ANOVA.
Figure S5. Dependence of TH+ neurons survival on DAcyt dose in VM cultures treated with rVMAT.
The dose of DAcyt exposure was calculated as described in Discussion, using IPE data from mouse VM neurons transfected with rVMAT2 or an empty vector (Figure 4A). Cell survival was determined in rat VM cultures transfected with empty vector or rVMAT2 and treated with 50, 100, 500 and 1000 μM L-DOPA for 7 days. Dotted lines and shadowed boxes represent mean ± SEM in untreated cells.
Acknowledgments
This article is dedicated to Barbara and Jeffry Picower, whose own dedication to this and other projects have provided extraordinary support and inspiration. We thank the Picower Foundation, the Parkinson's Disease Foundation, the National Parkinson Foundation, NINDS Udall Center of Excellence, and NIDA for support.
Abbreviations
- AADC
aromatic L-amino acid decarboxylase
- CV
cyclic voltammetry
- DA
dopamine
- DAcyt
cytosolic DA
- DAT
dopamine uptake transporter
- GFP
green fluorescent protein
- HA
hemagglutinin
- HPLC-EC
high performance liquid chromatography with electrochemical detection
- IPE
intracellular patch electrochemistry
- L-DOPA
L-3,4-dihydroxyphenylalanine
- METH
methamphetamine
- MAO
monoamine oxidase A
- MPP+
1-methyl-4-phenylpyridinium
- PD
Parkinson's Disease
- SN
substantia nigra
- α-syn
alpha-synuclein
- TH
tyrosine hydroxylase
- VM
ventral midbrain
- VMAT2
vesicular monoamine transporter 2
- rVMAT2
recombinant VMAT2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fischer D, Henze C, Strenzke C, Westrich J, Ferger B, Hoglinger GU, Oertel WH, Hartmann A. Characterization of the striatal 6-OHDA model of Parkinson's disease in wild type and alpha-synuclein-deleted mice. Exp Neurol. 2008;210:182–193. doi: 10.1016/j.expneurol.2007.10.012. Epub 2007 Nov 2001. [DOI] [PubMed] [Google Scholar]
- Baroudi G, Qu Y, Ramadan O, Chahine M, Boutjdir M. Protein kinase C activation inhibits Cav1.3 calcium channel at NH2-terminal serine 81 phosphorylation site. Am J Physiol Heart Circ Physiol. 2006;291:H1614–1622. doi: 10.1152/ajpheart.00095.2006. [DOI] [PubMed] [Google Scholar]
- Burke RE, Antonelli M, Sulzer D. Glial cell line-derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture. J Neurochem. 1998;71:517–525. doi: 10.1046/j.1471-4159.1998.71020517.x. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O'Dell M, Li SW, Pan Y, Chung HD, Galvin JE. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, Lampe P, Kumar VB, Franko M, Williams EA, Zahm DS. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology. 2004;25:101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Colebrooke RE, Emson PC, Miller GW. Altered vesicular dopamine storage in Parkinson's disease: a premature demise. Trends Neurosci. 2008;31:303–308. doi: 10.1016/j.tins.2008.02.010. Epub 2008 May 2009. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chang L, Alicata D, Ernst T, Volkow N. Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction. 2007;102:16–32. doi: 10.1111/j.1360-0443.2006.01782.x. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Swope DM, Dashtipour K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson's disease. Clin Ther. 2007;29:1825–1849. doi: 10.1016/j.clinthera.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, Hastings TG, Kang UJ, Zhuang X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28:425–433. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien JB, Wallingford RA, Ewing AG. Estimation of free dopamine in the cytoplasm of the giant dopamine cell of Planorbis corneus by voltammetry and capillary electrophoresis. J Neurochem. 1990;54:633–638. doi: 10.1111/j.1471-4159.1990.tb01918.x. [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- Colliver T, Hess E, Pothos EN, Sulzer D, Ewing AG. Quantitative and statistical analysis of the shape of amperometric spikes recorded from two populations of cells. J Neurochem. 2000;74:1086–1097. doi: 10.1046/j.1471-4159.2000.741086.x. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Duchemin AM, Berry MD, Neff NH, Hadjiconstantinou M. Phosphorylation and activation of brain aromatic L-amino acid decarboxylase by cyclic AMP-dependent protein kinase. J Neurochem. 2000;75:725–731. doi: 10.1046/j.1471-4159.2000.0750725.x. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Bobrovskaya L, Graham ME, von Nagy-Felsobuki EI, Dickson PW. Tyrosine hydroxylase phosphorylation: regulation and consequences. J Neurochem. 2004;91:1025–1043. doi: 10.1111/j.1471-4159.2004.02797.x. [DOI] [PubMed] [Google Scholar]
- Edwards RH. Neural degeneration and the transport of neurotransmitters. Ann Neurol. 1993;34:638–645. doi: 10.1002/ana.410340504. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson's disease. J Neurol. 2005;252:IV37–IV42. doi: 10.1007/s00415-005-4008-5. [DOI] [PubMed] [Google Scholar]
- Feigin A, Fukuda M, Dhawan V, Przedborski S, Jackson-Lewis V, Mentis MJ, Moeller JR, Eidelberg D. Metabolic correlates of levodopa response in Parkinson's disease. Neurology. 2001;57:2083–2088. doi: 10.1212/wnl.57.11.2083. [DOI] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer P, Uryu K, Leight S, Trojanowski J, Lee V. Neuroinflammation and Oxidation/Nitration of {alpha}-Synuclein Linked to Dopaminergic Neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Baimbridge KG, Miller JJ. The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc Natl Acad Sci U S A. 1985;82:8780–8784. doi: 10.1073/pnas.82.24.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Mytilineou C, Cohen G. L-DOPA up-regulates glutathione and protects mesencephalic cultures against oxidative stress. J Neurochem. 1996;66:501–510. doi: 10.1046/j.1471-4159.1996.66020501.x. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Lew MF, Hurtig HI, Ondo WG, Wojcieszek J, Fitzer-Attas CJ. Long-term outcome of early versus delayed rasagiline treatment in early Parkinson's disease. Mov Disord. 2008;11:11. doi: 10.1002/mds.22402. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- Isobe C, Abe T, Kikuchi T, Murata T, Sato C, Terayama Y. Cabergoline scavenges peroxynitrite enhanced by L-DOPA therapy in patients with Parkinson's disease. Eur J Neurol. 2006;13:346–350. doi: 10.1111/j.1468-1331.2006.01207.x. [DOI] [PubMed] [Google Scholar]
- Jaffe EH, Marty A, Schulte A, Chow RH. Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J Neurosci. 1998;18:3548–3553. doi: 10.1523/JNEUROSCI.18-10-03548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon MY, Lee WY, Kang HY, Chung EJ. The effects of L-3,4-dihydroxyphenylalanine and dopamine agonists on dopamine neurons in the progressive hemiparkinsonian rat models. Neurol Res. 2007;29:289–295. doi: 10.1179/174313206X153996. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci U S A. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz DE, Peter D, Liu Y, Edwards RH. Phosphorylation of a vesicular monoamine transporter by casein kinase II. J Biol Chem. 1997;272:6752–6759. doi: 10.1074/jbc.272.10.6752. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Li H, Waites CL, Staal RG, Dobryy Y, Park J, Sulzer DL, Edwards RH. Sorting of vesicular monoamine transporter 2 to the regulated secretory pathway confers the somatodendritic exocytosis of monoamines. Neuron. 2005;48:619–633. doi: 10.1016/j.neuron.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Liang CL, Nelson O, Yazdani U, Pasbakhsh P, German DC. Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: human midbrain dopamine neurons. J Comp Neurol. 2004;473:97–106. doi: 10.1002/cne.20098. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena MA, Davila V, Sulzer D. Neurotrophic effects of L-DOPA in postnatal midbrain dopamine neuron/cortical astrocyte cocultures. J Neurochem. 1997;69:1398–1408. doi: 10.1046/j.1471-4159.1997.69041398.x. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bove J, Prou D, Hananiya A, Markov D, Poulsen N, Larsen KE, Moore CM, Troyer MD, et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszczynska A, Fitzmaurice P, Ang L, Kalasinsky KS, Schmunk GA, Peretti FJ, Aiken SS, Wickham DJ, Kish SJ. Why is parkinsonism not a feature of human methamphetamine users? Brain. 2004;127:363–370. doi: 10.1093/brain/awh046. Epub 2003 Nov 2025. [DOI] [PubMed] [Google Scholar]
- Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol. 1993;466:727–747. [PMC free article] [PubMed] [Google Scholar]
- Nicholson C. Diffusion of ions and macromolecules in brain tissue. In: Rollema H, Abercrombie E, Sulzer D, Zackheim J, editors. Monitoring Molecules in Neuroscience. Newark, NJ: Rutgers Press; 1999. pp. 71–73. [Google Scholar]
- Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P, De Yebenes J, Gershanik O, Guttman M, Grandas F, et al. Levodopa in the treatment of Parkinson's disease: current controversies. Mov Disord. 2004;19:997–1005. doi: 10.1002/mds.20243. [DOI] [PubMed] [Google Scholar]
- Pardo B, Mena MA, Casarejos MJ, Paino CL, De Yebenes JG. Toxic effects of L-DOPA on mesencephalic cell cultures: protection with antioxidants. Brain Res. 1995;682:133–143. doi: 10.1016/0006-8993(95)00341-m. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–4118. doi: 10.1523/JNEUROSCI.18-11-04106.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadhyaksha A, Husson I, Satpute SS, Kuppenbender KD, Ren JQ, Guerriero RM, Standaert DG, Kosofsky BE. L-type Ca2+ channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci. 2004;24:7464–7476. doi: 10.1523/JNEUROSCI.0612-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadhyaksha AM, Kosofsky BE. Psychostimulants, L-type calcium channels, kinases, and phosphatases. Neuroscientist. 2005;11:494–502. doi: 10.1177/1073858405278236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport S, Sulzer D, Shi WX, Sawasdikosol S, Monaco J, Batson D, Rajendran G. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J Neurosci. 1992;12:4264–4280. doi: 10.1523/JNEUROSCI.12-11-04264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ, Greenfield SA. Characteristics of electrically evoked somatodendritic dopamine release in substantia nigra and ventral tegmental area in vitro. J Neurophysiol. 1997;77:853–862. doi: 10.1152/jn.1997.77.2.853. [DOI] [PubMed] [Google Scholar]
- Sampaio-Maia B, Serrao MP, Soares-da-Silva P. Regulatory pathways and uptake of L-DOPA by capillary cerebral endothelial cells, astrocytes, and neuronal cells. Am J Physiol Cell Physiol. 2001;280:C333–342. doi: 10.1152/ajpcell.2001.280.2.C333. [DOI] [PubMed] [Google Scholar]
- Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamoto K, Nakao N, Kobayashi K, Matsushita N, Takahashi H, Kakishita K, Yamamoto A, Yoshizaki T, Terashima T, Murakami F, et al. Visualization, direct isolation, and transplantation of midbrain dopaminergic neurons. Proc Natl Acad Sci U S A. 2001;98:6423–6428. doi: 10.1073/pnas.111152398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34:903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Bogulavsky J, Larsen KE, Behr G, Karatekin E, Kleinman MH, Turro N, Krantz D, Edwards RH, Greene LA, Zecca L. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci USA. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Mosharov E, Talloczy Z, Zucca FA, Simon JD, Zecca L. Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem. 2008;106:24–36. doi: 10.1111/j.1471-4159.2008.05385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Pothos EN. Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 2007;6:933–938. doi: 10.1016/S1474-4422(07)70246-6. [DOI] [PubMed] [Google Scholar]
- Thompson L, Barraud P, Andersson E, Kirik D, Bjorklund A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. J Neurosci. 2005;25:6467–6477. doi: 10.1523/JNEUROSCI.1676-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Saheki M, Yamazaki K, Murata T. Concentration of catecholamines and indoleamines in the cerebrospinal fluid of patients with vascular parkinsonism compared to Parkinson's disease patients. J Neural Transm. 1997;104:441–449. doi: 10.1007/BF01277662. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Yamazaki K, Saheki M, Takahashi S, Tsukamoto Y. Effects of the catechol-O-methyltransferase inhibitor tolcapone in Parkinson's disease: correlations between concentrations of dopaminergic substances in the plasma and cerebrospinal fluid and clinical improvement. Neurosci Lett. 1995;192:165–168. doi: 10.1016/0304-3940(95)11636-b. [DOI] [PubMed] [Google Scholar]
- Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, Hoener M, Rodrigues CM, Alfonso A, Steer C, et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005;280:42655–42668. doi: 10.1074/jbc.M505910200. Epub 42005 Oct 42619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto EJ, Jr, Viveros OH. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci U S A. 1991;88:10754–10758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, North RA, Shefner SA, Nishi S, Egan TM. Membrane properties of rat locus coeruleus neurones. Neuroscience. 1984;13:137–156. doi: 10.1016/0306-4522(84)90265-3. [DOI] [PubMed] [Google Scholar]
- Young EA, Duchemin AM, Neff NH, Hadjiconstantinou M. Parallel modulation of striatal dopamine synthetic enzymes by second messenger pathways. Eur J Pharmacol. 1998;357:15–23. doi: 10.1016/s0014-2999(98)00497-x. [DOI] [PubMed] [Google Scholar]
- Zecca L, Tampellini D, Gerlach M, Riederer P, Fariello RG, Sulzer D. Substantia nigra neuromelanin: structure, synthesis, and molecular behaviour. Mol Pathol. 2001;54:414–418. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IPE measurements from TH-GFP neurons.
A. Pseudo-3D image of a live ventral midbrain GFP-positive neuron from TH-GFP mice. Insets on the top and right show vertical sections of the neuron at planes indicated by the blue and red lines, respectively. An approximation of neuronal soma is shown as yellow oval.
B. Representative cyclic voltammetry trace from GFP-positive neuron treated with 100 μM L-DOPA for 1h; the current was sampled at 450 mV. Cell membrane rupture is indicated by the dotted line. The inset shows a typical catecholamine voltammogram taken at the maximum of DA oxidation wave.
Figure S2. Overexpression of VMAT2 in ventral midbrain neurons.
Representative images of mouse VM neurons (7-day post-plating) infected with rVMAT and double-stained for TH (green) and VMAT2 (red). Overlaid fluorescent images show complete colocalization of the two fluorescent markers in untreated neurons. Cultures exposed to rVMAT2 show increased VMAT2 immunoflourescence in the cell bodies of TH+ neurons, as well as an expression of the protein in non-dopaminergic cells.
As previously published1, expression of rVMAT2 was confirmed by immunostaining for both VMAT2 and the HA epitope placed in the lumenal loop between TMDs 1 and 2 that does not interfere with transporter activity or its subcellular localization. Overexpression of VMAT2 in VM neurons increased the overall transporter levels measured by western blot, and enhanced the depolarization-evoked release of DA and the total intracellular DA by ∼2-fold relative to control cells as assessed by HPLC-EC2. Transfection efficiency reached 80-90% with expression of VMAT2 protein observed in both dopaminergic and non-dopaminergic neurons.
1. Krantz, D. E., Peter, D., Liu, Y. & Edwards, R. H. Phosphorylation of a vesicular monoamine transporter by casein kinase II. J Biol Chem 272, 6752-6759 (1997).
2. Pothos, E. N. et al. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 20, 7297-306. (2000).
Figure S3. Immunocytochemistry of Cav1.3 channel in SN and VTA neurons.
Mouse VM neurons were fixed and stained for TH, calbindin and the α1D subunit of the L-type Ca2+ channels, which is uniquely present in the Cav1.3 channel. No difference in the Cav1.3 staining was observed between SN (calbindin-) and VTA (calbindin+) DA neurons. Scale bar is 20 microns.
Figure S4. Comparison of DAcyt in SN and VTA neurons before and after nimodipine treatment.
SN and VTA neurons were pre-treated with 10 μM nimodipine for 1h, and then exposed to 100 μM L-DOPA for 1h. * -p<0.05 vs. SN with L-DOPA only by one-way ANOVA.
Figure S5. Dependence of TH+ neurons survival on DAcyt dose in VM cultures treated with rVMAT.
The dose of DAcyt exposure was calculated as described in Discussion, using IPE data from mouse VM neurons transfected with rVMAT2 or an empty vector (Figure 4A). Cell survival was determined in rat VM cultures transfected with empty vector or rVMAT2 and treated with 50, 100, 500 and 1000 μM L-DOPA for 7 days. Dotted lines and shadowed boxes represent mean ± SEM in untreated cells.