

Abstract
Neurons require large amounts of energy to support their survival and function, and are therefore susceptible to excitotoxicity, a form of cell death involving bioenergetic stress that may occur in several neurological disorders including stroke and Alzheimer's disease. Here we studied the roles of NAD+ bioenergetic state, and the NAD+-dependent enzymes SIRT1 and PARP-1, in excitotoxic neuronal death in cultured neurons and in a mouse model of focal ischemic stroke. Excitotoxic activation of NMDA receptors induced a rapid decrease of cellular NAD(P)H levels and mitochondrial membrane potential. Decreased NAD+ levels and poly (ADP-ribose) polymer (PAR) accumulation in nuclei were relatively early events (<4 h) that preceded the appearance of propidium iodide-and TUNEL-positive cells (markers of necrotic cell death and DNA strand breakage, respectively) which became evident by 6 h. Nicotinamide, an NAD+ precursor and an inhibitor of SIRT1 and PARP1, inhibited SIRT1 deacetylase activity without affecting SIRT1 protein levels. NAD+ levels were preserved and PAR accumulation and neuronal death induced by excitotoxic insults were attenuated in nicotinamide-treated cells. Treatment of neurons with the SIRT1 activator resveratrol did not protect them from glutamate/NMDA-induced NAD+ depletion and death. In a mouse model of focal cerebral ischemic stroke, NAD+ levels were decreased in both the contralateral and ipsilateral cortex 6 h after the onset of ischemia. Stroke resulted in dynamic changes of SIRT1 protein and activity levels which varied among brain regions. Administration of nicotinamide (200 mg/kg, i.p.) up to 1 h after the onset of ischemia elevated brain NAD+ levels and reduced ischemic infarct size. Our findings demonstrate that the NAD+ bioenergetic state is critical in determining whether neurons live or die in excitotoxic and ischemic conditions, and suggest a potential therapeutic benefit in stroke of agents that preserve cellular NAD+ levels. Our data further suggest that, SIRT1 is linked to bioenergetic state and stress responses in neurons, and that under conditions of reduced cellular energy levels SIRT1 enzyme activity may consume sufficient NAD+ to nullify any cell survival-promoting effects of its deacetylase action on protein substrates.
Keywords: Excitotoxicity, Glutamate, NMDA, NAD+, NADH, SIRT1, PARP-1, PAR, Nicotinamide, MCAO, TUNEL
Introduction
Compromised energy metabolism in brain cells is believed to play a pivotal role in the dysfunction and degeneration of neurons that occurs in disorders such as stroke and Alzheimer's disease (Mattson and Liu 2002). Neurons are excitable cells which often survive the entire life of the organism and, during that time they consume large amounts of energy to support ion-motive ATPases and other enzymes to maintain ion homeostasis, electrochemical membrane potential, and signaling functions. The energy demand of neurons in the brain is increased when they are stimulated by glutamate, an excitatory neurotransmitter critical for cognition, motor function, and other behaviors (Antzoulatos and Byrne 2004). As a major excitatory neurotransmitter, glutamate and glutamate receptors are essential for the normal function of nervous system. However, during pathological conditions with impaired energy metabolism such as cerebral ischemia, membrane depolarization results in excessive release of glutamate from the synaptic vesicles of injured neurons. Impaired glutamate reuptake through glutamate transporters in astrocytes and neurons further exacerbates the extracellular accumulation of glutamate. Excessive or prolonged stimulation of glutamate receptors, particularly N-methyl-D-aspartate (NMDA) receptors, results in Ca2+ and Na+ influx and disruption of cellular ion homeostasis. Attempts to restore cellular ion homeostasis and membrane potential through ATP-dependent pumps increase ATP consumption. Subsequent bioenergetic and oxidative stress cause degeneration and death of neurons in a process called excitotoxicity (Mattson 2003).
A principle source of energy in neurons is ATP produced during mitochondrial oxidative phosphorylation, a process which also generates harmful reactive oxygen species (ROS). Alternatively, neurons can generate ATP by glycolysis, a pathway that may be particularly important when mitochondrial function is compromised (Liu et al. 2006; Hyun et al. 2007). In addition to the increased consumption of ATP during excitotoxicity, excessive activation of glutamate receptors may also deplete cellular nicotinamide adenine dinucleotide (NAD+), an important energy substrate and cofactor in multiple metabolic reactions (Bieganowski and Brenner 2004; Belenky et al. 2006). During excitotoxic and ischemic insults, the overloading of cytosol and mitochondria with Ca2+ results in altered mitochondrial permeability and impaired oxidative phosphorylation. In the latter conditions, NAD+ may be released from mitochondria and then cleaved by glycohydrolases (Belenky et al. 2006; Lisa et al. 2001; Soane et al. 2007). In addition, metabolic and oxidative stress results in DNA strand breakage and activation of poly (ADP-ribose) polymerase-1 (PARP-1) an enzyme that depletes intracellular NAD+ stores and induces massive poly (ADP-ribose) (PAR) polymer accumulation (Ha and Snyder 2000; Du et al. 2003; Dawson and Dawson 2004; Kauppinen and Swanson 2007; Pieper et al. 2000). NAD+ also serves as an adenosine donor and source of high energy phosphate for the synthesis of ATP. Depletion of NAD+ causes inhibition of glycolysis, the TCA cycle, and mitochondrial oxidative phosphorylation, processes necessary to restore ATP levels (Sheline et al. 2000).
The function of several enzymes that play important roles in the cellular responses to stress require NAD+ for their activity including NAD+-dependent class III histone deacetylases (HDAC) particularly sirtuin 1 (SIRT1) (Blander and Guarente 2004). SIRT1 is the mammalian homologue of yeast Sir2 (silent information regulator 2), a member of sirtuin family of NAD+-dependent histone deacetylase believed to play important roles in cellular stress resistance and longevity (Tanner et al. 2000; Landry et al. 2000; Imai et al. 2000; Schmidt et al. 2004). Studies of yeast and mammalian cells have suggested that Sir2/SIRT1 can promote cell survival (Howitz et al. 2003; Cohen et al. 2004) and may protect cells against oxidative stress by activating FOXO transcription factors resulting in the induction of genes that encode antioxidant enzymes (Brunet et al. 2004; Kobayashi et al. 2005). Increased NAD+ biosynthesis and SIRT1 activation can protect axons against degeneration (Araki et al. 2004). In addition, data suggest that SIRT1 can modulate gluconeogenic and glycolytic pathways (Rodgers et al. 2005). However, the observation that Class I/II HDAC inhibitors, even Class III HDAC inhibitor such as nicotinamide, confer neuroprotective effects in models of neurodegenerative diseases (Langley et al. 2005; Green et al. 2008) makes the roles of HDAC in neuronal survival more complicated. Since both SIRT1 and PARP1 consume NAD+ for their activity, depletion of cellular NAD+ due to PARP-1 activation could influence SIRT1 activity (Pillai et al. 2005; Kolthur-Seetharam et al. 2006). Moreover, because SIRT1 activity requires NAD+, it has the potential to consume cellular energy and render neurons vulnerable to ischemia and excitotoxicity, a possibility as yet untested.
The deaths of many neurons that occur following a stroke involves, in part, excitotoxicity which can manifest as necrosis or apoptosis (Beal 1992; Greene and Greenamyre 1996; Ankarcrona et al. 1995). Increased levels of extracellular glutamate occur in the penumbra region of a focal ischemic cerebral infarct (Winfree et al. 1996), which may induce depolarization-mediated spreading depression and increased metabolic activity in peri-infarct regions under reduced oxygen tension (Hossmann 2003). NMDA antagonists reduce infarct size in experimental stroke models supporting the role of glutamate excitotoxicity (Gill et al. 1992), although their effectiveness in human subject remains to be established (Lee et al. 2000). In the present study we evaluated the involvement of NAD+ bioenergetic state and the roles of SIRT1 during glutamate mediated excitotoxicity in rat cortical neurons. We found that treatment with nicotinamide, a NAD+ precursor as well as an inhibitor of SIRT1 and PARP-1, preserves cellular NAD+ levels and increases the resistance of neurons to excitotoxicity. The neuroprotective action of NAD+ is further supported in a mouse model of focal cerebral ischemic stroke, suggesting a potential therapeutic application of nicotinamide.
Materials and Methods
Neuronal Cell Culture
Procedures for preparation and maintenance of dissociated cerebral cortical cell cultures from embryonic rat (Sprague Dawley) embryos have been described previously (Mattson et al. 1995). Briefly, cerebral hemispheres were removed, pooled, cut into small pieces, and subjected to mild trypsinization and trituration. Dissociated neurons were seeded onto polyethyleneimine-coated plastic culture dishes (for biochemical assays) or glass coverslips (for imaging analyses) in MEM media with 15% serum for 4 h; the medium was then changed to Neurobasal Media (GIBCO) containing B-27 supplements (Invitrogen), 1 mM HEPES, and 2 mM gentamycin at 37°C (in a 6% CO2/94% air atmosphere). The medium contained 0.8 mM MgC12. All experiments were performed using 7-10 day-old rat cortical or hippocampal cultures.
Time-Lapse Confocal Imaging of Nad(P)H and ΔΨm
Nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH) (together abbreviated NAD(P)H) are fluorescent in their reduced forms and nonfluorescent in their oxidized forms (Eng et al. 1989; Brennan et al. 2006). Autofluorescence of NAD(P)H was monitored (excitation 360 nm and emission 450 nm) by time-lapse confocal imaging in a HEPES-buffer solution consisting of (in mM): 145 NaCl, 5 KCl, 1.8 CaCl2, 0.8 MgCl2, 10 D-glucose, 10 HEPES (295 mOsm, pH 7.2). The average pixel intensity of neuronal cell bodies was determined using the system software. Mitochondrial membrane potential (ΔΨm) was evaluated with the fluorescent probe tetramethylrhodamine ethyl ester (TMRE, Molecular Probes) using methods described previously (Liu et al. 2006). Cells were loaded with a low concentration of TMRE (25 nM) for 20 min at 37°C, washed, and maintained in HEPES-buffered saline with TMRE (25 nM) throughout the experiments. Images of TMRE fluorescence were acquired at excitation and emission wavelengths of 549 nm and 574 nm, respectively. The time-lapse images of NAD(P)H and TMRE fluorescence were acquired using a Zeiss LSM510 confocal laser scanning microscope; images were taken by alternative sequential scanning at 10 s intervals using multiple channel configuration settings. There was no detectable interference between TMRE and NAD(P)H fluorescence signals as determined by selective closing of one fluorescence channel. The data were normalized to the baseline intensity level (ΔF/F0).
NAD+ Determination
The total intracellular NAD+ concentrations in cells or brain tissue samples were measured using a modified enzymatic cycling method (Woodley and Gupta 1971; Hinz et al. 1973). Briefly, NAD+ was extracted from fresh cells or tissue by homogenization in HClO4 at 4°C, neutralized with KOH in Gly-Gly buffer and then centrifuged at 10,000×g for 20 min. NAD+ acid extracts in the supernatant were converted to NADH by enzymatic cycling with alcohol dehydrogenase (Boehringer Mannheim), which reduces MTT (3-[4, 5-dimethylthhylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) to formazan through an intermediate, phenazine methosulfate. The rate of reduction is proportional to the concentration of coenzyme. The optical absorbance was measured at 560 nm using a plate reader after incubation at 37°C; a standard curve and equation were generated using pure β-NAD (Sigma) which represents the correlation between NAD+ concentration and optical density (OD). Values of NAD+ concentrations in samples from cells or brain tissues were calculated using the equation of the standard curve and normalized to the cell number (107) or wet weight of brain tissue samples.
SIRT1 Deacetylase Activity Assay
SIRT1/Sir2 deacetylase activity was quantified using a fluorometric assay kit (Cyclex Co., Ltd, Japan). The kit provided a SIRT1 substrate with a fluorophore and quencher attached to amino and carboxyl terminals, respectively. Deacetylation of the substrate by SIRT1/Sir2 is coupled to the protease activity of lysly endepeptidase, which cleaves the quencher from the fluorophore and allows the substrate to emit fluorescence. All measurements were performed in the presence of Trichostatin A, a powerful inhibitor of histone deacetylases (HDAC) other than SIRT1/Sir2. The fluorescence intensities were measured with a microplate fluorometer (excitation wavelength = 360 nm, emission wavelength = 450 nm). For the measurement of cellular SIRT1 deacetylase activity, nuclear proteins were extracted from rat cortical cultures or mouse brains. The fluorescence intensities of SIRT1 deacetylase activity were normalized with protein levels measured in the cell or tissue samples.
Cell Survival Assay
Cell survival was evaluated using the dye Alamar blue (resazurin) and methods described previously (Liu et al. 2006). Briefly, neurons cultured in multi-well plates (1-2 × 105/well) were exposed to experimental treatments for designated time periods. The culture medium was removed and replaced with 0.5% Alamar blue diluted in HEPES-buffer, and incubated for 30-60 min at 37°C. Levels of fluorescence were measured using a fluorescence plate reader with excitation and emission wavelengths of 540 nm and 590 nm, respectively. Values were normalized to the mean value for vehicle-treated control cells and data were presented as percentage of the cell survival in control cultures. For excitotoxic insults, cells were exposed to L-glutamic acid (glutamate) and N-methyl-D-aspartate (NMDA).
TUNEL and Propidium Iodide Staining
TUNEL (terminal dUTP nick end-labeling) labeling was performed to detect cells with nicks in their DNA strands (free 3′ ends) using a terminal deoxynucleotidyl transferase kit (TdT, Trevigen, MD) and methods described previously (Liu et al. 1999). The DNA-binding dye Hoechst 33258 fluorochrome was used to label the nuclei of all cells. The membrane impermeant fluorescent DNA-binding dye propidium iodide (PI) was used to detect necrotic cell death; PI is only able to enter cells with damaged plasma membranes. The percentage of PI or TUNEL positive cells were evaluated from multiple images (Zeiss 510) collected from 3-5 culture dishes in each control and treatment group.
Immunocytochemistry and Immunoblots
Primary cortical neuronal cultures grown on glass coverslips and fresh frozen coronal cryosections of brains (16 μm) were fixed in 4% paraformaldehyde in PBS, washed, and permeabilized with 0.2% Triton X-100 (Sigma) in PBS, then incubated with primary antibodies overnight at 4°C. The secondary antibodies were conjugated to either FITC or Texas Red. The cells were counterstained with Hoechst dye to label nuclei. For immunoblots, proteins in cell lysates or brain tissue were separated by electrophoresis in 4-12% SDS gradient NuPAGE Novex gels (Invitrogen), then transferred to a nitrocellulose membrane. The membranes were blotted with antibodies against SIRT1 (Upstate, VA) or PARP-1 (Cell Signaling) overnight at 4°C. To confirm equal protein loading, the same blot was reprobed with a mouse actin antibody (Sigma). HRP-conjugated secondary antibodies were used in all immunoblots (Vector Laboratories) and visualized by enhanced chemiluminescence (Amersham).
Permanent Focal Cerebral Ischemia Animal Model
Three-month-old adult male C57BL/6 mice weighing 25-28 g were obtained from the National Cancer Institute and maintained on a 12-h light/12-h dark cycle with continuous access to food and water. Permanent focal cerebral ischemia was induced by cauterizing the left middle cerebral artery using methods described previously (Liu et al. 2002). Briefly, mice were anesthetized using isoflurane administered as a vapor; body temperature was maintained at 35°C using a heating pad throughout the surgical procedure. The left middle carotid artery was exposed through a 1 cm vertical incision between the left eye and ear. The temporal muscle was split and a portion of the skull was removed. Focal ischemia was produced by permanent occlusion of the left middle cerebral artery by electrocoagulation. Brains were removed at 24 h following middle cerebral artery occlusion (MCAO), rinsed in cold PBS, and cut into eight coronal sections (1 mm) for TTC (2,3,5-triphenyltetrazolium chloride) staining. The infarct border in each brain slice was outlined and the stained and unstained areas quantified as described previously. All animal procedures were approved by the National Institute on Aging Animal Care and Use Committee and complied with NIH guidelines.
In Situ Hybridization and Brain Histology
In situ hybridization analysis was performed with 35S-labeled antisense and sense (as control) riboprobes on sections of mouse brain as described previously (Liu et al. 1999). The mouse cDNA clone of glucose transporter 3 (GLUT3) was a gift from Dr. Graeme I. Bell. Cryosections (16 μm) were thaw-mounted onto charged slides, fixed in 4% paraformaldehyde in PBS, dehydrated, dilapidated, and air-dried. The hybridization solution contained 107 cpm/ml riboprobe in hybridization buffer. The slides were coverslipped and placed in a 55°C humidified incubator overnight. The slides were washed through graded salt solutions, dehydrated, exposed to hyperfilm-max (Amersham) for 2 weeks, and then developed. In situ terminal deoxynucleotidyl transferase (tdt)-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling was used to detect the occurrence of DNA nicks after ischemic injury using methods described previously (Liu et al. 2002). Briefly, cryosections were rehydrated, fixed with 4% formaldehyde, and incubated with deoxynucleotide triphosphate, biotinylated deoxyuridine triphosphate, and terminal deoxynucleotidyl transferase. The conjugates were viewed with fluorescein isothiocyanate conjugateavidin. Additional brain sections were immunostained with an antibody against the neuronal marker NeuN (Chemicon, CA) as described previously (Liu et al. 2002).
Results
Excitotoxic Insults Result in Reduced NAD+ Levels and SIRT1 Deacetylase Activity in Cultured Cortical Neurons
The bioenergetic changes induced by exposure to high (excitotoxic) concentrations of glutamate and NMDA were studied in 7-10 day cultured rat primary cortical neurons. Cellular NAD(P)H levels were monitored by NAD(P)H autofluorescence and mitochondrial membrane potential (ΔΨm) was monitored simultaneously using the fluorescent indicator TMRE and time-lapse imaging (Fig. 1a). Glutamate (100 μM) induced a rapid and progressive decrease of NAD(P)H fluorescence intensity and ΔΨm (Fig. 1a, b). Because glutamate stimulates respiration, which may increase NAD(P)H oxidation, the total cellular NAD+ levels were measured using an enzymatic cycling assay at several time points following excitotoxic insults. When N-methyl-D-aspartate (NMDA, 80 μM) was added in combination with glutamate (100 μM) to cortical neurons, a significant decrease of total cellular NAD+ levels occurred within 4 h; NAD+ levels continued to decrease through 6 h and remained depressed at 24 h (Fig. 1b). SIRT1 deacetylase activity, measured in nuclear protein extracts decreased by approximately 40% within 2 h of exposure of the neurons to glutamate/NMDA, and remained at this reduced level through 24 h (Fig. 1c). Cell viability decreased significantly to approximately 60% of the control level between 4 and 6 h of exposure to glutamate/NMDA (Fig. 1d), a time period soon after the drop in NAD+ levels (Fig. 1b), suggesting the possibility that NAD+ depletion was involved in the cell death process.
Fig. 1.

Activation of glutamate receptors results in decreased mitochondrial membrane potential, and reduced levels of NAD(P)H, NAD+, and SIRT1 deacetylase activity in cortical neurons. a Representative confocal images (top) and time-lapse traces (below) of NAD(P)H autofluorescence (blue) and the mitochondrial membrane potential (ΔΨm) indicator TMRE (red) prior to and during exposure to 100 μM glutamate. Values are average intensity change from baseline intensity (ΔF/Fo) (mean and SD of 4-5 independent experiments with 15-20 neurons analyzed in each experiment). b Total cellular NAD+ levels were measured by an enzymatic cycling assay at several time points after exposure to glutamate (100 μM) and NMDA (80 μM). c SIRT1 deacetylase activity at the indicated time points following exposure of neurons to glutamate and NMDA. d Survival of cortical neurons at the indicated time points following exposure of neurons to glutamate and NMDA. *P < 0.05, **P < 0.01 compared to the time 0 value
The percentages of TUNEL-positive cells (a marker of DNA strand breakage) and propidium iodide (PI)-positive cells (necrotic cell death) were evaluated by confocal imaging (Fig. 2a). A 6 h exposure to glutamate/NMDA resulted in a significant increases in the percentages of TUNEL- and PI-positive cells compared to control cultures (Fig. 2b, c), suggesting that many neurons were dying by excitotoxic necrosis. Since DNA strand breaks activate PARP-1, the levels of PAR polymers were examined. In neurons from control cultures there was only weak PAR polymer staining in the cytosol and no detectable PAR polymer immunoreactivity in the nucleus (Fig. 2d). In contrast, at 6 h and 24 h after exposure to glutamate/NMDA, there was a significant increase in the percentage of neurons that exhibited intense PAR polymer immunoreactivity in the nucleus (Fig. 2d, e). Collectively, these results demonstrate that excessive activation of NMDA receptors causes PARP1 activation and NAD+ depletion. We therefore performed additional experiments to investigate whether NAD+ depletion plays a pivotal role in excitotoxic and ischemic neuronal death.
Fig. 2.
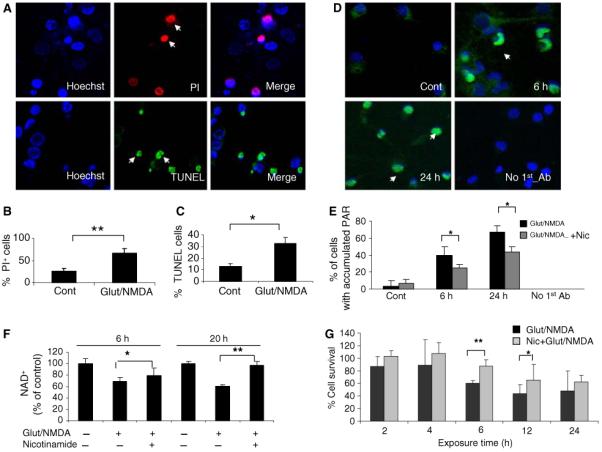
Excitotoxicity induced by glutamate and NMDA involves DNA damage and the accumulation of PAR polymers in the nuclei of cortical neurons. a Representative confocal images of TUNEL-positive (green) and PI-positive (red) cortical neurons at 6 h after exposure to glutamate (100 μM) and NMDA (80 μM). Cells were counterstained with Hoechst to show all cell nuclei. b and c Percentages of PI-positive and TUNEL-positive cells detected at 6 h after exposure to glutamate/NMDA. d Representative confocal images of poly (ADP-ribose) (PAR) polymer immunoreactivity in neurons in control cultures, and cultures that had been exposed to glutamate and NMDA for 6 h or 24 h. The lower right panel shows lack of staining of neurons (in a culture exposed to glutamate/NMDA for 24 h) when the primary antibody was omitted from the staining procedure. e Percentages of PAR polymer-positive neurons in control cultures (Con) and cultures that had been exposed to glutamate and NMDA alone or in combination with nicotinamide (2 mM) for 6 h or 24 h. f Cellular NAD+ levels in control cortical cultures, and cultures treated for 4 or 20 h with glutamate and NMDA (Glut/N) alone or in combination with 2 mM nicotinamide (Nic). *P < < 0.05, **P < 0.01
The NAD+ Precursor and SIRT1 Inhibitor Nicotinamide Prevents NAD+ Depletion and Attenuates Excitotoxic Neuronal Death
Previous findings (Ha and Snyder 2000; Mattson 2003; Dawson and Dawson 2004) and the data above show that excitotoxic insults induce compromised cellular ion homeostasis, oxidative stress, and bioenergetic deficits, which result in increased DNA strand breaks, PARP1 activation, and NAD+ depletion. We next asked whether preserving cellular NAD+ levels could protect neurons against excitotoxicity. Cultured cortical neurons were treated with nicotinamide, a precursor of NAD+ biosynthesis in the nuclear salvage pathway and also an inhibitor of SIRT1 and PARP1 activities (Grubisha et al. 2005; Bitterman et al. 2002; Virag and Szabo 2002; Klaidman et al. 2003). Treatment with nicotinamide (2 mM) attenuated PAR accumulation and preserved cellular NAD+ levels in cortical neurons exposed to glutamate and NMDA for 6 and 24 h (Fig. 2e, f). Cell survival was significantly enhanced in nicotinamide-treated cortical cultures at 6 and 12 h after exposure to glutamate and NMDA (Fig. 2g).
We next evaluated SIRT1 protein levels in cultured neurons that had been exposed to glutamate and NMDA for increasing time periods. Previous studies have shown that SIRT1 expression is induced by metabolic stress in non-neuronal cells (Brunet et al. 2004), but possible changes of SIRT1 upon glutamate receptor activation in neurons are unknown. Immunocytochemistry revealed that in untreated control cultures high levels of SIRT1 were present in the nucleus and lower levels in the cytoplasm of the majority of cortical (upper panel) and hippocampal (lower panel) neurons (Fig. 3a). As expected, SIRT1 deacetylase activity was strictly NAD+-dependent in cultured cortical neurons (Fig. 3b). SIRT1 deacetylase activity was significantly reduced in nuclear extracts of neurons exposed to glutamate and NMDA for 6 h, and was preserved in nicotinamide-treated cells, even though treatment with nicotinamide alone attenuated SIRT1 activity in neurons not exposed to glutamate and NMDA (Fig. 3b). We next performed immunoblots and densitometric quantification to determine levels of SIRT1 and PARP1 proteins (normalized to β-actin levels) in neurons that had been exposed to glutamate and NMDA for increasing time periods. Levels of both SIRT1 and full-length PARP-1 decreased during a 4 h time period in response to glutamate receptor activation (Fig. 3c). Nicotinamide treatment preserved the levels of SIRT1 and attenuated the decrease in full-length PARP-1 (Fig. 3c). However, by 6 h after exposure to glutamate and NMDA (a time point when necrotic cell death was occurring) levels of both SIRT1 and PARP-1 were greatly decreased in both vehicle and nicotinamide-treated neurons (Fig. 1d). It was previously reported that caspase-mediated cleavage or degradation of mammalian Sir2α and PARP-1 occur during apoptosis (Ohsawa and Miura 2006; Herceg and Wang 1999; Kaufmann et al. 1993). Proteolytic cleavage of PARP-1 is considered to be a cell defense response to prevent further energy depletion and cell death during metabolic insults, as mutants with caspase-resistant PARP increase rates of cell death (Boulares et al. 1999). The reduced level of SIRT1 induced by excitotoxic insults could also be a response to cellular bioenergetic crisis because its activity consumes NAD+.
Fig. 3.

Nicotinamide treatment preserves levels of cellular NAD+ and SIRT1 in neurons subjected to an excitotoxic insult. a SIRT1 immunoreactivity in cultured cortical (upper panel) and hippocampal (lower panel) neurons. SIRT1 immunoreactivity was concentrated in the nucleus and so colocalized with Hoechst (DNA-binding dye) fluorescence. b SIRT1 deacetylase activities detected in different cell-free reactions or in nuclear proteins extracted from cortical cells that had been subjected to the indicated treatments. G/N glutamate and NMDA, Nic nicotinamide (2 mM). c Immunoblot showing SIRT1 and PARP-1 (full length PARP-1 band is shown) levels in cortical neurons treated with glutamate and NMDA alone or in combination with nicotinamide for 4 or 6 h. Blots were probed with β-actin. d Densitometric analysis (lower) of full length of PARP-1 and SIRT1 levels (normalized to the β-actin level) in cortical cells that had been exposed to glutamate/NMDA for 6 h. Values are the mean and SD of determinations from 4-6 cultures. *P < 0.05, **P < 0.01
Dual Role of SIRT1 Deacetylase in Excitotoxic Neuronal Death
Our results indicate that nicotinamide can preserve cellular NAD+ levels and enhance the survival of neurons subjected to an excitotoxic insult. Nicotinamide was reported to be an inhibitor of SIRT1 (Bitterman et al. 2002; Sauve et al. 2005), and we did indeed find that levels of SIRT1 deacetylase activity decreased progressively in neurons in response to increasing concentrations of nicotinamide (Fig. 4a) and in nuclear protein extracts of neurons that had been treated with nicotinamide (Fig. 3b). Nicotinamide did not significantly alter SIRT1 protein levels at concentrations as high as 10 mM (Fig. 4b). As expected, resveratrol increased SIRT1 deacetylase activity in a concentration-dependent manner (Fig. 4a). Although nicotinamide can inhibit SIRT1 activity, it is also a precursor of NAD+ and so increases cellular NAD+ levels. To determine whether the excitoprotective action of nicotinamide might also be due, in part, to inhibition of SIRT1, we treated neurons with a specific inhibitor of SIRT1 called sirtinol. Sirtinol protected neurons from being killed by glutamate and NMDA (Fig. 4c). In addition, treatment of neurons with either NAD or NADH protected them from being killed by glutamate and NMDA (Fig. 4d), suggesting a key role for maintenance of NAD+ levels (in the absence of SIRT1 inhibition) in excitoprotection. At a low concentration (25 mM) resveratrol protected neurons from being killed by glutamate and NMDA, whereas high concentrations of resveratrol had either no effect or exacerbated excitotoxic neuronal death (Fig. 4e). In contrast to the preservation of cellular NAD+ levels in nicotinamide-treated neurons, resveratrol did not prevent glutamate/NMDA-induced NAD+ depletion (Fig. 4f). Collectively, our findings to this point suggested that cellular NAD+ levels are a critical determinant of neuronal survival during excitotoxic conditions, and that SIRT1 activity may contribute to NAD+ depletion and neuronal death under such conditions. In addition to being an NAD+ precursor, nicotinamide may preserve NAD+ levels by inhibiting NAD+ hydrolysis mediated by the activities of SIRT1 and PARP-1.
Fig. 4.
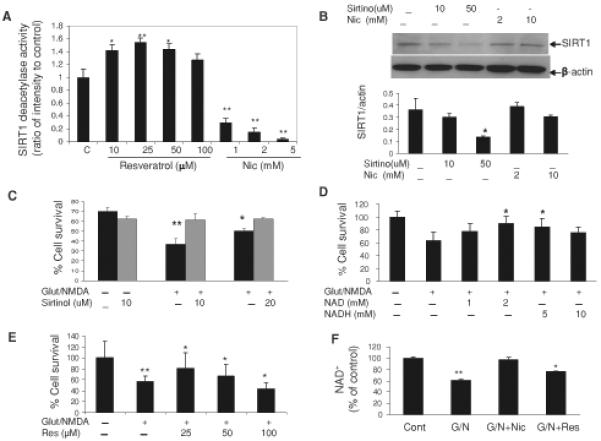
Evidence that SIRT1 activity contributes to excitotoxic neuronal death. a SIRT1 deacetylase activities in cell extracts of neurons that had been exposed to the indicated concentrations of resveratrol or nicotinamide for 2 h. b Immunoblot showing SIRT1 levels in cortical neurons that had been treated for 2 h with the indicated concentrations of nicotinamide and sirtinol. c-e Survival of cortical neurons in cultures that had been treated with the indicated concentrations of sirtinol, NAD, NADH, or resveratrol, and then exposed to glutamate/NMDA for 24 h. f NAD+ levels in cortical neurons that had been treated with indicated concentration of resveratrol for 24 h. Values are the mean and SD of determinations made in at least four separate cultures. *P< 0.05, **P < 0.01
Nicotinamide Increases Brain NAD+ Levels and is Neuroprotective in a Mouse Model of Focal Ischemic Stroke
Since glutamate-mediated excitotoxicity has been implicated in the death of neurons that occurs in ischemic stroke, we tested the potential therapeutic benefit of nicotinamide in a mouse model of focal permanent cerebral ischemia (Fig. 5a). Despite the fact that cellular ATP levels decrease significantly within 1 h of the onset of ischemia (Hata et al. 2000), levels of mRNA encoding the neuronal glucose transporter GLUT3 (detected by in situ hybridization using an 35S-labeled mouse GLUT3 riboprobe) were increased in the ipsilateral cortex (infarct area) at early time points (3 h) following middle cerebral artery occlusion (MCAO) (Fig. 5b), indicating that a large portion of neurons were still viable and responding to the metabolic stress during early post-stroke time period. The intensity of GLUT3 mRNA expression was reduced in the ischemic core of the infarct at 6 h post-MCAO, but remained elevated in the penumbra (Fig. 5b). There was a highly significant decrease in the NAD+ levels in the ipsilateral cortex collected at 6 h following MCAO, and a smaller decrease in NAD+ levels in the contralateral cortex, compared to the sham group (Table 1). The SIRT1 deacetylase activity level was significantly decreased in tissue samples from the ipsilateral cortex at 6 h post-MCAO (Fig. 5c), as neurons were dying (Fig. 5d). Interestingly, SIRT1 deacetylase activity then increased to or above basal levels at 12 h and 24 h post-MCAO, possibly as the result of its upregulation in activated glial cells (Liu et al. 1999). There was an increased level of SIRT1 protein during the first 3 h post-MCAO in both contralateral and ipsilateral regions of cerebral cortex (Fig. 5d), suggesting that SIRT1 is involved in the early cellular stress responses. SIRT1 protein levels were decreased at 6 h post-MCAO (Fig. 5e), corresponding to reduced brain NAD+ levels detected at the same time point (Table 1).
Fig. 5.

Levels of SIRT1 protein and enzymatic activity decrease transiently in the ischemic cerebral cortex following middle cerebral artery occlusion (MCAO). a Representative coronal sections stained with hematoxylin and eosin (H&E) showing the infarct, penumbra, and contralateral brain regions. b Autoradiographs showing the expression of glucose transporter 3 (GLUT3) mRNA detected by in situ hybridization with 35S labeled riboprobes in coronal brain sections from a sham-operated control mouse, and mice at 3 or 6 h after MCAO. Arrows point to the infarct area (3 h time point) and the penumbra (6 h time point). c SIRT1 deacetylase activities in the contralateral and ipsilateral cortex regions of brains collected at different time points after MCAO. d and e Representative immunoblots showing SIRT1 protein levels in the contralateral and ipsilateral regions of cerebral cortex at 3 h and 6 h post-MCAO, and densitometric quantification normalized to β-actin. There was an increase of SIRT1 levels in the contralateral and ipsilateral cortex compared to sham group, but the levels were decreased at 6 h post-MCAO. Values are the mean and SD of determinations made in at least three different experiments *P < 0.05, **P < 0.01
Table 1.
Brain tissue NAD+ levels following focal ischemia (MCAO) in mice
| Group | Time (h) | Region | NAD ± (nMoles/mg tissue) |
|---|---|---|---|
| Sham | 6 | Cortex | 696.23 ± 209.41 |
| Saline | 6 | Contralateral cortex | 459.04 ± 115.12a |
| Saline | 6 | Ipsilateral cortex | 149.69 ± 106.81a,b |
| Nicotinamide | 6 | Contralateral cortex | 1111.3 ± 394.96a,b |
| Nicotinamide | 6 | Ipsilateral cortex | 270.54 ± 197.87a,c |
Samples of cerebral cortex were collected from sham operated control mice, and saline or nicotinamide (250 mg/kg) treated mice that had been subjected to MCAO and killed 6 h later. NAD+ levels (nMoles/mg wet tissue) were measured using and enzymatic cycling assay. Values are the mean ± SD from 5-7 animals in each group
P < 0.05 compared to the sham value
P < 0.01 compared to the contralateral cortex of saline-treated mice
P < 0.05 compared to the ipsilateral cortex of saline-treated mice
To test the hypothesis that prevention of NAD+ depletion would have a therapeutic benefit in stroke, we administered nicotinamide (i.p. 200 mg/kg) or saline 1 h after MCAO. Measurements of infarct areas and volumes demonstrated that nicotinamide administration significantly reduced infarct area and infarct volume (Fig. 6a-c). Levels of NAD+ in the ischemic cerebral cortex were maintained at higher levels in nicotinamide-treated mice compared to saline-treated control mice (Table 1). In addition, there was a significant increase in NAD+ levels in the contralateral cortex of nicotinamide-treated mice compared to saline-treated control mice as detected at 6 h post-MCAO (Table 1). The reduced ischemic infarct size in nicotinamide-treated mice suggested that enhancing cellular NAD+ levels protects neurons from delayed neuronal death and thus prevents infarct expansion. Examination of NeuN- and Hoechst-stained coronal brain sections showed that neurons were dying at 6 h post-MCAO (loss of NeuN-positive cells; Fig. 6d). In situ TUNEL staining showed that following MCAO, DNA strand breaks (TUNEL-positive cells) were detected in the ipsilateral hemisphere and were especially abundant in the penumbra region (Fig. 6d). TUNEL-positive cells were relatively sparse at 6 h post-MCAO, but were increased at 12-48 h after the onset of ischemia (Liu et al. 1999, 2002).
Fig. 6.
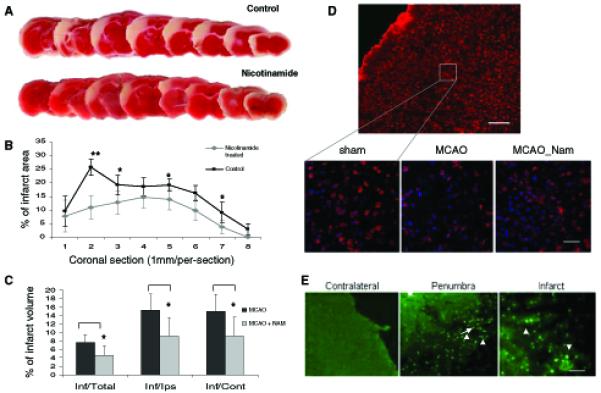
Nicotinamide treatment reduces ischemic brain injury in a permanent MCAO model of stroke. a Representative rostro-caudal series of TTC-stained coronal sections 24 after MCAO in a control mouse and a nicotinamide-treated mouse. b Quantification of infarct areas in saline-treated control mice and nicotinamide treated mice; values are the mean and SD (n = 5 or 6 mice per group). c Infarct volumes in saline-treated control mice and nicotinamide treated mice. *P < 0.05, **P 0.01. d The upper image shows a brain section from a sham-operated control mouse stained with neuronal marker NeuN (red) and counterstained with Hoechst (blue). The lower images are representative high magnification views of NeuN and Hoechst staining in the same region of the cerebral cortex from a sham-operated, a saline-treated, and a nicotinamide-treated mouse killed at 6 h after MCAO. Note that ischemia induced neuronal death (as indicated by decreased density of NeuN-positive cells) and nicotinamide treatment reduced the amount of neuronal loss. Scale bar = 50 μm. e Representative coronal sections showing TUNEL-positive cells (intense green fluorescent puncta) in the penumbra and infarct of the ipsilateral side of mouse brain 24 h after MCAO. Scale bar = 100 μm
Discussion
When glutamate receptors are overstimulated, particularly under conditions of metabolic or oxidative stress, the ability of neurons to maintain the function of vital ATP-dependent ion pumps and NAD+-dependent enzymes is compromised and the neurons may die. Previous findings suggested that glutamate-mediated excitotoxicity via NMDA receptors results in PARP-1 activation and cell death (Kauppinen and Swanson 2007; Pieper et al. 2000; Sheline et al. 2000). In the present study, we examined NAD+ related bioenergetic changes and the role of NAD+ linked histone deacetylase sirtuins, mainly SIRT1 on neuronal vulnerability, during glutamate induced excitotoxicity and cerebral ischemia in mice. Our findings suggest that cellular NAD+ levels play a critical role in determining the fate of neurons subjected to excitotoxic and ischemic conditions (Fig. 7). In response to excitotoxic insults, NAD+ levels drop in neurons subsequent to elevation of [Ca2+]c and [Ca2+]m, depolarization of ΔΨm, increased ROS production, and decreased NAD(P)H levels. NAD+ depletion occurs prior to concomitant to cell death. Preventing NAD+ depletion by treatment with nicotinamide attenuates excitotoxic neuronal death, preserves SIRT1 levels, and reduces PAR accumulation (marker of PARP-1 activation). In addition to nicotinamide, we found that treatment with NAD+ or its reduced form NADH+, or BDNF (a neurotrophin which increases glucose uptake and cellular NAD+ levels; Liu et al. 2008) also protects neurons against excitotoxicity.
Fig. 7.
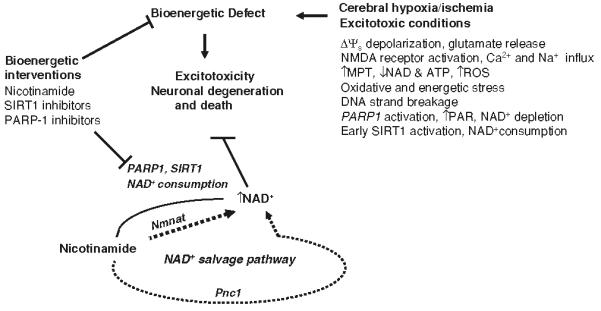
Model of the neurodegenerative cascade of events in excitotoxicity and ischemic brain injury with a focus on the neuroprotective role of NAD+. During cerebral ischemia, decreased energy and oxygen supply results in compromised brain energy metabolism and plasma membrane depolarization, which causes excessive release of the excitatory neurotransmitter glutamate. Activation of glutamate receptors, particularly NMDA receptors, causes Ca2+ and Na+ influx. Increased Ca2+ concentrations in the cytosol and mitochondria results in bioenergetic and oxidative stress, and mitochondrial membrane permeability transition (MPT) pore opening. As the result of increased levels of reactive oxygen species (ROS), DNA strand breaks, PARP-1 activation and PAR polymer formation occur. The activities of PARP-1 and SIRT1 further deplete cellular NAD+ levels. Bioenergetic interventions, including nicotinamide and PARP-1 and SIRT1 inhibitors, can preserve cellular NAD+ levels and protect neurons against ischemic/excitotoxic damage and death
Previous studies have shown that decreases in ATP levels occur within minutes in cultured neurons following excitotoxic insults (Ankarcrona et al. 1995) and within 1 h following cerebral ischemia in rodent brains (Hata et al. 2000). However, data from the present study and our previous studies of primary cortical neuronal cultures exposed to excitotoxic insults, and a mouse focal ischemic stroke model (Liu et al. 1999, 2002) suggest that many neurons survive more than 1 h as indicated by increased neuronal glucose transporter GLUT3 mRNA expression at early time points after MCAO. The latter “endangered” neurons, which are mainly located in the penumbra region in the stroke model, can be rescued by treatments aimed at preventing energetic depletion. We found that a significant drop of NAD+ levels in the ipsilateral cortex, and to a lesser extent in the contralateral cortex, was detected at 6 h following MCAO. The SIRT1 deacetylase activity level was reduced in nuclear protein extracts from ipsilateral cerebral cortical tissue collected at 6 h following MCAO, a result consistent with the decreased SIRT1 protein levels in the ipsilateral cortex detected in immunoblots. The depletion of NAD+ would be further exacerbated with the increase of DNA strand breakage (and associated PARP1 activation) as detected by increased TUNEL positive cells after 6 h. The neuroprotective effect of nicotinamide, as a NAD+ precursor in nuclear salvage pathway and proposed inhibitor of PARP1 and SIRT1, is associated with partial preservation of NAD+ levels due to its roles as both a precursor of NAD+ biosynthesis and an inhibitor of the NAD+-consuming enzymes SIRT1 and PARP1. In addition to its ability to protect neurons in models of stroke, nicotinamide has also been reported to be neuroprotective in animal models of Parkinson's disease (Schulz et al. 1995) and Alzheimer's disease (Green et al. 2008).
Studies of yeast and cultured mammalian cells indicate that Sir2/SIRT1 can increase replicative lifespan and cellular stress resistance (Blander and Guarente 2004), but may reduce the chronological lifespan of cells under certain conditions (Fabrizio et al. 2005). Previous findings concerning the influence of SIRT1 on neuronal stress resistance and survival are conflicting. SIRT1 activity plays an important role in maintaining the viability of at least some types of cells because most SIRT1 null mice die during the postnatal period and exhibit retardation in growth and defects in development (McBurney et al. 2003). SIRT1 deacetylase activity is dependent upon NAD+, and NAD+ depletion during excitotoxic and ischemic conditions would therefore be expected to decrease SIRT1 activity (Yang and Sauve 2006), although SIRT1 activity may not be affected by NAD+ fluctuations within the physiological range (Anderson et al. 2003). Our findings suggest that SIRT1 levels and activity in neurons are influenced by cellular NAD+ levels, and that nicotinamide treatment attenuates depletion of NAD+ and preserves SIRT1 activity in neurons under excitotoxic and ischemic conditions.
While SIRT1 may play a role in maintaining cell viability under conditions of sufficient energy availability, we found that the SIRT1-activating agent resveratrol could not rescue neurons under excitotoxic conditions, and even exacerbated excitotoxic neuronal death at higher concentrations (data not shown). Similar death-promoting effects of resveratrol and SIRT1 have been observed in models of zinc-induced cytotoxicity (Cai et al. 2006). The latter finding is of interest because zinc is released from excitatory synapses and may contribute to excitotoxicity and neuronal death following ischemia (Choi and Koh 1998). Other studies show that resveratrol can promote apoptosis of tumor cells and some types of normal mitotic cells (Clement et al. 1998; Gao et al. 2002). Consistent with our findings, a recent study showed that heart-specific overexpression of SIRT1 increases the vulnerability of cardiac myocytes to age-dependent apoptosis, whereas lower levels of SIRT1 overexpression were protective, possibly by activating an adaptive stress response pathway (Alcendor et al. 2007). We found that the SIRT1 inhibitors nicotinamide and sirtinol protected neurons against excitotoxicity, a finding consistent with previous data demonstrating neuroprotective effects of sirtuin inhibitors in models of Alzheimer's and Huntington's diseases (Green et al. 2008; Butler and Bates 2006). Pretreatment with resveratrol has been reported to protect neurons against ischemic cell death, apparently by inducing an adaptive stress response (Raval et al. 2006). Our findings suggest that SIRT1 may promote cell survival and be involved in adaptive stress responses under conditions where cells have sufficient NAD+; however, when NAD+ levels are limited, stimulating SIRT1 activity may render cells vulnerable to death.
Our findings indicate that cellular NAD+ bioenergetic state is a critical factor in determining the fate of neuronal survival in excitotoxic and ischemic conditions. Previous studies have supported the neuroprotective potential of treatments that increase cellular energy levels, with creatine being one prominent example (Matthews et al. 1998, 1999; Sullivan et al. 2000; Tarnopolsky and Beal 2001). Here we found that nicotinamide partially preserved cellular NAD+ levels and was effective in protecting neurons against ischemic injury in a mouse stroke model at a dose of 200 mg/kg administered 1 h after the onset of permanent MCAO ischemia. The latter findings are consistent with previous reports of neuroprotective effects of nicotinamide in other models of hypoxic/ischemic brain damage (Sadanaga-Akiyoshi et al. 2003; Yang et al. 2002; Feng et al. 2006). The neuroprotective effects of nicotinamide appear to result from both elevation of NAD+ levels and sirtuin inhibition. Preventing NAD+ depletion, either by increasing its biosynthesis or reducing its consumption, could be an important therapeutic strategy for protecting neurons against ischemia, excitotoxic insults involving DNA damage and age-related metabolic impairment.
Acknowledgments
We would like to thank Graeme I. Bell for providing us the mouse GLUT3 cDNA clone used for riboprobe preparation and in situ hybridization. This research was supported by the Intramural Research Program of the National Institute on Aging.
References
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation Research. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Latorre-esteves M, Neves AR, Lavu S, Medvedik O, Taylor C, et al. Yeast life-span extension by calorie restriction is independent of NAD+ fluctuation. Science. 2003;302:2124–2126. doi: 10.1126/science.1088697. doi:10.1126/science.1088697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. doi:10.1016/ 0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Antzoulatos EG, Byrne JH. Learning insights transmitted by glutamate. Trends in Neurosciences. 2004;27:555–560. doi: 10.1016/j.tins.2004.06.009. doi:10.1016/j.tins.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD+biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. doi:10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mechanisms of excitotoxicity in neurological diseases. The FASEB Journal. 1992;6:3338–3344. [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends in Biochemical Sciences. 2006;32:12–19. doi: 10.1016/j.tibs.2006.11.006. doi:10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. doi:10.1016/S0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. The Journal offf Biological Chemistry. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. doi:10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual Review of Biochemistry. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. doi:10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, et al. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis: Caspase 3-resistant parp mutant increases rates of apoptosis in transfected cells. The Journal of Biological Chemistry. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. doi:10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. Journal of Cerebral Blood Flow and Metabolism. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. doi:10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. doi:10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nature Reviews. Neuroscience. 2006;7:784–796. doi: 10.1038/nrn1989. doi:10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- Cai AL, Zipfel GJ, Sheline CT. Zinc neurotoxicity is dependent on intracellular NAD+ levels and the sirtuin pathway. The European Journal of Neuroscience. 2006;24:2169–2176. doi: 10.1111/j.1460-9568.2006.05110.x. doi:10.1111/j.1460-9568.2006.05110.x. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY. Zinc and brain injury. Annual Review of Neuroscience. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. doi:10.1146/annurev.neuro. 21.1.347. [DOI] [PubMed] [Google Scholar]
- Clement MV, Hirpara JL, Chawdhury SH, Pervaiz S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood. 1998;92:996–1002. [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wal NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. doi:10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Deadly conversations: Nuclearmitochondrial cross-talk. Journal of Bioenergetics and Biomembranes. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. doi:10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. The Journal of Biological Chemistry. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. doi:10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Eng J, Lynch RM, Balaban RS. Nicotinamide adenine dinucleotide fluorescence spectroscopy and imaging of isolated myocytes. Biophysical Journal. 1989;55:621–630. doi: 10.1016/S0006-3495(89)82859-0. doi:10.1016/ S0006-3495(89)82859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Gattazzo C, Battistella L, Wei M, Cheng C, McGrew K, et al. Sir2 blocks extreme life-span extension. Cell. 2005;18:655–667. doi: 10.1016/j.cell.2005.08.042. doi:10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- Feng Y, Paul IA, LeBlanc MH. Nicotinamide reduces hypoxic ischemic brain injury in the newborn rat. Brain Research Bulletin. 2006;69:117–122. doi: 10.1016/j.brainresbull.2005.11.011. doi:10.1016/j.brainresbull. 2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Xu YX, Divine G, Janakiraman N, Chapman RA, Gautam SC. Disparate in vitro and in vivo antileukemic effects of resveratrol, a natural polyphenolic compound found in grapes. The Journal of Nutrition. 2002;132:2076–2081. doi: 10.1093/jn/132.7.2076. [DOI] [PubMed] [Google Scholar]
- Gill R, Andine P, Hillerd L, Persson L, Hagberg H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischemia in the rat. Journal of Cerebral Blood Flow and Metabolism. 1992;12:371–379. doi: 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, et al. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. Journal of Neuroscience. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. doi:10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Progress in Neurobiology. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. doi:10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- Grubisha O, Smith BC, Denu JM. Small molecule regulation of sir2 protein deacetylase. FEBS. 2005;272:4607–4616. doi: 10.1111/j.1742-4658.2005.04862.x. doi:10.1111/j.1742-4658.2005.04862.x. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiology of Disease. 2000;7:225–239. doi: 10.1006/nbdi.2000.0324. doi:10.1006/nbdi.2000.0324. [DOI] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA. Dynamics of regional brain metabolism and gene expression after middle cerebral artery occlusion. Journal of Cerebral Blood Flow and Metabolism. 2000;20:306–315. doi: 10.1097/00004647-200002000-00012. doi:10.1097/00004647-200002000-00012. [DOI] [PubMed] [Google Scholar]
- Herceg Z, Wang ZQ. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Molecular and Cellular Biology. 1999;19:5124–5133. doi: 10.1128/mcb.19.7.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Katsilambros N, Maier V, Schatz H, Pfeiffer EF. Significance of streptozotocin induced nicotinamideadenine-dinucleotide (NAD+) degradation in mouse pancreatic islets. FEBS Letters. 1973;30:225–230. doi: 10.1016/0014-5793(73)80656-8. doi:10.1016/0014-5793(73) 80656-8. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Glutamate hypothesis of stroke. Fortschritte der Neurologie, Psychiatrie, und ihrer Grenzgebiete. 2003;71(Suppl 1):S10. doi: 10.1055/s-2003-40500. doi:10.1055/s-2003-40500. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. doi:10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. Journal of Neurochemistry. 2007;100:1364–1374. doi: 10.1111/j.1471-4159.2006.04411.x. doi:10.1111/j.1471-4159.2006.04411.x. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD? dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. doi:10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Research. 1993;53:3976–3985. [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience. 2007;145:1267–1272. doi: 10.1016/j.neuroscience.2006.09.034. doi:10.1016/j.neuroscience.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Klaidman L, Morales M, Kem S, Yang J, Chang ML, Adams JD. Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology. 2003;69:150–157. doi: 10.1159/000072668. doi:10.1159/000072668. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Furukawa-Hibi Y, Chen C, Horio Y, Isobe K, Ikeda K, et al. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. International Journal of Molecular Medicine. 2005;16:237–243. [PubMed] [Google Scholar]
- Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5:873–877. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, et al. The silencing protein SIR2 and its homologs are NAD+-dependent protein deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. doi:10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Current Drug Targets. CNS Neurological Disorders. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Lee JB, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. The Journal of Clinical Investigation. 2000;106:723–731. doi: 10.1172/JCI11003. doi:10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisa FD, Menabo R, Canton M, Baria M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytesin postischemic reperfusion of the heart. The Journal of Biological Chemistry. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. doi:10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, et al. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Medicine. 2006;8:389–414. doi: 10.1385/NMM:8:3:389. doi:10.1385/NMM:8:3:389. [DOI] [PubMed] [Google Scholar]
- Liu D, Lu C, Wan R, Auyeung WW, Mattson MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome C. Journal of Cerebral Blood Flow and Metabolism. 2002;22:431–433. doi: 10.1097/00004647-200204000-00007. doi:10.1097/00004647-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Liu D, Pitta M, Mattson M. Preventing NAD+ depletion protects neurons against excitotoxicity: Bioenergetic effects of mild mitochondrial uncoupling, caloric restriction. Annals of the New York Academy of Sciences. 2008;1147:275–282. doi: 10.1196/annals.1427.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, et al. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Molecular Brain Research. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. doi:10.1016/S0169-328X(99)00063-7. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, et al. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Experimental Neurology. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. doi:10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, et al. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington's disease. Journal of Neuroscience. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: Abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Medicine. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. doi:10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods in Cell Biology. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. doi:10.1016/ S0091-679X(08)61930-5. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Liu D. Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. Neuromolecular Medicine. 2002;2:215–231. doi: 10.1385/NMM:2:2:215. doi:10.1385/NMM:2:2:215. [DOI] [PubMed] [Google Scholar]
- McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Molecular and Cellular Biology. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. doi:10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa S, Miura M. Caspase-mediated changes in Sir2a during apoptosis. FEBS Letters. 2006;580:5875–5879. doi: 10.1016/j.febslet.2006.09.051. doi:10.1016/ j.febslet.2006.09.051. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Blackshaw S, Clements EE, Daniel J, Brat DJ, Krug DK, et al. Poly(ADP-ribosyl)ation basally activated by DNA strand breaks reflects glutamate-nitric oxide neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1845–1850. doi: 10.1073/pnas.97.4.1845. doi:10.1073/pnas.97.4.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced sir2α deacetylase activity. The Journal of Biological Chemistry. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. doi:10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Perez-Pinzon MA. Resveratrol mimics ischemic preconditioning in the brain. Journal of Cerebral Blood Flow and Metabolism. 2006;26:1141–1147. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1αnd SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. doi:10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Sadanaga-Akiyoshi F, Yao H, Tanuma S, Nakahara T, Hong JS, Ibayashi S, et al. Nicotinamide attenuates focal ischemic brain injury in rats: With special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochemical Research. 2003;28:1227–1234. doi: 10.1023/a:1024236614015. doi: 10.1023/A:1024236614015. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Moir RM, Schramm VL, Willis IM. Chemical activation of sir2-dependent silencing by relief of nicotinamide inhibition. Molecular Cell. 2005;17:595–601. doi: 10.1016/j.molcel.2004.12.032. doi: 10.1016/j.molcel.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: Implications for physiological regulation. The Journal of Biological Chemistry. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. doi:10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Henshaw DR, Matthews RT, Beal MF. Coenzyme Q10 and nicotinamide and a free radical spin trap protect against MPTP neurotoxicity. Experimental Neurology. 1995;132:279–283. doi: 10.1016/0014-4886(95)90033-0. doi:10.1016/0014-4886(95)90033-0. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD+ and inhibition of glycolysis. The Journal of Neuroscience. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soane L, Kahraman S, Kristian T, Fiskum G. Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. Journal of Neuroscience Research. 2007;85:3407–3415. doi: 10.1002/jnr.21498. doi:10.1002/jnr.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. Dietary supplement creatine protects against traumatic brain injury. Annals of Neurology. 2000;48:723–729. doi:10.1002/1531-8249(200011)48:5<723::AID-ANA5>3.0.CO;2-W. [PubMed] [Google Scholar]
- Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD-dependent histone/ protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. doi:10.1073/ pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky MA, Beal MF. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Annals of Neurology. 2001;49:561–574. doi: 10.1002/ana.1028. [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly (ADP-ribose) polymerase inhibitors. Pharmacological Reviews. 2002;54:375–429. doi: 10.1124/pr.54.3.375. doi:10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Winfree CJ, Baker CJ, Connoly ES, Fiore AJ, Solomon RA. Mild hypothermia reduces penumbral glutamate levels in the rat permanent focal cerebral ischemia model. Neurosurgery. 1996;38:1216–1222. doi: 10.1097/00006123-199606000-00034. doi:10.1097/00006123-19960 6000-00034. [DOI] [PubMed] [Google Scholar]
- Woodley CL, Gupta NK. New enzyme cycling method for determination of oxidized and reduced nicotinamide adenine dinucleotide. Analytical Biochemistry. 1971;43:341–348. doi: 10.1016/0003-2697(71)90262-4. [DOI] [PubMed] [Google Scholar]
- Yang J, Klaidman LK, Chang ML, Kem S, Sugawara T, Chan P, et al. Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacology, Biochemistry, and Behavior. 2002;73:901–910. doi: 10.1016/s0091-3057(02)00939-5. doi:10.1016/S0091-3057(02)00939-5. [DOI] [PubMed] [Google Scholar]
- Yang T, Sauve AA. NAD+ metabolism and sirtuins: Metabolic regulation of protein deacetylation in stress and toxicity. The AAPS Journal. 2006;8:632–643. doi: 10.1208/aapsj080472. doi:10.1208/ aapsj080472. [DOI] [PMC free article] [PubMed] [Google Scholar]