

Abstract
RNA-binding proteins play a key role in post-transcriptional regulation of mRNA stability and translation. We have identified that RBM3, a translation regulatory protein, is significantly upregulated in human tumors, including a stage-dependent increase in colorectal tumors. Forced RBM3 overexpression in NIH3T3 mouse fibro-blasts and SW480 human colon epithelial cells increases cell proliferation and development of compact multicellular spheroids in soft agar suggesting the ability to induce anchorage-independent growth. In contrast, down-regulating RBM3 in HCT116 colon cancer cells with specific siRNA decreases cell growth in culture, which was partially overcome when treated with prostaglandin E2, a product of cyclooxygenase (COX)-2 enzyme activity. Knockdown also resulted in the growth arrest of tumor xenografts. We have also identified that RBM3 knockdown increases caspase-mediated apoptosis coupled with nuclear cyclin B1, and phosphorylated Cdc25c, Chk1 and Chk2 kinases, implying that under conditions of RBM3 downregulation, cells undergo mitotic catastrophe. RBM3 enhances COX-2, IL-8 and VEGF mRNA stability and translation. Conversely, RBM3 knockdown results in loss in the translation of these transcripts. These data demonstrate that the RNA stabilizing and translation regulatory protein RBM3 is a novel proto-oncogene that induces transformation when overexpressed and is essential for cells to progress through mitosis.
Keywords: cyclooxygenase-2, RNA stability, transformation, mitosis, cell cycle
Introduction
Dysregulated expression of oncogenes and tumor suppressors is a critical regulator of tumorigenesis. Known targets that lead to a tumorigenic phenotype include cyclooxygenase (COX)-2, interleukin (IL)-8 and vascular endothelial growth factor (VEGF) (Dubois et al., 1998; Dixon et al., 2001; Wang et al., 2005). COX-2 is the rate-limiting enzyme in the production of prostaglandins, an important mediator of various cellular processes including increased proliferation, apoptosis resistance and enhanced angiogenesis (Mukhopadhyay et al., 2003b; Krysan et al., 2005). COX-2 overexpression occurs in multiple tumors, and can be observed at various stages of tumorigenesis (Eberhart et al., 1994). Although transcriptional activation of COX-2 is an early event, it is also regulated at the post-transcriptional levels of mRNA stability and translation (Dixon et al., 2000).
Distinct cis-acting AU-rich elements (ARE) sequences located within the 3′untranslated region (3′UTR) have been identified in the COX-2, IL-8 and VEGF mRNA that regulate mRNA stability and translation (Ristimaki et al., 1996; Cok and Morrison, 2001; Dixon et al., 2001). Specifically, the first 60 nucleotides in COX-2 3′UTR encode AREs, which regulate mRNA stability and translation (Cok and Morrison, 2001; Mukhopadhyay et al., 2003a). RNA-binding protein HuR interacts with these ARE sequences to regulate the stability and translation of COX-2 mRNA (Dixon et al., 2000; Cok and Morrison, 2001). HuR is also upregulated in various cancers (Nabors et al., 2001; Erkinheimo et al., 2003; Denkert et al., 2004, 2006).
Here, we demonstrate that RBM3 is a proto-oncogene, whose product binds COX-2 ARE and regulates COX-2 mRNA stability and translation (Danno et al., 1997). RBM3 mRNA and protein are upregulated in human tumors. RBM3 induces non-transformed cells to grow in an anchorage-independent manner. In contrast, downregulating RBM3 with short interfering RNA (siRNA) decreases HCT116 colon adenocarcinoma cell proliferation. Moreover, there is an increase in apoptosis and activation of checkpoint-related proteins that enhance cell cycle progression at the level of mitosis suggesting mitotic catastrophe. Furthermore, downregulating RBM3 in nude mice tumor xenografts decreased angiogenesis. We also demonstrate that RBM3 shuttles between nucleus and cytoplasm.
Results
RBM3 is induced in colon cancers
RBM3 regulates global mRNA translation by interacting with the 60S ribosome (Derry et al., 1995; Danno et al., 1997; Dresios et al., 2005). In addition, RBM3 was identified through its binding to AU-rich sequences in COX-2 3′UTR (Cok and Morrison, 2001). As COX-2 is significantly upregulated in cancers, we first examined RBM3 expression in colon cancers. There was a stage-dependent increase in RBM3 mRNA levels compared to the paired uninvolved tissues, with the highest levels observed in later stages (Figure 1a). Two novel isoforms, resulting from alternative splicing were recently identified in neurons (Smart et al., 2007). However, these isoforms were not observed in the colon cancer cells (data not shown). HuR was also significantly upregu-lated in the cancers, confirming previous reports (Dixon et al., 2001; Nabors et al., 2001; Erkinheimo et al., 2003). Details of colorectal adenocarcinoma and specific expression pattern for the three genes are presented in Supplementary Table 1. Western blot analyses for protein expression confirmed that both RBM3 and HuR are expressed at higher levels in the tumors, with at least a 10-fold increase in RBM3 (Figure 1b). Immuno-histochemistry analyses demonstrated RBM3 expression in a single cell within normal, human colonic crypts (Figure 1c). However, in the cancer tissues, the expression was widespread, and both nuclear and cytoplasmic, especially at the later stages of tumorigen-esis (Figure 1c). In addition, high levels of RBM3 expression was observed in other tumors, including breast, pancreas, colon, lung, ovary and prostate (Figure 1d). As a reference, the tumor presented for colon in Figure 1d is from an adenocarcinoma. High levels of HuR expression were also observed in breast, colon, lung and ovarian tumors (Figure 1d). Thus, expression of RBM3 is significantly induced in cancers and is localized in both the nucleus and cytoplasm.
Figure 1.

RBM3 and HuR are overexpressed in tumors. (a) RBM3 and HuR gene expression in tumor and surrounding uninvolved tissues. Significant induction of RBM3 mRNA expression was observed in stages 2–4, whereas HuR was induced only in stage 1. * denote statistically significant differences (*P<0.01). (b) Western blot analyses of total tissue extracts for RBM3 and HuR. Actin was determined as control for gel loading. RBM3 expression is significantly upregulated in the tumors. (c) Immunohistochemistry for RBM3 in normal and colon cancer tissues. Brown stain demonstrates the location of the RBM3 protein in the tissues. (d) Immunohistochemistry for HuR and RBM3 in various human tumors. Brown stain shows the location of the protein.
RBM3 overexpression induces anchorage-independent growth
To determine whether RBM3 overexpression affects growth rate, NIH3T3 cells stably expressing RBM3 were generated. There was a significantly higher level of proliferation in the RBM3-transfected cells when compared to the wild-type, vector-transfected controls (Figure 2a). Next, we tested whether RBM3 expressing cells can grow in an anchorage-independent manner, a characteristic of transformed cells. All the fast growing RBM3-expressing cells show anchorage-independent phenotype and grow in 0.3% agar (Figure 2b). More importantly, the cells exhibited obvious morphological differences. The NIH3T3-RBM3 cells formed tight, densely packed multicellular spheroids, where single cells could not be distinguished. Moreover, when the colony size was compared to that produced by HT-29 colon adenocarcinoma cells, the NIH3T3-RBM3 colonies were significantly bigger suggesting an aggressive phenotype (Figure 2c). To determine whether RBM3 overexpression affected growth of cells that were already transformed, SW480 colon cancer cells were stably transfected with RBM3. Cells overexpressing RBM3 had higher levels of proliferation when compared to vector transfected controls and formed larger colonies in soft agar (Figures 2e and f). Moreover, the colonies with RBM3 overexpression were significantly larger than those observed with vector transfected or untransfected controls (Figures 2f and g). Western blot analyses demonstrated that the expression of COX-2, VEGF and cyclin D1 was upregulated in both NIH3T3 and SW480 cells that had RBM3 overexpression (Figures 2d and h). Together, these data demonstrate that RNA-binding protein RBM3 is a proto-oncogene that induces anchorage-independent growth when overexpressed.
Figure 2.

RBM3 overexpression induces oncogenic transformation. (a) Proliferation of three independent NIH3T3-RBM3 clones were significantly higher than that observed with three independent NIH3T3-vector clones. (b) NIH3T3-RBM3 cells develop large colonies in soft agar, which are bigger than those formed by HT-29 cells. HuR overexpressing cells, on the other hand did not form any colonies in the soft agar. (c) Quantitative estimation of number of colonies formed in soft agar (*P<0.01). (d) Two clones of NIH3T3 cells stably expressing RBM3 were selected based on western blot analyses. Expression of COX-2, VEGF (p21 monomer and p42 dimer) and cyclin D1 increases in the RBM3 overexpressing cells. Clone 1 is the same one shown in b. (e) Proliferation of the SW480-RBM3 clones was significantly higher than that observed with SW480-vector clones. (f) SW480-RBM3 cells develop large colonies in soft agar, when compared to control, untransfected or vector transfected cells. (g) Quantitative estimation of number of colonies formed in soft agar (*P<0.01). (h) Two clones of SW480 cells stably expressing RBM3 were selected based on western blot analyses. Expression of COX-2, VEGF and cyclin D1 increases in the RBM3 overexpressing cells. Clone 2 is the same one as shown in f. *Nonspecific band. COX, cyclooxygenase; VEGF, vascular endothelial growth factor.
RBM3 is essential for tumor growth
Next, we determined the effect of downregulating RBM3 expression on proliferation. RBM3 protein levels in HCT116 colon adenocarcinoma cells were significantly downregulated after transfection with an RBM3-specific, but not scrambled siRNA (Figures 3a and b). Furthermore, COX-2 mRNA and proteins levels were decreased in these cells (Figures 3a and c). Next, we investigated the effect of RBM3 downregulation on HCT116 cell proliferation. Although the scrambled siRNA did not affect proliferation, siRNA-mediated downregulation of RBM3 significantly reduced it (Figure 3d). A 50% reduction in HCT116 cell proliferation was observed with 50 nM siRNA. To determine whether this was due to loss of COX-2, cells were also treated with 1 μM of prostaglandin E2 (PGE2), the product of COX-2 enzyme activity. There were significantly higher levels of proliferation in cells also treated with PGE2 for 24 h, suggesting that the decreased proliferation resulting from reducing RBM3 levels is, in part, due to decreased COX-2-mediated PGE2 synthesis. Similar results were observed with HT-29 cells (data not shown). Next, we determined the effect of RBM3 downregulation on the growth of HCT116 tumor cell xenografts in nude mice. After the tumors were allowed to develop (15 days), siRNA was injected a total of five times at an interval of 3 days. Tumors that received either liposome preparation without any siRNA or those that included the scrambled siRNA continued to grow, with tumor volume reaching 4 × 103 and 6 × 103 mm3, respectively (Figure 3e). On the other hand, tumors that received RBM3-specific siRNA were arrested in growth. RBM3 silencing in the tumors was confirmed by real-time PCR and immunohistochemistry analyses (Figures 3f and g). In addition, there was a fivefold decrease in COX-2 mRNA when compared to the controls in the xenografts where RBM3 was knocked down (Figure 3f). Furthermore, although COX-2 protein was widely expressed in the controls, the expression was significantly reduced in tumors lacking RBM3 (Figure 3g). These data suggest that RBM3 is essential for COX-2 expression in vivo. COX-2 derived PGE2 regulates expression of angiogenesis inducing factors VEGF and IL-8. Real-time PCR analyses demonstrated a significant decrease in VEGF and IL-8 mRNA in the RBM3 targeted tumors, the amounts similar to that seen with COX-2 (Figure 3f). Furthermore, staining for CD-31, a platelet endothelial cell adhesion molecule that marks the endothelial cells in blood vessels demonstrated a 70% reduction in micro-vessel density in the RBM3-targeted tumors (Figure 3g, Supplementary Figure S1). Together, these data suggest that targeting RBM3 effectively suppressed capillary formation through decreased expression of angiogenic factors, resulting in loss of tumor growth.
Figure 3.

RBM3 is essential for tumor growth. (a) RBM3 specific siRNA (si-RBM3), but not a scrambled siRNA (si-Scr) decreases RBM3 and COX-2 mRNA expression. * denote statistically significant differences (**P<0.01). (b and c) Western blot analysis demonstrated that RBM3 and COX-2 proteins were significantly reduced in the cells treated with RBM3-targeted siRNA. (d) Knockdown of RBM3 expression decreases colon cancer cell proliferation. HCT116 cells were transfected with increasing doses (0–100 nM) of either RBM3-specific or si-Scr. After 48 h, cells were incubated with 1 μM PGE2 for an additional 24 h. Cells transfected with RBM3-specific siRNA demonstrated significant reduction in proliferation, which was partially rescued in the presence of PGE2. * denote statistically significant differences (*P<0.05 and **P<0.01). (e) Antitumor activity of si-RBM3 in mice carrying HCT116 cell tumor xenografts. HCT116 cells were injected into the flanks of Ncr nude mice (five mice per group) and tumors were allowed to develop for 15 days. siRNA was injected directly into the tumors starting on day 15 and every third day for a total of five injections. Tumor sizes with standard error are shown from data collected at the time of every injection. si-Scr treated tumors were larger than the control carrier injected tumors, whereas si-RBM3 treated tumors were smaller. A representative excised tumor at day 28 is shown to the right. * denote statistically significant differences (*P<0.05 and **P<0.01). (f) Decreased gene expression in the si-RBM3 injected tumors. Real-time RT–PCR was performed with total RNA from the tissues and the expression of RBM3, COX-2, IL-8 and VEGF is plotted as relative to control, carrier injected tumors. (*P<0.01). (g) Immunohistochemistry for RBM3, COX-2 and CD31 in HCT116 xenografts. Presence of the protein in each panel is shown by brown stain. The sections were counterstained with hematoxylin (blue stain). There was reduced expression of RBM3 and COX-2 and decreased microvessel density. COX, cyclooxygenase; PGE2, prostaglandin E2; RT–PCR, reverse transcription–polymerase chain reaction; siRNA, short interfering RNA; VEGF, vascular endothelial growth factor.
RBM3 is necessary for overcoming mitotic catastrophe
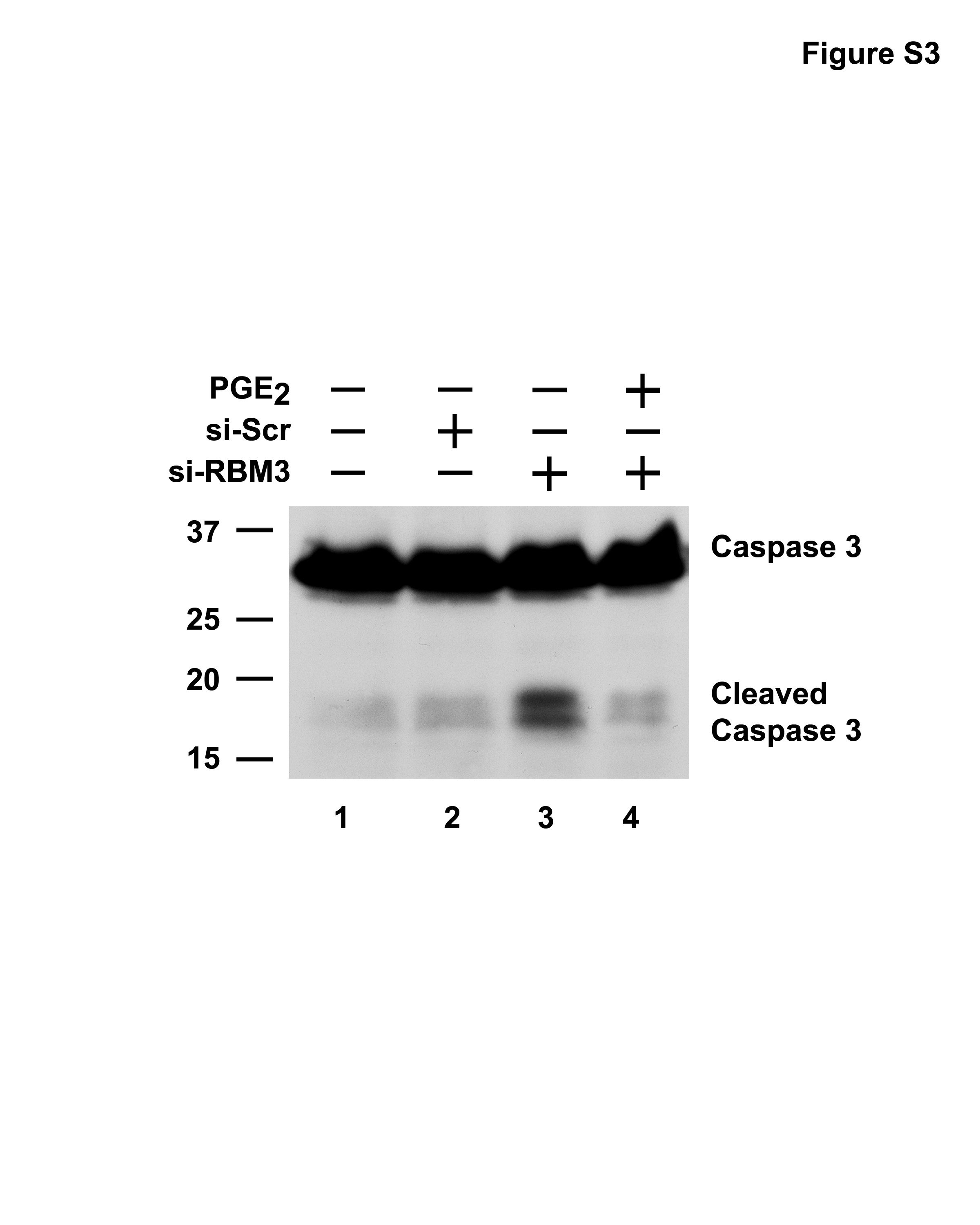
To characterize the inhibition of cell growth by RBM3 depletion, we next analysed cell cycle progression. While the scrambled siRNA did not affect the cell cycle profile 3 days after transfection, RBM3-specific siRNA increased the percentage of cells with 4N DNA content, suggesting that RBM3-depleted cells are blocked at G2/M (Figure 4a). While transfection of scrambled siRNA resulted in 12.7% G2/M, RBM3-targeted cells had 19% in this phase (Figure 4a, Supplementary Figure S2). However, treatment with 1 μM PGE2 in the setting of RBM3 suppression resulted in only 12.2% of the cells in G2/M phase, suggesting that PGE2 can override RBM3-mediated effects on cell cycle. To determine whether there was apoptosis occurring as well following RBM3 knockdown, the levels of TUNEL-positive cells were determined. TUNEL-positive cells following RBM3 knockdown were significantly higher than that seen with the scrambled siRNA (Figure 4b). However, treatment with 1 μM PGE2 resulted in significantly lower number of apoptotic cells, suggesting that PGE2 can protect the cells from undergoing apoptosis due to loss of RBM3. This further implies that COX-2 expression due to the action of RBM3 protects the cells from apoptosis in an autocrine manner through PGE2. Further conformation was obtained when western blot analyses were performed, which demonstrated increased caspase-3 activation in the siRBM3 transfected cells, which was suppressed by PGE2 (Supplementary Figures S3, S4). To gain further insight into the mechanism of G2/M cell-cycle-coupled apoptosis upon suppression of RBM3 expression, western blot analyses were performed for Cdc25c, a protein that catalyses the activation of the Cdk1:cyclin B1 kinase complex, which is believed to be a rate-limiting step for entry into mitosis (Maller, 1991; Millar and Russell, 1992). Suppressing RBM3 resulted in increased Cdc25c protein levels (Figure 4c). In addition, there was an increase in the level of cyclin B1, which was higher than that observed in the control or scrambled siRNA transfected cells (Figure 4c). Similar results were obtained with extracts from the tumor xenografts. There was increased cyclin B1 in RBM3-lacking tumors (Figure 4c). Furthermore, there were increased nuclear levels of cyclin B1 in these tumors (Figure 4d). Two kinases, Chk1 and Chk2, which are intermediaries of DNA damage checkpoint and are activated by phosphorylation on Ser-345/Ser-317 and Thr-68, respectively have been implicated in Ser-216 phosphorylation of Cdc25c (Canman, 2001; Walworth, 2001; Bulavin et al., 2002). In addition, phosphorylation of p53 protein at Ser-15 is critical for p53 protein stabilization and for activating its apoptotic function and G2/M checkpoint (Taylor and Stark, 2001). Immunoblots for phosphorylated forms of Chk1 (Ser-345), Chk2 (Thr-68) and p53 (Ser-15) showed increased phosphorylation of all the three proteins over control in the RBM3 depleted cells in culture and in the tumors (Figure 4c). Collectively, these data imply that RBM3-depleted cells undergo apoptosis while in the G2/M phase of the cell cycle, which is, in part, due to loss of COX-2 derived PGE2. To confirm that the cells were undergoing apoptosis while in mitosis, the xeno-graft cancer tissues were co-stained for phosphorylated histone H3 (Thr-11), a protein that is phosphorylated during mitosis, and for DNA damage by TUNEL. Many cells in the RBM3 suppressed tumors that were TUNEL positive also expressed phosphorylated histone H3 (Figure 4e). There was also an increase in phospho-H2AX staining in the RBM3-depleted tumors (Supplementary Figure S5). Taken together, these data suggest that the knockdown of RBM3 induces mitotic catastrophe, where the cells undergo apoptosis while in the process of mitosis.
Figure 4.

RBM3 downregulation results in mitotic catastrophe. (a) siRNA downregulation of RBM3 increased cells in the G2/M phase. HCT116 cells were transfected at the indicated dose of either scrambled (si-Scr) or RBM3-specific (si-RBM3) siRNA for 72 h. Cell-cycle profiles were analysed by flow cytometry following PI staining for DNA content. The percentage of cells in the G2/M phase following si-RBM3 transfection was increased compared to control and si-Scr cells. Addition of PGE2 partially suppressed the RBM3 siRNA-mediated effects. (b) Knockdown of RBM3 leads to apoptosis. HCT116 cells following siRNA transfection were stained by the TUNEL method. Arrows show the TUNEL-positive cells, which were higher in si-RBM3 transfected cells, but less in cells also treated with 1 μM PGE2. (c) Loss of RBM3 induces checkpoint proteins. Lysates from HCT116 cells treated with si-Scr (50 nM) or si-RBM3 (10 and 50 nM), and tumor xenografts from the various treatments were subjected to western blot analyses using specific antibodies for phospho-Ser345 Chk-1, phospho-Thr68 Chk-2, Cdc25C, phospho-Ser15 p53 and cyclin B1. Actin was used as an internal control for loading the gels. (d) Lack of RBM3 increases cyclin B1 translocation to nucleus. Tumor xenografts were subjected to immunohistochemical staining for cyclin B1. The arrows in the si-RBM3 treated tumors indicate cyclin B1-positive cells in the nucleus. Representative photographs are a magnification of × 400. (e) RBM3 depletion leads to mitotic catastrophe. Tumors treated with si-RBM3 were stained for TUNEL (green) and phosphorylated histone H3 (red). The cells positive for both are shown in the merged image with yellow stain. DAPI is used to stain the nucleus. COX, cyclooxygenase; PGE2, prostaglandin E2; RT–PCR, reverse transcription–polymerase chain reaction; siRNA, short interfering RNA; VEGF, vascular endothelial growth factor.
RBM3 is a nucleocytoplasmic shuttling protein that stabilizes COX-2, VEGF and IL-8 mRNA
Previous studies have demonstrated that COX-2-derived PGE2 induces cells to divide by enhancing mitosis (Andreis et al., 1981; Munkarah et al., 2002; Wu et al., 2005). Furthermore, treatment of colon cancer cells with NS-398, a COX-2 selective inhibitor increased the number of cells in the G2/M phase, whereas decreasing those in the G0/G1 phase (Yamashita et al., 2003). This suggests that mechanisms to increase COX-2 expression would result in protecting the cells from mitotic catastrophe. To identify the mechanism by which RBM3 inhibits mitotic catastrophe, we next determined the cellular functions of RBM3. Previous studies identified HuR and RBM3 as being in a complex bound to AU-rich sequences in COX-2 3′UTR (Cok and Morrison, 2001). Here, we observed that RBM3 interacts with HuR in a yeast two-hybrid analysis (Figure 5a). Moreover, RBM3 was isolated in a yeast two-hybrid screen using HuR as bait (data not shown). To confirm that the two proteins interact, HuR was generated by in vitro translation in the presence of 35S-methionine, and incubated with recombinant GST-HuR or GST-RBM3 fusion proteins, followed by affinity purification with a glutathione-.sepharose column. Radiolabeled HuR bound to RBM3, suggesting that the proteins can interact in solution in the absence of RNA (Figure 5b). To examine the interaction of HuR and RBM3 in mammalian cells, immunostaining was performed following transient transfection of HeLa cells with N-terminal myc epitope-tagged HuR and N-terminal FLAG epitope-tagged RBM3 plasmids. We found that the two proteins colocalize, predominantly in the nucleus (Figure 5c). HuR is primarily nuclear, but can be induced to redistribute from the nucleus to the cytoplasm (Fan and Steitz, 1998a). Furthermore, cytoplasmic HuR expression in cancer cells has been suggested to be a prognostic marker for cancers (Erkinheimo et al., 2003, 2005; Lopez de Silanes et al., 2003; Heinonen et al., 2005; Denkert et al., 2006). Given the strong nuclear colocalization of HuR and RBM3, and that there is increased cytoplasmic localization of the protein in cancers, we next determined whether RBM3 also exhibits shuttling activity. We performed a heterokaryon assay, where we transfected Flag-tagged RBM3 or HuR in human HeLa cells and fused the cells with the murine NIH3T3 cells. RBM3, like HuR was found in both the human and murine nuclei implying that RBM3 is a nucleocytoplasmic shuttling protein (Figure 5d). HuR shuttles to the cytoplasm where it can stabilize certain transcripts such as COX-2 (Fan and Steitz, 1998a, b; Peng et al., 1998; Nabors et al., 2001; Lopez de Silanes et al., 2003). As knockdown of RBM3 decreased COX-2 mRNA levels, and RBM3 is a nucleocytoplasmic protein, we next determined whether RBM3 was able to regulate COX-2 expression. For this, we determined the effect of ectopic transient FLAG-tagged RBM3 and FLAG-tagged HuR on COX-2, IL-8 and VEGF mRNA levels in HCT116 cells. Both RBM3 and HuR alone significantly induced the endogenous expression of COX-2 mRNA (Figure 5e). While the steady-state levels of endogenous COX-2 mRNA were increased by approximately 7-fold in the presence of RBM3 and HuR, there was further increase to 10-fold when RBM3 and HuR were coexpressed (Figure 5e). Furthermore, western blot analyses demonstrated increased COX-2 protein expression in the cells that expressed FLAG-tagged RBM3 (Figure 5f). Similar results were obtained with IL-8 (Figure 5e). However, while both RBM3 and HuR induced VEGF expression when expressed alone, there was no additive effect when the two proteins were coexpressed (Figure 5e). These data demonstrate that RBM3 and HuR coordinate their functions to induce COX-2, VEGF and IL-8 gene expression.
Figure 5.

RBM3 and HuR interact and enhance stability. (a) Yeast two-hybrid interaction of RBM3 with HuR. RBM3 and HuR expressed as bait and test proteins interact in the yeast by the colonies formed on quadruple dropout media. Breakdown of the X-α-gal results in a blue colony. Tumor suppressor protein p53 and SV40 T antigen (RecT) were used as positive control for interaction, but negative for interaction with either RBM3 or HuR. (b) GST pull-down assay. 35S-methionine labeled in vitro translated HuR (35S-HuR) was incubated with either GST-RBM3 or GST-HuR. The GST-proteins were immobilized on to glutathione sepharose beads. The immobilized proteins were separated by SDS–PAGE and subjected to phosphorimager analyses. Pure GST served as negative control. (c). Colocalization of HuR and RBM3. HeLa cells were transiently transfected with plasmids expressing myc-epitope tagged HuR and FLAG-epitope tagged RBM3. Immunocytochemistry was performed for the myc and FLAG epitopes. Images for the HuR and RBM3 were merged demonstrating colocalization. Nucleus was stained by DAPI. (d) Nuclear-cytoplasmic shuttling of HuR and RBM3. Plasmids encoding FLAG-epitope tagged HuR or RBM3 were transiently transfected into human HeLa cells and subsequently fused with mouse NIH3T3 cells. The proteins were immunostained for the FLAG tag, and the nuclei by Hoescht stain to differentiate human and mouse nuclei. Mouse nuclei, seen as punctuate staining are denoted by an arrow. (e) RBM3 and HuR induce COX-2, IL-8 and VEGF mRNA expression. Ectopic expression of Flag epitope-tagged RBM3 and HuR resulted in significant increase in endogenous COX-2, IL-8 and VEGF mRNA in HCT116 cells. There was a trend for even higher levels when proteins were coexpressed (**P<0.01). (f) COX-2 protein increased in cells expressing RBM3 and HuR. COX, cyclooxygenase; GST, glutathione S-transferase; P, GST-bound fraction; S, supernatant; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; T, total input or whole cell extract; VEGF, vascular endothelial growth factor.
RBM3 enhances COX-2, VEGF and IL-8 mRNA stability and translation
HuR is an RNA-binding protein that mediates nucleo-cytoplasmic transport, mRNA stability and translation functions following binding to ARE sequences in the 3′UTR of rapidly degraded transcripts such as COX-2, VEGF and IL-8. RBM3 also encodes a RNA-binding domain suggesting that it might also modulate RNA stability function (Derry et al., 1995; Sutherland et al., 2005). Electrophoretic mobility shift assays demonstrated that recombinant GST-RBM3 protein binds to ARE sequences located in the first 60 nucleotides of the COX-2 3′UTR (Figure 6a). Furthermore, the coupled immunoprecipitation–RT–PCR demonstrated a significantly higher level of RBM3-bound COX-2 mRNA in RBM3 overexpressing cells (Figure 6b). Similar results were obtained with VEGF and IL-8. Next, we determined whether RBM3 is involved in regulating the mRNA stability. RBM3 and HuR were transiently overexpressed in HCT116 cells, following which actino-mycin D was added to inhibit de novo mRNA synthesis. In cells transfected with either RBM3 or HuR alone, there was increased stability of COX-2 mRNA, the half-life increased from 1 h in control cells to 5 h in the RBM3 or HuR expressing cells (Figure 6c). Furthermore, when RBM3 and HuR were coexpressed, COX-2 mRNA stability increased to 8 h (Figure 6c). Similarly, the half-life of IL-8 mRNA increased from 0.5 to 1 h with either RBM3 or HuR, which was further increased to 4 h when the two proteins were coexpressed (Figure 6c, middle panel). The half-life of VEGF mRNA was also increased from 0.5 h to 8 h with either RBM3 or HuR, which was further increased to >8 h with both proteins (Figure 6c, right panel). These data, taken together suggest that although RBM3 and HuR are mutually exclusive, they can also synergize to increase the mRNA stability of key oncogenic proteins.
Figure 6.

RBM3 overexpression increases COX-2 mRNA stability and translation. (a) RBM3 is an ARE-binding protein. The first 60 nucleotides of COX-2 3′UTR containing many ARE sequences was transcribed in vitro in the presence of 32P-UTP. Purified recombinant GST-RBM3 was allowed to interact with the radiolabeled RNA and subsequently separated by native PAGE. Presence of the RBM3 bound RNA is shown by a mobility shift as indicated to the right. (b) Increased binding of COX-2, IL-8 and VEGF mRNA to RBM3 following overexpression. Whole cell extracts (T) from vector transfected or RBM3 overexpressing cells were prepared after cross-linking, and subjecting to immunoprecipitation with anti-RBM3 antibody. RNA present in the immunoprecipitate (P) and supernatant (S) were isolated after reversing the cross-link and subjected to RT–PCR for COX-2, IL-8 and VEGF mRNA. Data demonstrates increased COX-2, IL-8 and VEGF mRNA in the pellet of RBM3 overexpressing cells. (c) RBM3 and HuR increase COX-2, IL-8 and VEGF mRNA stability. HCT116 cells were transfected with Flag epitope-tagged RBM3 and/or HuR and the stability of endogenous transcripts was determined following addition of actinomycin D. Both RBM3 and HuR increased COX-2 (left panel), IL-8 (middle panel) and VEGF (right panel) mRNA stability on their own, which was further increased when the two were coexpressed. (d) Schematic representation of control luciferase mRNA (Luc) and luciferase mRNA containing the full-length COX-2 3′UTR (Luc-COX) that is encoded in the plasmid under the control of the CMV promoter. (e) RBM3 and HuR increase the translation of Luc mRNA containing COX-2 3′UTR. HCT116 cells transiently overexpressing RBM3, HuR or both were co-transfected with plasmids encoding either the Luc-COX and luciferase activity was measured. Luciferase activity of Luc-COX is shown in black bars and is relative to control cells. * denote statistically significant differences (**P<0.01). (f) RBM3 and HuR increases the translation of Luc mRNA containing COX-2 3′UTR. HCT116 cells transiently overexpressing RBM3, HuR or both were co-transfected with plasmids encoding either the Luc-COX or Luc control mRNA and luciferase activity was measured. Luciferase activity of Luc-COX is shown in black bars and that of Luc in gray bars. *indicate statistically significant differences (**P<0.01). ARE, AU-rich elements; COX, cyclooxygenase; GST, glutathione S-transferase; PAGE, polyacrylamide gel electrophoresis; RT–PCR, reverse transcription–polymerase chain reaction; VEGF, vascular endothelial growth factor.
To determine whether RBM3-mediated COX-2 mRNA translation occurs through binding to COX-2 3′UTR, we next determined the levels of a chimeric luciferase-COX-2 3′UTR mRNA (Figure 6d). Both RBM3 and HuR significantly increased the steady-state levels of luciferase mRNA, which was further increased when both RBM3 and HuR were coexpressed (Figure 6e). In addition, there was an increase in luciferase activity when RBM3 and HuR were co-transfected (Figure 6f). In contrast, neither RBM3 nor HuR affected luciferase levels expressed from the control transcript (Figure 6f). These data demonstrate the ability of RBM3 and HuR to increase the translation of COX-2 mRNA via its 3′UTR, either alone or in partnership with each other.
Discussion
Although, it is apparent that post-transcriptional events of mRNA stability and translation contribute to the development and progression of malignant tumors, the mechanisms that regulate these processes especially in inflammation and cancer are not clearly understood. One element that is present in the 3′UTR of transcripts encoding oncoproteins, cytokines and transcription factors is the ARE (Chen and Shyu, 1995; Chen et al., 1995). However, little is known about the RNA-binding proteins that bind these elements. HuR, the best characterized factor regulates the stability of COX-2, IL-8 and VEGF mRNAs, resulting in their increased expression, thereby enhancing tumorigenesis (Myer et al., 1997; Fan and Steitz, 1998b; Peng et al., 1998; Blaxall et al., 2000; Dixon et al., 2001; Nabors et al., 2001; Denkert et al., 2004, 2006; Erkinheimo et al., 2005; Heinonen et al., 2005). However, more recently it is believed that cytoplasmic localization, rather than total amount, governs HuR activity (Blaxall et al., 2000; Erkinheimo et al., 2003, 2005; Denkert et al., 2004, 2006; Heinonen et al., 2005). Here, we have identified RBM3 as a novel HuR interacting partner, whose expression is not only significantly upregulated in tumors, but is also localized in the cytoplasm. A more comprehensive study to determine whether there is a correlation between protein localization and tumor aggressiveness is required. Nevertheless, our studies demonstrate a similar trend in RBM3 and HuR expression, implying a related if not redundant function.
Nucleocytoplasmic shuttling of RBM3 is similar to that of hnRNP proteins that export mRNA from the nucleus (Pinol-Roma, 1997; Shyu and Wilkinson, 2000; Hacker and Krebber, 2004; Carpenter et al., 2006). Previously, Steitz and colleagues have speculated that HuR may bind to ARE-containing mRNAs and remain associated during transit through the nuclear envelope (Fan and Steitz, 1998b). A similar possibility also exists for RBM3. In addition, as RBM3 is a global translation inducer (Dresios et al., 2005), it is tempting to speculate that RBM3 not only transports the mRNAs, but also loads them on to ribosomes to induce translation.
The observation that the increase in RBM3 expression is dependent on the tumor stage suggests that it may play a role in tumorigenesis. Indeed, there is a significantly higher level of RBM3 expression in HCT116 cells in the tumor xenograft as compared to the cells in culture (data not shown). In addition, RBM3 overexpression induced NIH3T3 cells to grow in an anchorage-independent manner. Furthermore, when RBM3 expression was suppressed, there was a complete shut down of tumor xenograft growth. Previous studies from the Gorospe group have shown that knockdown of HuR results in smaller tumors (Lopez de Silanes et al., 2003). However, the inhibition was not as pronounced with HuR suppression as that observed here with RBM3 suppression. This might be because HuR levels were only decreased by 35–50% in that study. In this regard, it should be noted that both proteins influence the expression of COX-2, IL-8 and VEGF expression. These factors play a major role in tumor cell growth and in angiogenesis. Hence, further characterization of the role of HuR and RBM3 in tumorigenesis will be useful in understanding their contributions to the aggressive phenotype.
Importantly, it should be noted that PGE2 not only partially reverses the effects of RBM3 knockdown, but also affects baseline proliferation rate. Previous studies have demonstrated that PGE2 induces growth of colon cancer cells. PGE2 activated the cyclic AMP/protein kinase A pathway to induce the expression of amphiregulin, which then exerted a mitogenic effect on the cells (Shao et al., 2003). PGE2 has also been shown to activate the phosphatidylinositol 3-kinase/AKT pathway resulting in changes in cell motility and colony morphology (Sheng et al., 2001). PGE2 inhibits a COX-2 inhibitor-mediated apoptosis of colon cancer cells by inducing Bcl2 expression (Sheng et al., 1998). HuR is known to bind to AU-rich sequences in Bcl2 mRNA and enhances its expression. This suggests that PGE2 may affect Bcl2 expression by regulating RBM3 and/or HuR.
Not much is known about the pathways that regulate mitotic catastrophe and the role of apoptosis in the process. Previous studies have suggested that apoptosis is an early event in the mitotic catastrophic process, in such cases there was an arrest at the G2/M phase (Wahl et al., 1996; Chen et al., 1999; Ning and Knox, 1999). G2/M transition is regulated by the mitosis-promoting factor, cyclin B and cdc2 kinase (Ohi and Gould, 1999; Doree and Hunt, 2002; Stark and Taylor, 2004). Activation of the cdc2 requires cyclin B binding, which is positively regulated through dephosphorylation by cdc25. In turn, Cdc25c can be inhibited by phosphorylation by Chk1 and Chk2. Activated cyclin B then translocates to the nucleus where it greatly reduces the damage-induced G2 arrest. We observed that there was high level of DNA damage in the RBM3 lacking cells, based on staining for H2AX. Furthermore, we observed high levels of nuclear cyclin B1. Coupled with this, both Chk1 and Chk2 were phosphorylated and there was increased Cdc25C phosphorylation. At the same time, there was significant activation of caspase-3 and TUNEL. These data suggest a novel process of mitotic catastrophe, where cells lacking RBM3 undergo apoptosis at the same time when they are in G2/M transition, rather than during G2/M arrest.
Together, these data imply that the product of the novel RBM3 proto-oncogene is required for preventing mitotic catastrophe. RBM3 exerts its effects by increasing mRNA stability and translation of otherwise rapidly degraded transcripts. Furthermore, RBM3 is the central regulator of tumorigenesis, depletion of which enhances the regression of tumors. Hence, RBM3 may represent a potential target for chemotherapeutic and chemopreventive strategies.
Materials and methods
Yeast two-hybrid screening
We cloned full-length HuR cDNA into the yeast vector pGBKT7 (Clontech, Mountain View, CA, USA) as bait at the EcoR1 and Sal1 restriction sites, and performed yeast two-hybrid screening with the human cDNA liver library according to the manufacturer’s protocols. For confirmation, we cloned the full-length RBM3 cDNA in to yeast vectors pGBKT7 and pGADT7 at the EcoR1 and BamH1 restriction sites, and HuR in pGADT7 at the EcoR1 and Xho1 restriction sites.
Cell culture and treatment
HCT116, SW480 human colon adenocarcinoma, HeLa cervical carcinoma and NIH3T3 mouse fibroblast cells (all from American Type Culture Collection) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. To examine RNA stability, HeLa cells were transiently transfected with pCMV-Tag2B plasmids (Stratagene, La Jolla, CA, USA) expressing FLAG-tagged RBM3 and/or FLAG-tagged HuR. Twenty-four hours after transfection, the cells were treated with actinomycin D (10 μg/ml) to prevent the de novo mRNA synthesis, and total RNA was isolated using Trizol reagent. For stable expression, NIH3T3 and SW480 cells were stably transfected and colonies isolated following incubation in 800 μg/ml geneticin.
Anchorage-independent growth
NIH3T3 and SW480 cells transfected with plasmid vector (Vec) or stably expressing RBM3 were suspended in a 0.3% Sea Plaque agarose overlay in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum. The overlay (1.0 ml), consisting of cells, agarose, and medium, was plated at 2000 cells/well in Nunc 10 cm plates over bottom layers of soft agarose (0.8%) containing only Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum. Plates were incubated at 37 °C for 7 days. Colonies were counted and photographed.
Recombinant proteins
Recombinant RBM3 and HuR were expressed as N-terminal glutathione S-transferase (GST) fusion proteins from the plasmid pGEX-4T3 (Amersham-Pharmacia, Piscataway, NJ, USA) at BamHI and XhoI. Electrophoretic mobility shift assays were performed as described previously (Deschenes-Furry et al., 2005), using an in vitro transcribed 32P-labeled cRNA encoding the first 60 nucleotides of COX-2 3′UTR.
siRNA
RBM3 siRNA sequence targeting the coding region of RBM3 (nucleotides 470–488, Accession no. nm_006743) was GGGTATGGATATGGATATG and a scrambled control siRNA not matching any of the human genes were obtained from Ambion Inc., Austen, TX, USA and transfected using Transfectol (Ambion Inc., Austin, TX, USA).
Immunoprecipitation coupled RT–PCR
HCT116 whole cell lysates were prepared following cross-linking with formaldehyde, and immunoprecipitated with anti-RBM3 IgG using Seize X protein A purification kit (Pierce, Rockford, IL, USA). The pellet and supernatant was subsequently incubated at 70 °C for 1 h to reverse the cross-links. RNA was isolated and subjected to RT–PCR for COX-2, IL-8 and VEGF. In vitro protein interaction studies were performed as reported earlier with recombinant GST-HuR or GST-RBM3 (Sureban et al., 2007).
Flow cytometric analysis
Procedure for flow cytometric analysis was described in Supplementary procedures.
Real-time PCR analyses
Total RNA from cells or colorectal tumor samples was subjected to mRNA analysis for various genes wherever indicated. β-actin was used as internal control. Primer sequences are given in the Supplementary procedures.
Western blotting
Cells and tumor xenograft tissues were lysed and the concentration of protein was determined by BCA protein assay (Pierce). Forty micrograms of protein was subjected to western blot analysis. RBM3 antibody was generated in a rabbit against a peptide (Sigma-Genosys, The Woodlands, TX, USA). Other antibodies were obtained from Cell Signaling (Danvers, MA, USA) and Santa Cruz (Santa Cruz, CA, USA). Actin was used as an internal control for loading.
Immunocytochemistry
HeLa cells transfected with plasmid encoding FLAG and Myc tags were stained by immunofluoresence. Tumor xenografts and human multiple tissue slides were immunostained as described in the Supplementary section.
Luciferase reporter gene assay
Luciferase reporter assay was performed with plasmid encoding RBM3 and HuR as described earlier (Sureban et al., 2007).
Cell proliferation assay
RBM3-targeted siRNA was transfected with 1 × 105 HCT116 cells and plated simultaneously in a 96-well plate. Cell numbers were estimated after 48 h transfection as described earlier (Landegren, 1984).
Xenograft tumor model
siRNA was administered into the xenografts after incorporation into DOPC (1,2-Dioleoyl-sn-Glycero-3-Phosphocholine) (Avanti Polar Lipids, Alabaster, AL, USA) (Landen et al., 2005) generated by injecting HCT116 cells (6 × 106 cells) subcutaneously into the flanks of female athymic nude mice (NCr-nu) and housed in specific pathogen-free conditions. Tumors were measured with calipers and the volume calculated as (length × width2) × 0.5. The tumors reached 1000 mm3 after 15 days of injection of cells. These tumors were injected with 50 μl (5 μM) on every third day from day 15 for a total of 5 doses. We included five mice per group for the xenograft study and the experiment was repeated thrice. Data are represented as mean±s.e.m.
Statistics
All the experiments were performed in triplicate. The data was analysed by Student’s t-test. Where indicated, the data is presented as mean±s.e.m.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Stephen Prescott for helpful suggestions and Joan Steitz for the Flag-HuR plasmid. This work was supported by NIH Grants DK-62265 and CA-109269.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Andreis PG, Whitfield JF, Armato U. Stimulation of DNA synthesis and mitosis of hepatocytes in primary cultures of neonatal rat liver by arachidonic acid and prostaglandins. Exp Cell Res. 1981;134:265–272. doi: 10.1016/0014-4827(81)90425-0. [DOI] [PubMed] [Google Scholar]
- Blaxall BC, Dwyer-Nield LD, Bauer AK, Bohlmeyer TJ, Malkinson AM, Port JD. Differential expression and localization of the mRNA binding proteins, AU-rich element mRNA binding protein (AUF1) and Hu antigen R (HuR), in neoplastic lung tissue. Mol Carcinog. 2000;28:76–83. [PubMed] [Google Scholar]
- Bulavin DV, Amundson SA, Fornace AJ. p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr Opin Genet Dev. 2002;12:92–97. doi: 10.1016/s0959-437x(01)00270-2. [DOI] [PubMed] [Google Scholar]
- Canman CE. Replication checkpoint: preventing mitotic catastrophe. Curr Biol. 2001;11:R121–R124. doi: 10.1016/s0960-9822(01)00057-4. [DOI] [PubMed] [Google Scholar]
- Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, Murray GI. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer. 2006;95:921–927. doi: 10.1038/sj.bjc.6603349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen CY, Xu N, Shyu AB. mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol Cell Biol. 1995;15:5777–5788. doi: 10.1128/mcb.15.10.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Hsieh JT, Yao F, Fang B, Pong RC, Cipriano SC, et al. Induction of apoptosis and G2/M cell cycle arrest by DCC. Oncogene. 1999;18:2747–2754. doi: 10.1038/sj.onc.1202629. [DOI] [PubMed] [Google Scholar]
- Cok SJ, Morrison AR. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J Biol Chem. 2001;276:23179–23185. doi: 10.1074/jbc.M008461200. [DOI] [PubMed] [Google Scholar]
- Danno S, Nishiyama H, Higashitsuji H, Yokoi H, Xue JH, Itoh K, et al. Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem Biophys Res Commun. 1997;236:804–807. doi: 10.1006/bbrc.1997.7059. [DOI] [PubMed] [Google Scholar]
- Denkert C, Koch I, von Keyserlingk N, Noske A, Niesporek S, Dietel M, et al. Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclo-oxygenase-2. Mod Pathol. 2006;19:1261–1269. doi: 10.1038/modpathol.3800645. [DOI] [PubMed] [Google Scholar]
- Denkert C, Weichert W, Winzer KJ, Muller BM, Noske A, Niesporek S, et al. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin Cancer Res. 2004;10:5580–5586. doi: 10.1158/1078-0432.CCR-04-0070. [DOI] [PubMed] [Google Scholar]
- Derry JM, Kerns JA, Francke U. RBM3, a novel human gene in Xp11.23 with a putative RNA-binding domain. Hum Mol Genet. 1995;4:2307–2311. doi: 10.1093/hmg/4.12.2307. [DOI] [PubMed] [Google Scholar]
- Deschenes-Furry J, Belanger G, Mwanjewe J, Lunde JA, Parks RJ, Perrone-Bizzozero N, et al. The RNA-binding protein HuR binds to acetylcholinesterase transcripts and regulates their expression in differentiating skeletal muscle cells. J Biol Chem. 2005;280:25361–25368. doi: 10.1074/jbc.M410929200. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doree M, Hunt T. From Cdc2 to Cdk1: when did the cell cycle kinase join its cyclin partner? J Cell Sci. 2002;115:2461–2464. doi: 10.1242/jcs.115.12.2461. [DOI] [PubMed] [Google Scholar]
- Dresios J, Aschrafi A, Owens GC, Vanderklish PW, Edelman GM, Mauro VP. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc Natl Acad Sci USA. 2005;102:1865–1870. doi: 10.1073/pnas.0409764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- Erkinheimo TL, Lassus H, Sivula A, Sengupta S, Furneaux H, Hla T, et al. Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res. 2003;63:7591–7594. [PubMed] [Google Scholar]
- Erkinheimo TL, Sivula A, Lassus H, Heinonen M, Furneaux H, Haglund C, et al. Cytoplasmic HuR expression correlates with epithelial cancer cell but not with stromal cell cyclooxygenase-2 expression in mucinous ovarian carcinoma. Gynecol Oncol. 2005;99:14–19. doi: 10.1016/j.ygyno.2005.04.047. [DOI] [PubMed] [Google Scholar]
- Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci USA. 1998a;95:15293–15298. doi: 10.1073/pnas.95.26.15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998b;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker S, Krebber H. Differential export requirements for shuttling serine/arginine-type mRNA-binding proteins. J Biol Chem. 2004;279:5049–5052. doi: 10.1074/jbc.C300522200. [DOI] [PubMed] [Google Scholar]
- Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, et al. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005;65:2157–2161. doi: 10.1158/0008-5472.CAN-04-3765. [DOI] [PubMed] [Google Scholar]
- Krysan K, Reckamp KL, Dalwadi H, Sharma S, Rozengurt E, Dohadwala M, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65:6275–6281. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- Landegren U. Measurement of cell numbers by means of the endogenous enzyme hexosaminidase. Applications to detection of lymphokines and cell surface antigens. J Immunol Methods. 1984;67:379–388. doi: 10.1016/0022-1759(84)90477-0. [DOI] [PubMed] [Google Scholar]
- Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- Lopez de Silanes I, Fan J, Yang X, Zonderman AB, Potapova O, Pizer ES, et al. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene. 2003;22:7146–7154. doi: 10.1038/sj.onc.1206862. [DOI] [PubMed] [Google Scholar]
- Maller JL. Mitotic control. Curr Opin Cell Biol. 1991;3:269–275. doi: 10.1016/0955-0674(91)90151-n. [DOI] [PubMed] [Google Scholar]
- Millar JB, Russell P. The cdc25 M-phase inducer: an unconventional protein phosphatase. Cell. 1992;68:407–410. doi: 10.1016/0092-8674(92)90177-e. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell. 2003a;11:113–126. doi: 10.1016/s1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Jung J, Murmu N, Houchen CW, Dieckgraefe BK, Anant S. CUGBP2 plays a critical role in apoptosis of breast cancer cells in response to genotoxic injury. Ann N Y Acad Sci. 2003b;1010:504–509. doi: 10.1196/annals.1299.093. [DOI] [PubMed] [Google Scholar]
- Munkarah AR, Morris R, Baumann P, Deppe G, Malone J, Diamond MP, et al. Effects of prostaglandin E(2) on proliferation and apoptosis of epithelial ovarian cancer cells. J Soc Gynecol Investig. 2002;9:168–173. [PubMed] [Google Scholar]
- Myer VE, Fan XC, Steitz JA. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 1997;16:2130–2139. doi: 10.1093/emboj/16.8.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- Ning S, Knox SJ. G2/M-phase arrest and death by apoptosis of HL60 cells irradiated with exponentially decreasing low-dose-rate gamma radiation. Radiat Res. 1999;151:659–669. [PubMed] [Google Scholar]
- Ohi R, Gould KL. Regulating the onset of mitosis. Curr Opin Cell Biol. 1999;11:267–273. doi: 10.1016/s0955-0674(99)80036-2. [DOI] [PubMed] [Google Scholar]
- Peng SS, Chen CY, Xu N, Shyu AB. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinol-Roma S. HnRNP proteins and the nuclear export of mRNA. Semin Cell Dev Biol. 1997;8:57–63. doi: 10.1006/scdb.1996.0122. [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J. 1996;318(Pt 1):325–331. doi: 10.1042/bj3180325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Lee SB, Guo H, Evers BM, Sheng H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003;63:5218–5223. [PubMed] [Google Scholar]
- Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58:362–366. [PubMed] [Google Scholar]
- Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- Shyu AB, Wilkinson MF. The double lives of shuttling mRNA binding proteins. Cell. 2000;102:135–138. doi: 10.1016/s0092-8674(00)00018-0. [DOI] [PubMed] [Google Scholar]
- Smart F, Aschrafi A, Atkins A, Owens GC, Pilotte J, Cunningham BA, et al. Two isoforms of the cold-inducible mRNA-binding protein RBM3 localize to dendrites and promote translation. J Neurochem. 2007;101:1367–1379. doi: 10.1111/j.1471-4159.2007.04521.x. [DOI] [PubMed] [Google Scholar]
- Stark GR, Taylor WR. Analyzing the G2/M checkpoint. Methods Mol Biol. 2004;280:51–82. doi: 10.1385/1-59259-788-2:051. [DOI] [PubMed] [Google Scholar]
- Sureban SM, Murmu N, Rodriguez P, May R, Maheshwari R, Dieckgraefe BK, et al. Functional antagonism between RNA binding proteins HuR and CUGBP2 determines the fate of COX-2 mRNA translation. Gastroenterology. 2007;132:1055–1065. doi: 10.1053/j.gastro.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem. 2005;94:5–24. doi: 10.1002/jcb.20204. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW, et al. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med. 1996;2:72–79. doi: 10.1038/nm0196-72. [DOI] [PubMed] [Google Scholar]
- Walworth NC. DNA damage: Chk1 and Cdc25, more than meets the eye. Curr Opin Genet Dev. 2001;11:78–82. doi: 10.1016/s0959-437x(00)00160-x. [DOI] [PubMed] [Google Scholar]
- Wang D, Mann JR, DuBois RN. The role of prostaglandins and other eicosanoids in the gastrointestinal tract. Gastroenterology. 2005;128:1445–1461. doi: 10.1053/j.gastro.2004.09.080. [DOI] [PubMed] [Google Scholar]
- Wu G, Yi J, Di F, Zou S, Li X. Celecoxib inhibits proliferation and induces apoptosis via cyclooxygenase-2 pathway in human pancreatic carcinoma cells. J Huazhong Univ Sci Technolog Med Sci. 2005;25:42–44. doi: 10.1007/BF02831383. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Osaki M, Honjo S, Yoshida H, Teshima R, Ito H. A selective cyclooxygenase-2 inhibitor, NS-398, inhibits cell growth by cell cycle arrest in a human malignant fibrous histiocytoma cell line. Anticancer Res. 2003;23:4671–4676. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.