

Abstract
Although long recognized in microvascular research, an increasing body of evidence suggests that inflammatory markers are present in human diseases. Since the inflammatory cascade serves as a repair mechanism, the presence of inflammatory markers in patient groups has raised an important question about the mechanisms that initiate the inflammatory cascade, i.e. the mechanisms that cause tissue injury. Using a severe forms of inflammation, shock and multi-organ failure, for which there is no accepted injury mechanism, we summarize studies which suggest that the powerful pancreatic digestive enzymes play a central role in destruction of the intestine and other tissues if their compartmentalization in the lumen of the intestine and in the pancreas is compromised. Furthermore, we summarize evidence that uncontrolled degrading enzyme activity in plasma causes proteolytic cleavage of the extracellular domain of membrane receptors and loss of associated cell functions. For example, in a model of metabolic disease with Type II diabetes proteolytic cleavage of the insulin receptor causes the inability of insulin to signal glucose transport across membranes. The evidence suggests that uncontrolled proteolytic and lipolytic enzyme activity may trigger mechanism for tissue injury. The significance of such mechanisms remain to be explored in human diseases.
Keywords: Microcirculation, inflammation, pancreatic enzymes, matrix metalloproteinases, shock, multi-organ failure, hypertension
Introduction
With deep gratitude I accept the 2008 Landis Award by the Microcirculatory Society. I thank President Cindy Meininger, the Members of the Awards Committee and the individuals who proposed my name for the award. Professor Landis’ seminal contributions to microvascular research have been guiding principles to our understanding and analysis of the microvascular phenomena, and I am honored to have this association with him although I never met him in person. My roots are firmly embedded in this Society having been introduced to the field of microcirculation by Benjamin W. Zweifach, one of the founders and pioneer and for decades one of the major proponents of the field.
Microcirculation and Inflammation
I learned the first time about the inflammatory cascade from the Benjamin W. Zweifach, reading about the pioneers. For decades advancements in microvascular research and in the understanding of inflammation went hand in hand and many of the main discoveries in the inflammatory processes were made by studying directly the living microcirculation. Introduction of microvascular techniques made it possible to document and measure the early elevation of endothelial permeability, cell degranulation, leukocyte and platelet adhesion to the endothelium, diapedesis, red cell aggregation, thrombosis, endothelial apoptosis. These were the basis for a modern description of the inflammatory process in form of cell signaling studies, transmembrane transport, pro- and anti-inflammatory gene expression, and systems biology descriptions.
The term “inflammation” was originally introduced to characterize symptoms of tissue injury (e.g. redness, swelling, sensation). But today the term is used in a broader context with reference to an entire cascade of events that starts with an infectious or non-infectious injury and eventually terminates with healing of tissue, i.e. “resolution of the inflammation”.
Inflammation as Injury as well as Tissue Repair Mechanism
The inflammation cascade is the only known mechanism to repair an injured living tissue. Depending on circumstances the repair results in a scar with reduced cell functions. In the presence of stem cells the repair may reconstitute original tissue functions present before injury. One can divide the inflammatory cascade into two general stages. The first stage serves to remove injured cells and tissue, e.g. by apoptosis and/or infiltration of tissue neutrophils and macrophages and phagocytosis. The second stage consists of new tissue growth facilitated by release of growth factors, mitosis and angiogenesis, production of an extracellular matrix by means of biosynthesis processes that are also part of embryonic development. Both stages of inflammation are required to resolve tissue injury.
Decades of research, especially in the 1980th, into the various steps of the inflammatory cascade have brought to light that leukocyte adhesion and leukocyte cytotoxic activity, e.g. oxygen free radical formation, are part of the inflammatory cascade, and may actually serve to injure innocent bystander cells and organs, like obstruction of capillaries. This observation led to explorations designed to attenuate the injury side of the inflammatory cascade. Several of these ideas (e.g. oxygen free radical scavenging, leukocyte adhesion blockade) were tested in clinical trials but emerged with only limited success as a protective measure in patients (43) even though preclinical studies gave indications to the contrary (27, 113, 126). This disappointment was a wakeup call in our thinking about inflammation and motivated us to approach the inflammatory cascade in an alternative fashion.
Clinical Evidence for Markers of Inflammation in Human Disease
Clinical evidence for markers of inflammation in patient plasma and urine samples (e.g. in form of leukocyte activation, oxygen free radical production, levels of C-reactive protein and several others), have now been documented for more than a decade in a variety of chronic and acute disease (97). The evidence is consistently supported.
In fact, today we are justified to ask whether there exists any disease in which markers of inflammation may not be detected in clinical samples. The consistent presence of inflammatory markers is in line with the microvascular evidence that had shown evidence for hallmarks of inflammation in many models of diseases. Thus, if one recognizes that the inflammatory cascade represents a tissue repair mechanism, then we need to ask an important question: What mechanisms cause tissue injury that evokes a repair cascade? The answer to this question is essential to develop new strategies for prevention of inflammation.
Normal cell function without inflammation depends on a multitude of factors, from the microenvironment to the supply of nutrients and removal of metabolites in the microcirculation. One can list a large number of mechanisms that cause tissue injury, e.g. trauma, ischemia and reperfusion, depletion of metabolites, exposure to toxins, infections by bacteria or fungi, viruses, hypoxia, exposure to prions. But there exists important medical conditions in which no clear link has been established with any of these particular tissue injury mechanisms.
Inflammation in Shock
Nowhere is the lack of a mechanism that causes tissue injury more apparent than in the case of physiological shock and multi-organ failure, one of the most important medical conditions from a mortality point of view. A severe form of inflammation accompanies physiological shock (70, 82, 123) irrespective how physiological shock is generated, e.g. by hypovolemia, cardiogenic mechanisms, trauma, anesthesia, sepsis, tumors, radiation, blood flow obstructions, anaphylaxis, or by ischemia in the intestinal circulation. Typical for early inflammation, shock is accompanied by rapid onset of cellular dysfunctions, microvascular perfusion deficiencies, elevated permeability, diathesis of red cells, mast cell degranulation, attenuation of autoregulatory responses, capillary stasis, coagulation, thrombus and embolus formation, tissue edema, leukocyte-endothelial interaction, oxygen free radical formation, apoptosis, and eventual end organ failure (99).
Progress has been made in the description of these phenomena at the microvascular level (45, 61, 98, 118), but there is no consensus of how this tissue destruction occurs at the molecular level. At issue are mechanisms in shock that lead to the rapid progression of perfusion failure in organs that appear to be innocent bystanders and secondary victims to a primary insult.
Traditional Theories for Inflammation in Shock
Many interventions against shock have been explored, including endotoxin or cytokine blockade, endotoxin binding protein receptor, blockade of leukocyte adhesion molecules, complement depletion, oxygen free radical scavengers, nitric oxide modulators, and fluid resuscitation to name just a few (32, 34, 47, 49, 68, 76, 94, 104, 123, 125, 127). None lead to clinically effective interventions. The use of hypertonic resuscitation, designer crystalloids and hemoglobin carrying solutions that preserve the fluid shear stress on the capillary endothelium are more recent treatments under investigation (17, 44, 124). Current interventions are largely targeted to reduce symptoms of shock/multi-organ failure and do not target the root cause or intervene at the “beginning of the cascade”.
How is it possible that normal functioning organs even in young healthy individuals can progress within hours into complete dysfunction during multi-organ failure? We have embarked several years ago on an analysis to identify one of the most elusive targets in cardiovascular research, the actual trigger mechanisms for multi-organ failure.
The Auto-digestion Hypothesis
Compelling experiments confirm a long held clinical impression that the intestine plays a central role in shock (e.g. with actual removal of the intestine) (13). A unique feature of the intestine is the presence of the digestive enzymes from the pancreas. Digestive enzymes are discharged from the pancreas in the form of zymogens and are activated by enterokinases in the duodenal segments of the upper ileum (41). The digestive enzymes in the ileum are fully activated as part of normal digestion until they are degraded and/or auto-degrade as they pass through the lumen of the ileum. Even though food protein, lipid and complex carbohydrates are digested as part of normal nutrition, digestion of the intestinal tissue itself is prevented. This protection against auto-digestion is provided to a large extent by the special barrier characteristics of the villus structure and epithelial brush border. Two major mechanisms are known to prevent transport of digestive enzymes into the wall of the intestine, (a) the barrier formed by the epithelium and (b) mucus secretion from goblet cells that lead to a net outward transport away from the brush border barrier.
Under ischemic conditions, however, the usually tight epithelial barrier may be significantly compromised with major morphological damage (Figure 1) and high molecular weight molecules may penetrate the tight junction regions in the inter-epithelial gaps and enter into the interstitial space of the intestinal wall (14, 37, 90, 93, 95, 122). There may also be a lack of mucin secretion from goblet cells reducing a potentially significant diffusion barrier for active digestive enzymes. Instead, fully activated digestive enzymes enter into the submucosal space and initiate self-digestion of an otherwise unprotected villus structure, a process that leads to complete destruction of their tissue matrix (75). Digestive enzymes may be carried even into deeper muscle tissue layers of the intestinal wall (92) (Figure 1); they can enter the portal venous circulation, the intestinal lymphatics, and may escape across the serous coat into the peritoneal fluid.
Figure 1. Auto-digestion of the intestine by pancreatic enzymes during ischemia.

Micrographs of rat intestinal wall morphology (semi-thin section stained with toluidine blue) and zymographic image with trypsin fluorescently quenched substrate (green fluorescence) before (Panels A, B, C, respectively) and after 45 min intestinal ischemia (D, E, F). Note the extensive damage to the microvilli and mucosal epithelium (D, E, arrows) and penetration of activated trypsin across the full thickness of the intestinal wall (F) with activation of trypsin activity (bright green fluorescence). Adapted from (29, 92).
Thus failure of the mucosal barrier in the intestine is a process of special pathophysiological significance. The fully activated and powerful digestive enzymes in the lumen of the intestine, usually prevented from entry into the mucosal barrier, will rapidly enter into the wall of the intestine under any conditions that compromises this important barrier. The barrier may open due to reduced ATP production, e.g. due to reduced oxygen supply, since ATP is a requirement for maintenance of the junctional complexes and epithelial barrier properties (12, 39). But there are several other pathways by which the intestinal barrier may also be compromised, the presence of inflammatory mediators, e.g. derived from microbial colonies. We also found inflammatory mediators in the lumen of the intestine generated from food items after exposure to digestive enzymes, such as unbound free fatty acids (84). This observation has far-reaching implications for understanding the sources of inflammatory mediators in the intestine.
Blockade of Pancreatic Digestive Enzymes in Shock
The fact that digestive enzymes can generate powerful inflammatory mediators and that they themselves are cytotoxic if given access to the circulation, raises the question whether blockade of the pancreatic digestive enzymes may serve to attenuate the inflammation in multi-organ failure. There are few previous studies that used blockade of pancreatic digestive enzymes in the lumen of the intestine as an experimental tool. We started to explore this approach in different models of shock and multi-organ failure (summarized in recent Reviews (2, 31)).
One approach along these lines proposed in the literature is acute pancreatectomy or occlusion of the pancreatic duct (40, 58, 65). But this approach yielded so far no significant reduction in shock symptoms (74) in part due to the fact that the digestive enzymes that are already present in the lumen of the intestine were not blocked. As part of normal digestive activity, the pancreatic enzymes in the lumen of the intestine are usually sufficient to cause autodigestion of the intestinal wall without the need for further enzyme enrichment by discharge from the pancreatic duct.
Instead, we started to explore the hypothesis that blockade of digestive enzymes in the lumen of the intestine reduces the level of inflammation in shock and attenuates typical symptoms of multi-organ failure. In a model with splanchnic artery occlusion, blockade of serine proteases and lipases in the intestinal lumen, leads to a significant reduction of the inflammatory mediator production and dramatically reduced the destruction of the mucosal barrier (Figure 2), a hallmark of most models of shock (74, 75), and the long term survival rate (23). These observations were confirmed in an alternative model of shock with combined trauma and hemorrhage (20).
Figure 2. Blockade of Digestive Enzymes Preserves Villi.

Intestinal villi in the rat before (Panel A) and after 90 min shock by occlusion of the superior mesentery artery without (B) and with (C) serine proteases blockade with ANGD in the lumen (L) of the intestine. Length of crossbar 100 μm. Note a significant protection of the intestinal villi structure by blockade of luminal pancreatic proteases. Similar protection of villi is observed in severe porcine shock after blockade of pancreatic enzymes in the intestinal lumen. Adapted from (74).
One important consequence of the blockade of the digestive enzymes in the lumen of the intestine was that markers for inflammation and tissue apoptosis in remote organs were eliminated as well, i.e. there is a reduction of the organ failure in peripheral organs. For example, the characteristic enhancement of leukocyte rolling and attachment on the endothelium in the cremaster muscle microcirculation and the tissue apoptosis is essentially eliminated when the digestive enzymes in the lumen of the intestine are inhibited with a serine protease inhibitor (29) (Figure 3). There was no need for intravenous anti-inflammatory treatment to reduce the peripheral inflammation.
Figure 3. Blockade of Digestive Enzymes Prevents Peripheral Inflammation.

Micrograph of postcapillary venules in cremaster muscle at 80 min ischemia by superior mesentery artery occlusion (left column) followed by 80 min reperfusion. (Panels A, B) Non-ischemic control (sham), (C,D) with buffer as fluid for intestinal lavage, (E,F) with the serine protease inhibitor FOY (0.37 mM) in the lumen of the intestine (remote from the microvascular observation site). Note, the characteristic leukocyte adhesion to microvascular endothelium during shock in the post-capillary venules (arrows in panel D) is absent after blockade of the digestive enzymes in the lumen of the intestine (29). Bar equals 20 μm.
In contrast, if digestive enzyme blockers are administered only intravenously, without simultaneous blockade in the lumen of the intestine, no significant reduction of the inflammation is observed (20, 74). The same is observed when elastase is blocked only in the circulation (116). The evidence points to the important role of the digestive enzymes in the intestine.
A reduction of the central inflammation may be observed even if the blockade of the digestive enzymes in the intestine is carried out after ischemia and reperfusion (30). But such time-delayed treatment is only expected to be effective as a therapeutic approach if the damage to the intestine by the digestive enzymes is minimal and, in addition, the cellular damage in the liver remains limited so that inflammatory mediators from the intestine passing through the portal venous system are still absorbed in the liver. As increasing concentrations of digestive enzymes infiltrate the wall of the intestine and generate inflammatory mediators any further delay in the blockade of the digestive enzymes is associated with reduced protection against autodigestion and increased apoptosis, leukocyte adhesion and many other markers for inflammation in the peripheral microcirculation.
Pancreatic Digestive Enzymes in Endotoxin Shock
The evidence presented so far points towards the critical importance of the mucosal barrier as the main protection mechanism against transport of digestive enzymes from the lumen of the intestine into its wall and tissue autodigestion. There are other mechanisms that may affect the mucosal barrier in the intestine producing shock-like symptoms. An example is inflammatory mediators generated by bacterial infections and endotoxin release. In the past the hypothesis has been that endotoxin serves as the sole mediator generating for example septic shock. But this hypothesis has not lead to a new treatment for shock. Even though endotoxin itself is a well-recognized inflammatory mediator, no conclusive evidence has been advanced that shows that the endotoxin is singly responsible for the lethal progression following exposure to endotoxin.
Instead, we tested the hypothesis that while endotoxin may cause an initial elevation of the mucosal barrier properties (just like it increases endothelial permeability) the progression of the inflammatory cascade in endotoxic shock depends on the digestive enzymes penetrating into the wall of the intestine, similar to the case of shock produced by an ischemic intestine. Accordingly, we blocked the digestive enzymes in the intestine and administered a lethal dose (for the controls) of Gram-negative endotoxin (i.v.). During blockade of the digestive enzymes, the endotoxin causes a transient elevation of markers for inflammation (e.g. leukocyte adhesion to the endothelium, elevated endothelial permeability) but is followed within less than an hour by return to a state with low levels of inflammation and no further progression to multi-organ failure as in controls without blockade of digestive enzymes (28). These observations suggest that endotoxic shock, as a model of sepsis, is associated with auto-digestion by pancreatic enzymes as the primary reason for progression into multi-organ failure; the initial endotoxin serves to elevate the mucosal permeability and therefore merely starts the autodigestion process.
Inflammatory Mediators in Shock
The inflammation caused by pancreatic digestive enzymes may involve multiple pathways: damage to the tissue directly by degrading enzymes, activation and generation of inflammatory mediators by reaction of the digestive enzymes with a variety of tissue components. After intestinal ischemia and reperfusion, as well as in shock, plasma contains factors that exhibit a variety of biological activities, including activation of circulating leukocytes, depression of T-lymphocyte proliferation, depression of cardiac muscle activity (designated as myocardial depressant factor), carcinogenic (clastogenic) activity (26) and others (58, 71, 114). There are many reports in the literature of pancreatic enzymes releasing active factors affecting heart tissue (57) with the hypothesis that the MDF is released from ischemic pancreas during shock. A chemotactic factor can be released from self-digested spleen (73).
Consequently repeated attempts sought to identify the inflammatory mediators in shock (see summary in (99)). Among several mediators, platelet-activating factor, generated by phospholipase A2 in the post-ischemic gut, was proposed as active in priming neutrophils as well as cytokines, e.g. Interleukin-6, as stimulator for neutrophil-mediate post-ischemic injury (8). After trauma and hemorrhagic shock both lipid and aqueous factors in mesenteric lymph are toxic to endothelial cells in culture (19 ) with a putative factor greater than 100 kDa but independent of endotoxin (3). The investigators demonstrated a cationic peptide of rat serum albumin present in post-trauma/hemorrhagic lymph, but not in lymph from control animals subjected to trauma without hemorrhagic-shock. However, this peptide was not toxic to endothelial cells (48).
Bioactive factors in digested foods could contribute to the inflammatory mediator load in the intestine (83, 84). Fully characterized bioactive peptides from milk and casein have a range of functions (e.g. antimicrobial, immunostimulatory, immunosuppressive, antihypertensive, opioid-like and mineral binding) (53, 72, 80, 81). Cytotoxic peptides have been identified in trypsin digests of casein (66, 67). Consequently, the luminal content of the intestine is a mixture of factors that are derived from digested food items and possible autolytic breakdown of pancreatic enzymes themselves.
One of the reasons why previous approaches to isolate and identify the inflammatory mediators in-vivo remained inconclusive is due to the fact that the active compounds in plasma, and also in lymphatic fluid, derived from animals or patients in shock, while potent, are present only in low concentrations. Therefore separation and purification techniques lead to loss of the biological activity. We therefore developed an alternative approach to gain insight into potential mediators and tested tissue homogenates from a spectrum of organs (54). This work showed that most organ homogenates can generate a low level of cell activation, for example measured by pseudopod formation in neutrophils due to actin polymerization and/or by oxygen free radical formation. Only the pancreas and the intestine in the presence of digestive enzymes, but not in their absence, produced high levels of inflammatory and even cytotoxic mediators. Inflammatory factors derived from the pancreatic enzymes are highly toxic and may cause rapid death (50). The high toxicity is preceded by extensive oxygen free radical production in microvascular endothelium, adherent leukocyte interaction with the endothelium, as well as mast cell degranulation and parenchymal cells death with or without the presence of leukocytes (51).
The evidence suggests that when pancreatic digestive enzymes come into contact with healthy tissue components, such as the extracellular matrix proteins and cell membranes, proinflammatory mediators are formed and released that exhibit significant biological activity (50, 54). We hypothesize that the ‘shock factors’ are strongly pro-inflammatory in nature and may include besides proteinaceous also lipid fragments.
Inflammatory Mediators Derived from Intestine
After ischemia the small intestine generates inflammatory mediators that have the ability to kill naive cells within minutes (84). To shed light on these mediators we exposed intestinal tissue after homogenization to purified pancreatic trypsin, chymotrypsin and elastase and tested the products generated after an incubation period and separation into aqueous protein and lipid fractions. In spite of the fact that the intestine was initially exposed only to proteases, only the lipid fraction exhibited the high cytotoxicity generated by an ischemic intestine. In fact, the lipid fraction of even a homogenized non-ischemic intestine, not exposed to pancreatic proteases, is highly cytotoxic even though the unseparated homogenate of such control intestine exhibits no detectable levels of cytotoxicity. Recombination with the proteinaceous fraction greatly attenuates the lipid cytotoxicity, but only if the proteinaceous fraction was not previously exposed to proteases.
The lipid fraction of homogenized intestinal tissue with and without exposure to purified pancreatic serine proteases was found to contain cytotoxic levels of unbound free fatty acids. Addition of a free fatty acid binding protein, such as albumin (18), prevented the lipid cytotoxicity. Thus, the evidence indicates that a major contribution to the cytotoxicity of ischemic intestine may be derived from unbound free fatty acids. Breakdown of free fatty acid proteins by proteases causes release of free fatty acids to act as cytotoxic mediators (85).
The most important mediators that generate an acute inflammatory reaction are derived from the digestive proteases and lipases (54, 119). This information is important in the design of interventions against the pancreatic digestive enzymes.
Chronic Inflammation and Autodigestion
The question arises whether there are other circumstances in which an autodigestion process may play a role in organ dysfunction. At issue are conditions, perhaps less severe than shock, but still accompanied by cell dysfunction and organ disease. We started to explore a chronic version of cardiovascular disease, the spontaneously hypertensive rat (SHR).
While recognized for its elevated blood pressure, an extensive microvascular research has brought to light that the SHR strain suffers also from a multitude of cardiovascular and cellular complications (106). The list of complications is quite extensive and individual complications are in most cases not correlated or spatially associated with the elevated blood pressure on the arterial side of the circulation (131). Examples of such vascular abnormalities are a life-long elevated level of circulating leukocyte counts (100), leukocyte activation (103), immune suppression with a deficiency for leukocyte adhesion to the endothelium (107, 109) due to deficient CD18 expression and P-selectin (5, 107), an elevated level of free radical production (22, 110) with enhanced levels of xanthine oxidase and NADPH oxidase (24), enhanced apoptosis in microvascular endothelium (63) and in lymphocytes (108), capillary rarefaction (87, 115), elevated plasma glucose levels, glycosylated hemoglobin and insulin resistance (11, 46, 102), and multiple forms of lesion formation at young age (111). The SHR is thus a genetic model of hypertension with deficiencies similar to those encountered in the metabolic syndrome (129). Challenging questions arise how to put these multiple cell dysfunctions under a unified conceptual roof; e.g. how an elevated arterial blood pressure may be associated with a depressed adhesion of leukocytes to postcapillaries, microvessels with normal blood pressure (130).
Measurement of the proteolytic activity in fresh plasma with a broadly responsive, fluorescently quenched substrates shows about a twofold increase in unchecked proteolytic activity in the SHR compared to its normotensive control, the Wistar-Kyoto (WKY) rat strain. The enhanced proteolytic activity is due in part to serine proteases and in part due to matrix metalloproteinases (25). Endothelial cells, in both high and low pressure regions of the microcirculation also exhibit an increased expression level and increased activity (Figure 4), which can be traced at least in part to MMP-9 (Figure 5). The source of the plasma serine protease activity and other MMPs in the SHR remain to be explored.
Figure 4. Enhanced MMP activity in the SHR Microcirculation.
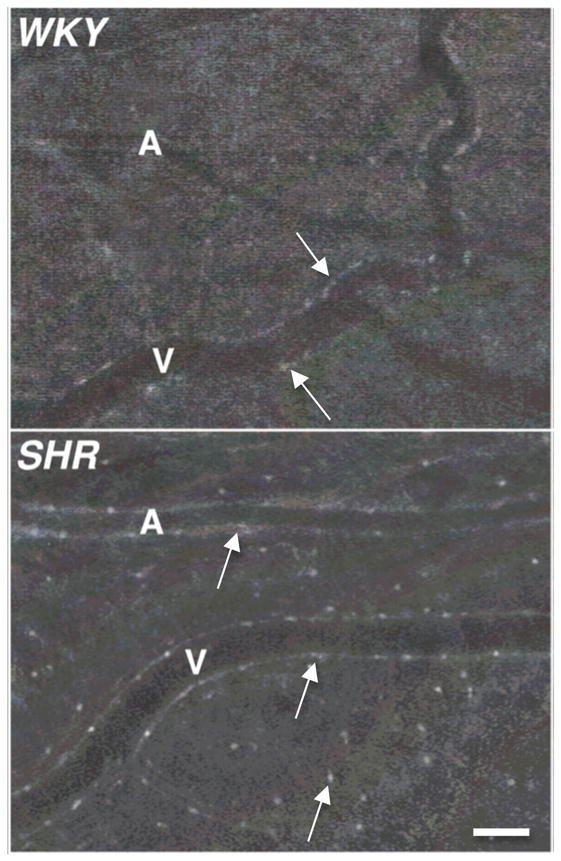
Digital fluorescent micrographs of WKY and SHR mesenteric microvessels labeled with fluorogenic peptide substrate showing matrix metalloproteinase (MMP-2, 9) enzymatic activity. Arterioles (A) and venules (V) are visible. Note the enhanced fluorescent emission over the endothelial cells and mast cells in the SHR, an affect that is less detectable after the doxycycline treatment. Adapted from (25). Bar equals 60 μm.
Figure 5. Elevated MMP-9 protein levels in SHR Tissue.
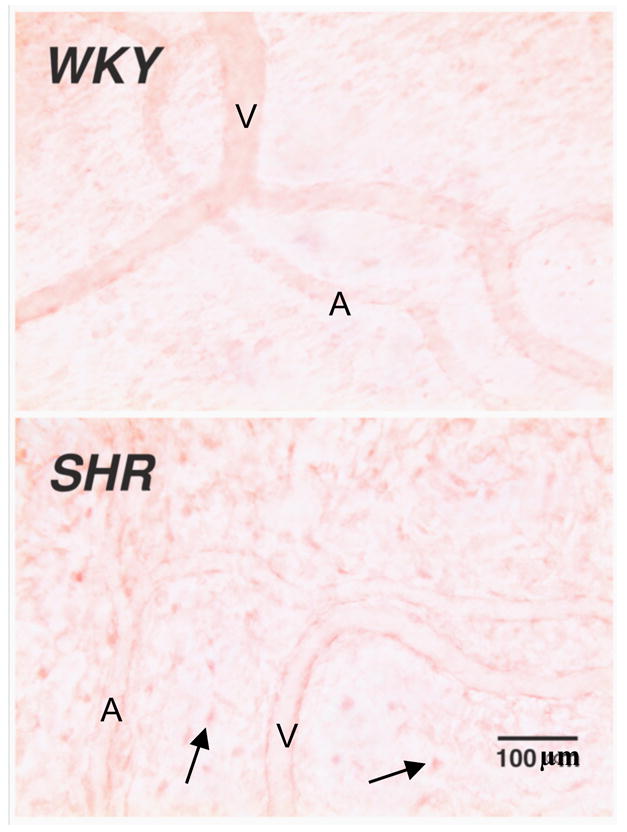
Selected micrographs of microvessels and interstitium of WKY and SHR mesentery after MMP-9 immunolabeling (with Vector NovaRED substrate) (25). Note the pronounced labeling in SHR endothelial cells of arterioles (A) and venules (V) as well as in interstitial mast cells and fibroblasts (arrows).
Proteolytic Activity and Receptor Cleavage: Cause of Insulin Resistance
To check whether such unchecked proteolytic activity in SHR plasma cleaves besides fluorescently quenched substrates also important proteins, we checked whether the extracellular domain of the insulin receptor might be subject to proteolytic cleavage. Immunolabeling with an antibody directed against the extracellular domain of the receptor shows that the SHR has indeed a reduced density of the extracellular domain of insulin receptor in many tissues, including the mesentery and on circulating leukocytes (25), both of which are exposed only in small part to elevated blood pressure. Thus, even though insulin levels may be near normal, the ability to signal glucose transport by insulin is compromised since the receptor is proteolytically cleaved, i.e. a characteristic sign of insulin resistance. Further evidence for cleavage of the extracellular domain of the insulin receptor comes from the fact that incubation of naïve control cells with fresh plasma from the SHR leads within a period of only 30 min to a reduced density (~ 15 %) of the extracellular domain of the insulin receptor. No significant receptor cleavage is generated by plasma from control rats. The receptor cleavage by the SHR plasma reduces the transport of the glucose into the cell cytoplasm. In addition, the SHR suffers from cleavage of other receptors, e.g. the integrin CD18 on leukocytes (25), in line with the defective adhesion to the endothelium and elevated leukocyte counts in the circulation. The enzymatic cleavage in the SHR is not limited to the membrane receptors, in fact major parts of the glycocalyx in the SHR are cleaved as well (96).
Chronic blockade of the protease activity in the SHR with a broad acting MMP inhibitor (e.g. Doxycycline) leads to a dramatic reduction of enzyme expression levels and plasma protease activities, concomitant with a restoration of the transmembrane glucose transport, reduction of the blood glucose levels and the level of glycosylated hemoglobin. The cleavage of other receptors is also reduced including CD18. With normalized expression of CD18 the adhesion to the endothelium and the circulating leukocyte counts are restored to control levels (25).
This evidence then serves as the first direct indication that autodigestion may be an underlying cause of cell and organ dysfunction not only in severe conditions, like shock, but in chronic conditions such as hypertension and diabetes. The involvement of degrading proteases is at a lower level in chronic conditions, yet autodigestion still progresses with direct cleavage of extracellular membrane receptors.
A key unresolved issue is the origin of the degrading enzyme activity in the plasma of the SHR. The enhancement of the MMP levels in endothelium are observed even in isolated endothelial cells, suggesting a genetic origin in the SHR (unpublished results). The SHR has a strong dependence on adrenal hormones (63) possibly associated with an abnormal expression of chromogranin expression, granule storage, and release of secretory hormones (78, 101, 112).
Unchecked Protease Activity and Disease
There are numerous indications in the literature that degrading enzyme activity due to MMPs and other proteolytic or lipolytic enzymatic activities may be associated with disease. The evidence is seen in form of tissue degradation, in form of zymographic evidence and in part supported by blocker studies, including studies in the heart (36, 42, 91, 105), lung (4, 6, 89, 117), renal dysfunction and hypertension (16), atherosclerosis (7, 35, 62, 64, 77, 79, 121), the skin (55, 86), in vascular remodeling and diseases, e.g. aortic aneurysm and venous disease (59, 88), multiple sclerosis (10, 128), to name just a selected few. Brain injury may be mediated by proteases (1, 15, 21, 33, 38, 52, 56, 60). Trypsin-like serine proteases and MMPs may be involved in neural development, learning and memory, as well as in chronic degenerative diseases. Protease-activated receptors (PARs) are activated by serine proteases, e.g. trypsin and thrombin and also mesotrypsin, P22, and neurosin (120). Thrombin at low concentrations protects neurons from damage by ischemic injury via activation of PAR-1, -2, whereas at higher concentrations, thrombin causes neurodegeneration and brain insults. Analysis of the blockade of trypsin by serpins, their interaction with PARs, and its significance for proteolytic brain injury provides insight into neural and other disorders.
The sources of extracellular degrading enzymes may be constitutive expression, e.g. in neutrophils or macrophages, MMPs on the extracellular matrix, as well as sources of degrading enzymes from specialized organs like the pancreas or the salivary gland. We don’t know the exact distribution and activity of degrading enzymes in any tissue at the level of the microcirculation and we don’t know the regulation of endogenous protease inhibitors. The degree of posttranslational protease activation of proenzymes and their inhibition by endogenous inhibitors (e.g. serpins in the case of serine proteases or endogenous tissue inhibitors of metalloproteinases) will determine the activity of a degrading enzyme. Normal plasma has a considerable ability to block proteolytic activity. The expression of inhibitory proteins may be limited in the course of a disease and their blocking activity may be exhausted, as we see in acute shock when high concentrations of digestive enzymes enter into the central circulation. Thus, it is essential that enzyme activity be studies in living cells and tissues in the context of an intact tissue and microcirculation (9). Synthetic chromogenic or fluorogenic substrates. in conjunction with in-vivo versions of zymography provide important tools (9, 25).
Conclusion
Since the discovery of superoxide dismutase thirty years ago (69), an extraordinary effort has been under way to explore the role of oxygen free radicals in disease and normal physiological function. It is evident that oxygen and nitrogen radicals have the ability to compromise cell function and damage proteins and lipids. But there is currently only limited evidence that interventions against free radicals have significant clinical benefits. The limited evidence supporting a role for free radicals raises the question whether there are other major organ injury mechanisms.
The evidence presented here supports the hypothesis that degrading enzymes may be a fundamental pathogenic factor. Proteolytic tissue degradation may compromise the ability of free radical scavenging proteins to be compromised, and therefore free radicals to be released. Future research needs to clarify interactions between the two damage mechanisms. Direct quantitative studies in the intact living microcirculation will play a critical role in elucidating the mechanisms and pathways leading to dysfunctions associated with degrading enzymes and free radicals. Identification of the trigger mechanisms is essential to design effective interventions.
In the case of acute physiological shock we have identified a major contribution to organ damage by the digestive enzymes (Figure 7A), the same enzymes that are the requirement for a lifetime of nutrient breakdown and delivery. Shock and multi-organ failure appear to be tied to one of the most fundamental mechanisms, i.e. nutrition over lifetime. Escape of the powerful pancreatic digestive enzymes from the lumen of the intestine or even from a pancreas (e.g. in pancreatitis, pancreatic tumor) can generate severe cell and tissue damage and generate an autodigestion process. In chronic diseases, such as Type II diabetes in the SHR, we see a more subtle, but still significant auto-digestion process with proteolytic activity that involves besides serine proteases and MMPs, which causes cleavage and therefore destruction of key membrane receptors in the microcirculation. Consequently the particular cell functions carried out by such cleaved receptors go into failure. Since proteolytic activity in the plasma tends to cleave several receptors, a multitude of cell dysfunctions arise, which is a characteristic of the metabolic syndrome. The full consequences of an auto-digestion process need to be documented from its earliest to its most severe stage and may have a major impact on our understanding of the origin of disease and the feasibility to use inhibitors of degrading enzyme as therapeutic intervention.
Figure 7.

(A) Schematic Diagram of Autodigestion by Pancreatic Enzymes in the Intestine. The fully activated pancreatic digestive enzymes are during normal digestion contained within the lumen of the intestine by the mucosal epithelial barrier (Left Panel). Compromise of the mucosal barrier, e.g. due to ischemia or infection of the mucosal barrier, permits entry of fully activated pancreatic enzymes into the wall of the intestine (Right Panel). This creates two complications, generation if proinflammatory and cytotoxic mediators, and morphological destruction of the mucosal barrier allowing further entry of digestive enzymes into the wall of the intestine. Pancreatic enzymes as well as inflammatory mediators are carried out of the intestinal wall via the intestinal venous system, the intestinal lymphatics and by leakage across the outer coat of intestine connective tissue (serosa) into the peritoneum and into the central circulation. Appearance of pancreatic enzymes and these inflammatory mediators leads to central inflammation and innocent bystander organ damage. i.e. multi-organ failure.
(B) Schematic Diagram of Protease Activity, Insulin Receptor Cleavage, and Insulin Resistance. The normal transmembane transport of glucose into the cell cytoplasm via the glucose transporter GLUT-4 as well as gene expression and growth factor synthesis requires intracellular signaling from the activated (phosphorylated) insulin receptor after binding of insulin to its extracellular domain (left panel). The appearance of uncontrolled degrading enzyme activity (e.g. due to MMPs, serine proteases) leads to cleavage of the extracellular domain of the insulin receptor (right panel), a lack of insulin binding sites on the insulin receptor, reduced intracellular signaling with attenuated glucose transport by GLUT-4, i.e. type II diabetes with insulin resistance due to reduced insulin receptor binding sites.
Acknowledgments
The research summarized here has been supported in part by NHLBI HL10881-42, HL67825 and HL76180, by the Max Kade Foundation, and an unrestricted educational gift by Leading Ventures, San Diego California. My most profound thanks goes to the following colleagues and collaborators for many discussions and for their devotion to the research in this review, including Drs. Jay J. Doucet, Florian Fitzal, Erik B. Kistler, David B. Hoyt, Tony Hugli, Nobuhiro Kobayashi, Hui Miao, Hiroshi Mitsuoka, Lee W. Murfee, Alexander Penn, Henrique Rosario, Hainsworth Shin, Ayako Makino, and Edward D. Tran. My long-term colleague Frank A. DeLano maintained the laboratory, explored diverse experimentation, completed numerous studies, and maintained uncompromised devotion to the highest standards. He prepared Figures 4 to 6. Special thanks to Scott A. Becker, Alex Hu, Steve Waldo, Randy Balete, Hubert Lim, and Corey Young for their extraordinary effort in the laboratory as undergraduate students in inflammation research. All animal research in our laboratory that is summarized here was reviewed and approved by the University of California San Diego Animals Subjects Committee.
Figure 6. Extracellular Insulin Receptor Cleavage and Insulin Resistance.

Panel A: Typical micrographs of immunolabel (Vector NovaRed) for the extracellular domain binding site of the insulin receptor α on fresh leukocytes (neutrophils and monocytes) from WKY and SHR. Note the reduced density of the insulin binding sites on the SHR leukocytes associated with about 20% average reduction of the receptor density on the plasma membrane and with reduced transport of glucose (shown with fluorescence-tagged analog of glucose in panel B) into the cell cytoplasm (25). Panel C: Glucose transport into naïve leukocytes from the normotensive Wistar strain before and after a 30 min incubation in fresh plasma of the Wistar (Wistar and Wistar-W in panel C) and in plasma from WKY and SHR (Wistar-WKY and Wistar-SHR in panel C). For quantitative measurements see (25).
References
- 1.Abraham CR, Potter H. The protease inhibitor, alpha 1-antichymotrypsin, is a component of the brain amyloid deposits in normal aging and Alzheimer’s disease. Ann Med. 1989;21:77–81. doi: 10.3109/07853898909149188. [DOI] [PubMed] [Google Scholar]
- 2.Acosta JA, Hoyt DB, Schmid-Schönbein GW, Hugli TE, Anjaria DJ, Frankel DA, Coimbra R. Intraluminal pancreatic serine protease activity, mucosal permeability, and shock: a review. Shock. 2006;26:3–9. doi: 10.1097/01.shk.0000209557.31457.ae. [DOI] [PubMed] [Google Scholar]
- 3.Adams CA, Jr, Xu DZ, Lu Q, Deitch EA. Factors larger than 100 kd in post-hemorrhagic shock mesenteric lymph are toxic for endothelial cells. Surgery. 2001;129:351–63. doi: 10.1067/msy.2001.111698. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad E, Steinberg SM, Goldin L, Hess CJ, Caporaso N, Kreitman RJ, Wiestner A, Wilson W, White T, Marti G, Stetler-Stevenson M. Immunophenotypic features distinguishing familial chronic lymphocytic leukemia from sporadic chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2008;74:221–6. doi: 10.1002/cyto.b.20423. [DOI] [PubMed] [Google Scholar]
- 5.Arndt H, Smith CW, Granger DN. Leukocyte-endothelial cell adhesion in spontaneously hypertensive and normotensive rats. Hypertension. 1993;21:667–73. doi: 10.1161/01.hyp.21.5.667. [DOI] [PubMed] [Google Scholar]
- 6.Arroyo AG, Genis L, Gonzalo P, Matias-Roman S, Pollan A, Galvez BG. Matrix metalloproteinases: new routes to the use of MT1-MMP as a therapeutic target in angiogenesis-related disease. Curr Pharm Des. 2007;13:1787–802. doi: 10.2174/138161207780831284. [DOI] [PubMed] [Google Scholar]
- 7.Beaudeux JL, Giral P, Bruckert E, Foglietti MJ, Chapman MJ. Matrix metalloproteinases, inflammation and atherosclerosis: therapeutic perspectives. Clin Chem Lab Med. 2004;42:121–31. doi: 10.1515/CCLM.2004.024. [DOI] [PubMed] [Google Scholar]
- 8.Biffl WL, Moore EE, Moore FA. Gut-derived mediators of multiple organ failure: platelet-activating factor and interleukin-6. Br J Hosp Med. 1995;54:134–8. [PubMed] [Google Scholar]
- 9.Boonacker E, Van Noorden CJ. Enzyme cytochemical techniques for metabolic mapping in living cells, with special reference to proteolysis. J Histochem Cytochem. 2001;49:1473–86. doi: 10.1177/002215540104901201. [DOI] [PubMed] [Google Scholar]
- 10.Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125:1297–308. doi: 10.1093/brain/awf133. [DOI] [PubMed] [Google Scholar]
- 11.Bursztyn M, Ben-Ishay D, Gutman A. Insulin resistance in spontaneously hypertensive rats but not in deoxycorticosterone-salt or renal vascular hypertension. Journal of Hypertension. 1992;10:137–42. doi: 10.1097/00004872-199202000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Bush KT, Tsukamoto T, Nigam SK. Selective degradation of E-cadherin and dissolution of E-cadherin-catenin complexes in epithelial ischemia. Am J Physiol Renal Physiol. 2000;278:F847–52. doi: 10.1152/ajprenal.2000.278.5.F847. [DOI] [PubMed] [Google Scholar]
- 13.Chang TW. Improvement of survival from hemorrhagic shock by enterectomy in rats: finding to implicate the role of the gut for irreversibility of hemorrhagic shock. J Trauma. 1997;42:223–30. doi: 10.1097/00005373-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Childs EW, Udobi KF, Hunter FA. Hypothermia reduces microvascular permeability and reactive oxygen species expression after hemorrhagic shock. J Trauma. 2005;58:271–7. doi: 10.1097/01.ta.0000119203.24601.7e. [DOI] [PubMed] [Google Scholar]
- 15.Cho KO, La HO, Cho YJ, Sung KW, Kim SY. Minocycline attenuates white matter damage in a rat model of chronic cerebral hypoperfusion. J Neurosci Res. 2006;83:285–91. doi: 10.1002/jnr.20727. [DOI] [PubMed] [Google Scholar]
- 16.Chung AW, Booth AD, Rose C, Thompson CR, Levin A, van Breemen C. Increased matrix metalloproteinase 2 activity in the human internal mammary artery is associated with ageing, hypertension, diabetes and kidney dysfunction. J Vasc Res. 2008;45:357–62. doi: 10.1159/000119755. [DOI] [PubMed] [Google Scholar]
- 17.Coimbra R, Hoyt D, Winchell R, Simons R, Fortlage D, Garcia J. The ongoing challenge of retroperitoneal vascular injuries. Am J Surg. 1996;172:541–4. doi: 10.1016/S0002-9610(96)00231-0. discussion 545. [DOI] [PubMed] [Google Scholar]
- 18.Curry S, Brick P, Franks NP. Fatty acid binding to human serum albumin: new insights from crystallographic studies. Biochim Biophys Acta. 1999;1441:131–40. doi: 10.1016/s1388-1981(99)00148-1. [DOI] [PubMed] [Google Scholar]
- 19.Dayal SD, Hauser CJ, Feketeova E, Fekete Z, Adams JM, Lu Q, Xu DZ, Zaets S, Deitch EA. Shock mesenteric lymph-induced rat polymorphonuclear neutrophil activation and endothelial cell injury is mediated by aqueous factors. J Trauma. 2002;52:1048–55. doi: 10.1097/00005373-200206000-00005. discussion 1055. [DOI] [PubMed] [Google Scholar]
- 20.Deitch ED, Shi HP, Lu Q, Feketeova E, Xu DZ. Serine proteases are involved in the pathogenesis of trauma-hemorrhagic shoch-induced gut and lung injury. Shock. 2003;19:542–456. doi: 10.1097/01.shk.0000048899.46342.f6. [DOI] [PubMed] [Google Scholar]
- 21.del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–51. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- 22.DeLano FA, Balete R, Schmid-Schönbein GW. Control of oxidative stress in microcirculation of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;288:H805–12. doi: 10.1152/ajpheart.00696.2004. [DOI] [PubMed] [Google Scholar]
- 23.DeLano FA, Hoyt DB, Schmid-Schönbein GW. The Auto-Digestion Hypothesis: Blockade of Pancreatic Digestive Enzymes in the Lumen of the Intestine during Hemorrhagic Shock Reduces Mortality. FASEB Journal. 2007;21:421. (Abstract) [Google Scholar]
- 24.DeLano FA, Parks DA, Ruedi JM, Babior BM, Schmid-Schönbein GW. Microvascular display of xanthine oxidase and NADPH oxidase in the spontaneously hypertensive rat. Microcirculation. 2006;13:551–66. doi: 10.1080/10739680600885152. [DOI] [PubMed] [Google Scholar]
- 25.DeLano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage: mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52:415–23. doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emerit I, Garban F, Vassy J, Levy A, Filipe P, Freitas J. Superoxide-mediated clastogenesis and anticlastogenic effects of exogenous superoxide dismutase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:12799–804. doi: 10.1073/pnas.93.23.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eppihimer MJ, Granger DN. Ischemia/reperfusion-induced leukocyte-endothelial interactions in postcapillary venules. Shock. 1997;8:16–25. doi: 10.1097/00024382-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Fitzal F, Delano FA, Young C, Rosario HS, Junger WG, Schmid-Schönbein GW. Pancreatic enzymes sustain systemic inflammation after an initial endotoxin challenge. Surgery. 2003;134:446–56. doi: 10.1067/s0039-6060(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 29.Fitzal F, DeLano FA, Young C, Rosario HS, Schmid-Schönbein GW. Pancreatic protease inhibition during shock attenuates cell activation and peripheral inflammation. J Vasc Res. 2002;39:320–9. doi: 10.1159/000065544. [DOI] [PubMed] [Google Scholar]
- 30.Fitzal F, DeLano FA, Young C, Schmid-Schönbein GW. Improvement in early symptoms of shock by delayed intestinal protease inhibition. Arch Surg. 2004;139:1008–16. doi: 10.1001/archsurg.139.9.1008. [DOI] [PubMed] [Google Scholar]
- 31.Fitzal F, Kistler E, Mitsuoka H, Hugli TE, Schmid-Schönbein GW. A new hypothesis for the origin of shock: Self-digestion of the ischemic intestine by pancreatic enzymes. In: Pitzer JA, editor. Progress in Inflammation. Nova Science Pub; 2005. [Google Scholar]
- 32.Fruchterman TM, Spain DA, Wilson MA, Harris PD, Garrison RN. Selective microvascular endothelial cell dysfunction in the small intestine following resuscitated hemorrhagic shock. Shock. 1998;10:417–22. doi: 10.1097/00024382-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004;35:998–1004. doi: 10.1161/01.STR.0000119383.76447.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuda T, Hata N. Mechanisms of endotoxin shock in rats and the anti-endotoxic effect of glucocorticoids and endotoxin-conditioning. Japanese Journal of Physiology. 1969;19:509–20. doi: 10.2170/jjphysiol.19.509. [DOI] [PubMed] [Google Scholar]
- 35.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62. [PubMed] [Google Scholar]
- 36.Garcia RA, Pantazatos DP, Gessner CR, Go KV, Woods VL, Jr, Villarreal FJ. Molecular interactions between matrilysin and the matrix metalloproteinase inhibitor doxycycline investigated by deuterium exchange mass spectrometry. Mol Pharmacol. 2005;67:1128–36. doi: 10.1124/mol.104.006346. [DOI] [PubMed] [Google Scholar]
- 37.Garcia Soriano F, Liaudet L, Marton A, Hasko G, Batista Lorigados C, Deitch EA, Szabo C. Inosine improves gut permeability and vascular reactivity in endotoxic shock. Crit Care Med. 2001;29:703–8. doi: 10.1097/00003246-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Gasche Y, Soccal PM, Kanemitsu M, Copin JC. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front Biosci. 2006;11:1289–301. doi: 10.2741/1883. [DOI] [PubMed] [Google Scholar]
- 39.George SK, Meyer TN, Abdeen O, Bush KT, Nigam SK. Tunicamycin preserves intercellular junctions, cytoarchitecture, and cell-substratum interactions in ATP-depleted epithelial cells. Biochem Biophys Res Commun. 2004;322:223–31. doi: 10.1016/j.bbrc.2004.07.097. [DOI] [PubMed] [Google Scholar]
- 40.Glenn TM, Herlihy BL, Ferfuson WW, Lefer AM. Protective effect of pancreatic duct ligation in splanchnic ischemia shock. Am J Physiol. 1972;222:1278–84. doi: 10.1152/ajplegacy.1972.222.5.1278. [DOI] [PubMed] [Google Scholar]
- 41.Go VLW, Dimagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA. Biology, Pathobiology, and Disease. Raven Press; New York: 1993. The Pancreas; pp. 1–1176. [Google Scholar]
- 42.Griffin MO, Jinno M, Miles LA, Villarreal FJ. Reduction of myocardial infarct size by doxycycline: a role for plasmin inhibition. Mol Cell Biochem. 2005;270:1–11. doi: 10.1007/s11010-005-2540-3. [DOI] [PubMed] [Google Scholar]
- 43.Harlan JM, Winn RK. Leukocyte-endothelial interactions: clinical trials of anti-adhesion therapy. Crit Care Med. 2002;30:S214–9. doi: 10.1097/00003246-200205001-00007. [DOI] [PubMed] [Google Scholar]
- 44.Healey MA, Samphire J, Hoyt DB, Liu F, Davis R, Loomis WH. Irreversible shock is not irreversible: a new model of massive hemorrhage and resuscitation. J Trauma. 2001;50:826–34. doi: 10.1097/00005373-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 45.Hinshaw LB. Sepsis/septic shock: participation of the microcirculation: an abbreviated review. Crit Care Med. 1996;24:1072–8. doi: 10.1097/00003246-199606000-00031. [DOI] [PubMed] [Google Scholar]
- 46.Hulman S, Falkner B, Freyvogel N. Insulin resistance in the conscious spontaneously hypertensive rat: euglycemic hyperinsulinemic clamp study. Metabolism: Clinical and Experimental. 1993;42:14–8. doi: 10.1016/0026-0495(93)90165-k. [DOI] [PubMed] [Google Scholar]
- 47.Jarrar D, Wang P, Cioffi WG, Bland KI, Chaudry IH. Critical role of oxygen radicals in the initiation of hepatic depression after trauma hemorrhage. J Trauma. 2000;49:879–85. doi: 10.1097/00005373-200011000-00015. [DOI] [PubMed] [Google Scholar]
- 48.Kaiser VL, Sifri ZC, Senthil M, Dikdan GS, Lu Q, Xu DZ, Deitch EA. Albumin peptide: A molecular marker for trauma/hemorrhagic-shock in rat mesenteric lymph. Peptides. 2005 doi: 10.1016/j.peptides.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Kien ND, Antognini JF, Reilly DA, Moore PG. Small-volume resuscitation using hypertonic saline improves organ perfusion in burned rats. Anesthesia and Analgesia. 1996;83:782–8. doi: 10.1097/00000539-199610000-00022. [DOI] [PubMed] [Google Scholar]
- 50.Kistler EB, Hugli TE, Schmid-Schönbein GW. The pancreas as a source of cardiovascular cell activating factors. Microcirculation. 2000;7:183–92. [PubMed] [Google Scholar]
- 51.Kistler EB, Lefer AM, Hugli TE, Schmid-Schönbein GW. Plasma activation during splanchnic arterial occlusion shock. Shock. 2000;14:30–4. doi: 10.1097/00024382-200014010-00006. [DOI] [PubMed] [Google Scholar]
- 52.Kolev K, Skopal J, Simon L, Csonka E, Machovich R, Nagy Z. Matrix metalloproteinase-9 expression in post-hypoxic human brain capillary endothelial cells: H2O2 as a trigger and NF-kappaB as a signal transducer. Thromb Haemost. 2003;90:528–37. doi: 10.1160/TH03-02-0070. [DOI] [PubMed] [Google Scholar]
- 53.Korhonen H, Pihlanto A. Food-derived bioactive peptides--opportunities for designing future foods. Curr Pharm Des. 2003;9:1297–308. doi: 10.2174/1381612033454892. [DOI] [PubMed] [Google Scholar]
- 54.Kramp WJ, Waldo S, Schmid-Schönbein GW, Hoyt D, Coimbra R, Hugli TE. Characterization of two classes of pancreatic shock factors: functional differences exhibited by hydrophilic and hydrophobic shock factors. Shock. 2003;20:356–62. doi: 10.1097/01.shk.0000082442.66379.90. [DOI] [PubMed] [Google Scholar]
- 55.Labat-Robert J, Kern P, Robert L. Biomarkers of connective tissue aging: biosynthesis of fibronectin, collagen type III, and elastase. Ann N Y Acad Sci. 1992;673:16–22. doi: 10.1111/j.1749-6632.1992.tb27431.x. [DOI] [PubMed] [Google Scholar]
- 56.Lee CZ, Yao JS, Huang Y, Zhai W, Liu W, Guglielmo BJ, Lin E, Yang GY, Young WL. Dose-response effect of tetracyclines on cerebral matrix metalloproteinase-9 after vascular endothelial growth factor hyperstimulation. J Cereb Blood Flow Metab. 2006;26:1157–64. doi: 10.1038/sj.jcbfm.9600268. [DOI] [PubMed] [Google Scholar]
- 57.Lefer AM. Myocardial depressant factor and circulatory shock. Klinische Wochenschrift. 1974;52:358–70. doi: 10.1007/BF01468434. [DOI] [PubMed] [Google Scholar]
- 58.Lefer AM. Role of a myocardial depressant factor in the pathogenesis of circulatory shock. Federation Proceedings. 1970;29:1836–47. [PubMed] [Google Scholar]
- 59.Lehoux S, Lemarie CA, Esposito B, Lijnen HR, Tedgui A. Pressure-induced matrix metalloproteinase-9 contributes to early hypertensive remodeling. Circulation. 2004;109:1041–7. doi: 10.1161/01.CIR.0000115521.95662.7A. [DOI] [PubMed] [Google Scholar]
- 60.Leonardo CC, Eakin AK, Ajmo JM, Collier LA, Pennypacker KR, Strongin AY, Gottschall PE. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J Neuroinflammation. 2008;5:34. doi: 10.1186/1742-2094-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ley K, Gaehtgens P. Microcirculation disorders in shock. Klin Anasthesiol Intensivther. 1987;33:19–36. [PubMed] [Google Scholar]
- 62.Lijnen HR. Extracellular proteolysis in the development and progression of atherosclerosis. Biochem Soc Trans. 2002;30:163–7. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 63.Lim HH, DeLano FA, Schmid-Schönbein GW. Life and death cell labeling in the microcirculation of the spontaneously hypertensive rat. J Vasc Res. 2001;38:228–36. doi: 10.1159/000051051. [DOI] [PubMed] [Google Scholar]
- 64.Luttun A, Dewerchin M, Collen D, Carmeliet P. The role of proteinases in angiogenesis, heart development, restenosis, atherosclerosis, myocardial ischemia, and stroke: insights from genetic studies. Curr Atheroscler Rep. 2000;2:407–16. doi: 10.1007/s11883-000-0079-z. [DOI] [PubMed] [Google Scholar]
- 65.Manabe T, Suzuki T, Honjo I. Role of the pancreas in organ blood flow during shock. Surgery, Gynecology and Obstetrics. 1978;146:577–82. [PubMed] [Google Scholar]
- 66.Martin MA, Monnnai M, Otani H. Isolation and characterization of a cytotoxic pentapeptide, k-casecidin, from bovine K-casein digested with bovine trypsin. Anim Sci J. 2000;71:197–207. [Google Scholar]
- 67.Martin MA, Otani H. Antimicrobial and cytotoxic peptides released from milk proteins by the action of mammalian gastrointestinal proteinases. Cur Res Adv Agric Biol Chem. 2001;1:23–36. [Google Scholar]
- 68.Mathison JC, Wolfson E, Ulevitch RJ. Participation of tumor necrosis factor in the mediation of gram negative bacterial lipopolysaccharide-induced injury in rabbits. J Clin Invest. 1988;81:1925–37. doi: 10.1172/JCI113540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCord JM, Fridovich I. Superoxide dismutase: an enzymatic function for erthryocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 70.McCuskey RS. Hepatic and splanchnic microvascular responses to inflammation and shock. Hepato-Gastroenterology. 1999;46(Suppl 2):1464–7. [PubMed] [Google Scholar]
- 71.McMillen MA, Huribal M, Sumpio B. Common pathway of endothelial-leukocyte interaction in shock, ischemia, and reperfusion. Am J Surg. 1993;166:557–62. doi: 10.1016/s0002-9610(05)81153-5. [DOI] [PubMed] [Google Scholar]
- 72.Meisel H, FitzGerald RJ. Biofunctional peptides from milk proteins: mineral binding and cytomodulatory effects. Curr Pharm Des. 2003;9:1289–95. doi: 10.2174/1381612033454847. [DOI] [PubMed] [Google Scholar]
- 73.Migeotte I, Riboldi E, Franssen JD, Gregoire F, Loison C, Wittamer V, Detheux M, Robberecht P, Costagliola S, Vassart G, Sozzani S, Parmentier M, Communi D. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J Exp Med. 2005;201:83–93. doi: 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1772–7. doi: 10.1073/pnas.97.4.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Protease inhibition in the intestinal lumen: attenuation of systemic inflammation and early indicators of multiple organ failure in shock. Shock. 2002;17:205–9. doi: 10.1097/00024382-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 76.Mota-Filipe H, McDonald MC, Cuzzocrea S, Thiemermann C. A membrane-permeable radical scavenger reduces the organ injury in hemorrhagic shock. Shock. 1999;12:255–61. doi: 10.1097/00024382-199910000-00002. [DOI] [PubMed] [Google Scholar]
- 77.Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 78.O’Connor DT, Takiyyuddin MA, Printz MP, Dinh TQ, Barbosa JA, Rozansky DJ, Mahata SK, Wu H, Kennedy BP, Ziegler MG, Wright FA, Schlager G, Parmer RJ. Catecholamine storage vesicle protein expression in genetic hypertension. Blood Press. 1999;8:285–95. doi: 10.1080/080370599439508. [DOI] [PubMed] [Google Scholar]
- 79.Ooyama T, Sakamato H. Elastase in the prevention of arterial aging and the treatment of atherosclerosis. Ciba Found Symp. 1995;192:307–17. doi: 10.1002/9780470514771.ch16. discussion 318–20. [DOI] [PubMed] [Google Scholar]
- 80.Otani H, Hata I. Inhibition of proliferative responses of mouse spleen lymphocytes and rabbit Peyer’s patch cells by bovine milk caseins and their digests. J Dairy Res. 1995;62:339–48. doi: 10.1017/s0022029900031034. [DOI] [PubMed] [Google Scholar]
- 81.Parker F, Migliore-Samour D, Floc’h F, Zerial A, Werner GH, Jolles J, Casaretto M, Zahn H, Jolles P. Immunostimulating hexapeptide from human casein: amino acid sequence, synthesis and biological properties. Eur J Biochem. 1984;145:677–82. doi: 10.1111/j.1432-1033.1984.tb08609.x. [DOI] [PubMed] [Google Scholar]
- 82.Peitzman AB, Billiar TR, Harbrecht BG, Kelly E, Udekwu AO, Simmons RL. Hemorrhagic shock. Current Problems in Surgery. 1995;32:925–1002. doi: 10.1016/s0011-3840(05)80008-5. [DOI] [PubMed] [Google Scholar]
- 83.Penn AH. Ph.D. Thesis. University of California; San Diego: 2005. Digestive enzymes in the generation of cytotoxic mediators during shock. [Google Scholar]
- 84.Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. 2007;27:296–304. doi: 10.1097/01.shk.0000235139.20775.7f. [DOI] [PubMed] [Google Scholar]
- 85.Penn AH, Schmid-Schönbein GW. The intestine as source of cytotoxic mediators in shock: free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol. 2008;294:H1779–92. doi: 10.1152/ajpheart.00902.2007. [DOI] [PubMed] [Google Scholar]
- 86.Pillai S, Oresajo C, Hayward J. Ultraviolet radiation and skin aging: roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation - a review. Int J Cosmet Sci. 2005;27:17–34. doi: 10.1111/j.1467-2494.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 87.Prewitt RL, Cardoso SS, Wood WB. Prevention of arteriolar rarefaction in the spontaneously hypertensive rat by exposure to simulated high altitude. J Hypertens. 1986;4:735–40. doi: 10.1097/00004872-198612000-00008. [DOI] [PubMed] [Google Scholar]
- 88.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–59. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roghanian A, Sallenave JM. Neutrophil elastase (NE) and NE inhibitors: canonical and noncanonical functions in lung chronic inflammatory diseases (cystic fibrosis and chronic obstructive pulmonary disease) J Aerosol Med Pulm Drug Deliv. 2008;21:125–44. doi: 10.1089/jamp.2007.0653. [DOI] [PubMed] [Google Scholar]
- 90.Rollwagen FM, Li YY, Pacheco ND, Dick EJ, Kang YH. Microvascular effects of oral interleukin-6 on ischemia/reperfusion in the murine small intestine. Am J Pathol. 2000;156:1177–82. doi: 10.1016/S0002-9440(10)64987-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Romero-Perez D, Fricovsky E, Yamasaki KG, Griffin M, Barraza-Hidalgo M, Dillmann W, Villarreal F. Cardiac uptake of minocycline and mechanisms for in vivo cardioprotection. J Am Coll Cardiol. 2008;52:1086–94. doi: 10.1016/j.jacc.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosario HS, Waldo SW, Becker SA, Schmid-Schönbein GW. Pancreatic trypsin increases matrix metalloproteinase-9 accumulation and activation during acute intestinal ischemia-reperfusion in the rat. Am J Pathol. 2004;164:1707–16. doi: 10.1016/S0002-9440(10)63729-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roumen RM, Hendriks T, Wevers RA, Goris JA. Intestinal permeability after severe trauma and hemorrhagic shock is increased without relation to septic complications. Archives of Surgery. 1993;128:453–7. doi: 10.1001/archsurg.1993.01420160095016. [DOI] [PubMed] [Google Scholar]
- 94.Rubanyi GM. Nitric oxide and circulatory shock. Advances in Experimental Medicine and Biology. 1998;454:165–72. doi: 10.1007/978-1-4615-4863-8_20. [DOI] [PubMed] [Google Scholar]
- 95.Russell DH, Barreto JC, Klemm K, Miller TA. Hemorrhagic shock increases gut macromolecular permeability in the rat. Shock. 1995;4:50–5. doi: 10.1097/00024382-199507000-00008. [DOI] [PubMed] [Google Scholar]
- 96.Scheidler N. Ph.D. University of California; San Diego: 2007. The Impact of Endothelial Mechanotransduction on Capillary Network Perfusion. [Google Scholar]
- 97.Schmid-Schönbein GW. Analysis of inflammation. Annu Rev Biomed Eng. 2006;8:93–131. doi: 10.1146/annurev.bioeng.8.061505.095708. [DOI] [PubMed] [Google Scholar]
- 98.Schmid-Schönbein GW, Hugli TE, Kistler EB, Sofianos A, Mitsuoka H. Pancreatic enzymes and microvascular cell activation in multiorgan failure. Microcirculation. 2001;8:5–14. [PubMed] [Google Scholar]
- 99.Schmid-Schönbein GW, Kistler EB, Hugli TE. Mechanisms for cell activation and its consequences for biorheology and microcirculation: Multi-organ failure in shock. Biorheology. 2001;38:185–201. [PubMed] [Google Scholar]
- 100.Schmid-Schönbein GW, Seiffge D, DeLano FA, Shen K, Zweifach BW. Leukocyte counts and activation in spontaneously hypertensive and normotensive rats. Hypertension. 1991;17:323–30. doi: 10.1161/01.hyp.17.3.323. [DOI] [PubMed] [Google Scholar]
- 101.Schober M, Howe PR, Sperk G, Fischer-Colbrie R, Winkler H. An increased pool of secretory hormones and peptides in adrenal medulla of stroke-prone spontaneously hypertensive rats. Hypertension. 1989;13:469–74. doi: 10.1161/01.hyp.13.5.469. [DOI] [PubMed] [Google Scholar]
- 102.Sechi LA, Griffin CA, Giacchetti G, Zingaro L, Catena C, Bartoli E, Schambelan M. Abnormalities of insulin receptors in spontaneously hypertensive rats. Hypertension. 1996;27:955–61. doi: 10.1161/01.hyp.27.4.955. [DOI] [PubMed] [Google Scholar]
- 103.Shen K, Sung KL, Whittemore DE, DeLano FA, Zweifach BW, Schmid-Schönbein GW. Properties of circulating leukocytes in spontaneously hypertensive rats. Biochem Cell Biol. 1995;73:491–500. doi: 10.1139/o95-054. [DOI] [PubMed] [Google Scholar]
- 104.Smedegård G, Cui LX, Hugli TE. Endotoxin-induced shock in the rat. A role for C5a. American Journal of Pathology. 1989;135:489–97. [PMC free article] [PubMed] [Google Scholar]
- 105.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–30. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 106.Suematsu M, Suzuki H, Delano FA, Schmid-Schönbein GW. The inflammatory aspect of the microcirculation in hypertension: oxidative stress, leukocytes/endothelial interaction, apoptosis. Microcirculation. 2002;9:259–76. doi: 10.1038/sj.mn.7800141. [DOI] [PubMed] [Google Scholar]
- 107.Suematsu M, Suzuki H, Tamatani T, Iigou Y, DeLano FA, Miyasaka M, Forrest MJ, Kannagi R, Zweifach BW, Ishimura Y, Schmid-Schönbein GW. Impairment of selectin-mediated leukocyte adhesion to venular endothelium in spontaneously hypertensive rats. J Clin Invest. 1995;96:2009–2016. doi: 10.1172/JCI118248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suzuki H, DeLano FA, Jamshidi N, Katz D, Mori M, Kosaki K, Gottlieb RA, Ishii H, Schmid-Schönbein GW. Enhanced DNA fragmentation in the thymus of spontaneously hypertensive rats. Am J Physiol. 1999 doi: 10.1152/ajpheart.1999.276.6.H2135. [DOI] [PubMed] [Google Scholar]
- 109.Suzuki H, Schmid-Schönbein GW, Suematsu M, DeLano FA, Forrest MJ, Miyasaka M, Zweifach BW. Impaired leukocyte-endothelial cell interaction in spontaneously hypertensive rats. Hypertension. 1994;24:719–27. doi: 10.1161/01.hyp.24.6.719. [DOI] [PubMed] [Google Scholar]
- 110.Suzuki H, Swei A, Zweifach BW, Schmid-Schönbein GW. In vivo evidence for microvascular oxidative stress in spontaneously hypertensive rats. Hydroethidine microfluorography. Hypertension. 1995;25:1083–9. doi: 10.1161/01.hyp.25.5.1083. [DOI] [PubMed] [Google Scholar]
- 111.Suzuki T, Motoyama T, Sato R. Periarteritis nodosa in spontaneously hypertensive rats. Acta Pathol Jpn. 1980;30:907–912. doi: 10.1111/j.1440-1827.1980.tb03279.x. [DOI] [PubMed] [Google Scholar]
- 112.Takiyyuddin MA, De Nicola L, Gabbai FB, Dinh TQ, Kennedy B, Ziegler MG, Sabban EL, Parmer RJ, O’Connor DT. Catecholamine secretory vesicles. Augmented chromogranins and amines in secondary hypertension. Hypertension. 1993;21:674–9. doi: 10.1161/01.hyp.21.5.674. [DOI] [PubMed] [Google Scholar]
- 113.Thiagarajan RR, Winn RK, Harlan JM. The role of leukocyte and endothelial adhesion molecules in ischemia-reperfusion injury. Thromb Haemost. 1997;78:310–4. [PubMed] [Google Scholar]
- 114.Tompkins SD, Gregory S, Hoyt DB, Ozkan AN. In vitro inhibition of IL-2 biosynthesis in activated human peripheral blood mononuclear cells by a trauma-induced glycopeptide. Immunol Lett. 1990;23:205–9. doi: 10.1016/0165-2478(90)90193-t. [DOI] [PubMed] [Google Scholar]
- 115.Tran ED, Schmid-Schönbein GW. An In-Vivo Analysis of Capillary Stasis and Endothelial Apoptosis in a Model of Hypertension. Microcirculation. 2007:1–12. doi: 10.1080/10739680701419992. [DOI] [PubMed] [Google Scholar]
- 116.Uchiba M, Okajima K, Murakami K, Okabe H, Takatsuki K. Endotoxin-induced pulmonary vascular injury is mainly mediated by activated neutrophils in rats. Thromb Res. 1995;78:117–25. doi: 10.1016/0049-3848(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 117.Vender RL. Therapeutic potential of neutrophil-elastase inhibition in pulmonary disease. J Investig Med. 1996;44:531–9. [PubMed] [Google Scholar]
- 118.Vollmar B. Microcirculation and oxygen supply to the liver in hemorrhagic shock and sepsis. Anasthesiol Intensivmed Notfallmed Schmerzther. 1995;30(Suppl 1):S52–4. doi: 10.1055/s-2007-996562. [DOI] [PubMed] [Google Scholar]
- 119.Waldo SW, Rosario HS, Penn AH, Schmid-Schönbein GW. Pancreatic digestive enzymes are potent generators of mediators for leukocyte activation and mortality. Shock. 2003;20:138–43. doi: 10.1097/01.shk.0000073866.47824.ae. [DOI] [PubMed] [Google Scholar]
- 120.Wang Y, Luo W, Reiser G. Trypsin and trypsin-like proteases in the brain: proteolysis and cellular functions. Cell Mol Life Sci. 2008;65:237–52. doi: 10.1007/s00018-007-7288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Watanabe N, Ikeda U. Matrix metalloproteinases and atherosclerosis. Curr Atheroscler Rep. 2004;6:112–20. doi: 10.1007/s11883-004-0099-1. [DOI] [PubMed] [Google Scholar]
- 122.Wattanasirichaigoon S, Menconi MJ, Delude RL, Fink MP. Effect of mesenteric ischemia and reperfusion or hemorrhagic shock on intestinal mucosal permeability and ATP content in rats. Shock. 1999;12:127–33. doi: 10.1097/00024382-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 123.Waxman K. Shock: ischemia, reperfusion, and inflammation. New Horiz. 1996;4:153–60. [PubMed] [Google Scholar]
- 124.Wettstein R, Tsai AG, Erni D, Winslow RM, Intaglietta M. Resuscitation with polyethylene glycol-modified human hemoglobin improves microcirculatory blood flow and tissue oxygenation after hemorrhagic shock in awake hamsters. Crit Care Med. 2003;31:1824–30. doi: 10.1097/01.CCM.0000069340.16319.F2. [DOI] [PubMed] [Google Scholar]
- 125.Winn RK, Mihelicic D, Vedder NB, Sharar SR, Harlan JM. Monoclonal antibodies to leukocyte and endothelial adhesion molecules attenuate ischemia-reperfusion injury. Behring Institute Mitteilungen. 1993;119:229–37. [PubMed] [Google Scholar]
- 126.Winn RK, Sharar SR, Vedder NB, Harlan JM. Leukocyte and endothelial adhesion molecules in ischaemia/reperfusion injuries. Ciba Found Symp. 1995;189:63–71. doi: 10.1002/9780470514719.ch6. discussion 72–6, 77–8. [DOI] [PubMed] [Google Scholar]
- 127.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–3. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 128.Yong VW, Zabad RK, Agrawal S, Goncalves Dasilva A, Metz LM. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259:79–84. doi: 10.1016/j.jns.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 129.Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. 2005;12:295–300. doi: 10.5551/jat.12.295. [DOI] [PubMed] [Google Scholar]
- 130.Zweifach BW, Kovalcheck S, DeLano F, Chen P. Micropressure-flow relationships in a skeletal muscle of spontaneously hypertensive rats. Hypertension. 1981;3:601–614. doi: 10.1161/01.hyp.3.5.601. [DOI] [PubMed] [Google Scholar]
- 131.Zweifach BW, Lipowsky HH. Pressure-flow relations in blood and lymph microcirculation. In: Renkin EM, Michel CC, editors. Handbook of Physiology, Section 2: The Cardiovascular System. American Physiological Society; Bethesda, M.D: 1984. pp. 251–307. [Google Scholar]