

Abstract
In Sprague-Dawley rats, renal AT2Rs mediate natriuresis in response to renal interstitial (RI) D1-like receptor (D1R) stimulation or RI Ang III infusion. After D1R activation, apical membrane (AM) but not total renal proximal tubule cell (RPTC) AT2R expression is increased, suggesting that AM AT2R translocation may be important for natriuresis. The onset of hypertension in spontaneously hypertensive rats (SHR) is preceded by defects in renal sodium excretion. The present study examines AT2R-mediated natriuresis in response to RI Ang III infusion in Wistar-Kyoto rats (WKY) and SHR. WKY and SHR received RI Ang III infusion after 24 h of systemic AT1R blockade with candesartan (CAND). In WKY, urine sodium excretion rate (UNaV) increased from 0.043±0.01 to 0.191±0.06 μmol/min (P<0.05) in response to Ang III infusion, but identical conditions failed to increase UNaV in SHR. The increase in UNaV was blocked by co-infusion of PD-123319, a selective AT2R antagonist. On confocal microscopy images, Ang III-infused WKY demonstrated greater RPTC AM AT2R fluorescence intensity compared to SHR (5385±725 vs. 919±35, P<.0001), and Western blot analysis demonstrated increased AM (0.050±0.003 vs. 0.038±0.003, P<0.01) but not total cell AT2R expression in WKY. In SHR, AM AT2R expression remained unchanged in response to RI Ang III infusion. Thus, RI Ang III infusion elicits natriuresis and RPTC AT2R translocation in WKY. Identical manipulations fail to induce natriuresis or AT2R translocation in SHR, suggesting that defects in AT2R-mediated natriuresis and trafficking may be important to the development of hypertension in SHR.
Keywords: sodium, natriuresis, angiotensin III, AT2 receptor, AT1 receptor, translocation
INTRODUCTION
The renin–angiotensin system (RAS) is a coordinated hormonal cascade of crucial importance in cardiovascular and renal function. In recent years, an emphasis has been placed on delineating the role of the intrarenal RAS in the regulation of blood pressure (BP)1, 2 and sodium (Na+) balance3-5. The majority of the effects of the intrarenal RAS are mediated by two angiotensin receptors, AT1 (AT1R) and AT2 (AT2R). Renal AT1Rs, as a consequence of their antinatriuretic actions, are required for the development of angiotensin II (Ang II)-dependent hypertension, since the presence of systemic extrarenal AT1Rs alone is not sufficient to sustain hypertensive responses to Ang II infusion1. Furthermore, AT1Rs in renal proximal tubule cells (RPTCs), as opposed to other sites along the nephron, are primarily responsible for this response [Gurley GB, Aleen AM, Haase VH, Snouwaert JN, Koller BH, Le TH, and Coffman TM. AT1Rs in the Proximal Tubule of the Kidney Are Essential for Blood Pressure Regulation, AHA Council For High Blood Pressure Research Annual Meeting, 2008; Abstract 062]. In Sprague-Dawley rats, renal AT2Rs have been reported to mediate natriuresis in response to renal interstitial (RI) AT1R blockade or angiotensin III (Ang III) infusion3. Inhibition of the conversion of Ang II to Ang III in the kidney abolishes natriuresis mediated by renal AT2Rs, indicating that Ang III is the preferred agonist of this response4. Thus, RPTC AT1Rs and AT2Rs are major determinants of BP and Na+ responses in normal and hypertensive animals.
The intrarenal dopaminergic system also plays an important role in the regulation of Na+ balance. Dopamine, synthesized by the RPTCs, mediates diuresis and natriuresis via D1-like receptor (D1R) activation6, 7. A physiological interaction between the intrarenal RAS and dopaminergic systems has been reported in normal Sprague-Dawley rats. In response to high salt diet, RI D1R activation with fenoldapam (FEN), results in natriuresis and diuresis that is abolished by selective pharmacological inhibition of renal AT2Rs with PD-123319 (PD)8. Furthermore, FEN-induced natriuresis is accompanied by an increase in apical plasma membrane (AM) but not total RPTC AT2R expression as quantified by Western blot analysis8. Thus, D1R-mediated natriuresis is dependent on functional renal AT2Rs, and one of the mechanisms involves RPTC AT2R translocation.
Spontaneously hypertensive rats (SHR) develop hypertension as they age and are widely employed as a model to study the development and maintenance of human primary (essential) hypertension9. Prior to the onset of hypertension, SHR demonstrate inappropriately increased RPTC Na+ reabsorption, which is not accompanied by an increase in glomerular filtration rate or renal blood flow10-16. These observations suggest a primary defect in the renal tubule function rather than in renal hemodynamics. Over time, however, increased renal perfusion pressure is required by the kidneys to continue to excrete Na+, and this adaptation is fundamental to the development and maintenance of hypertension17, 18.
Previous studies examining the Na+ excretory defects in SHR that ultimately lead to hypertension have focused on alterations in renal dopaminergic or AT1R-mediated effects. However, as mentioned previously, natriuresis due to renal D1R stimulation and AT1R blockade is mediated, at least in part, by renal AT2Rs. Thus, the present study was undertaken to determine whether another strain of normal rat, Wistar-Kyoto rats (WKY), demonstrates AT2R-mediated natriuresis in response to RI Ang III infusion, and if so, whether such natriuresis is demonstrable in SHR. Because one of the mechanisms of AT2R-mediated natriuresis involves AT2R translocation from the cytoplasm to the AM of RPTCs, AT2R localization and expression after pharmacological stimulation of natriuresis was determined via confocal microscopy and Western blot analysis in both WKY and SHR.
METHODS
Animal Preparation
The experiments, which were approved by the University of Virginia Animal Care and Use Committee, were conducted in 12 week-old female WKY and SHR rats (Harlan, Teklad), in accordance with the NIH Guide for Care and Use of Laboratory Animals. Rats (N=18) were placed under general anesthesia with pentobarbital (50 mg/mL) given 5 mg/100 g body weight intraperitoneally. A tracheostomy was performed, and arterial access was achieved by direct cannulation of the right carotid artery. Renal cortical interstitial infusion catheters were placed as previously reported3-5. When two substances were simultaneously infused into the kidney, separate interstitial catheters were used. Intravenous access was obtained via cannulation of the right internal jugular vein. Rats were housed under controlled conditions (temperature, 21±1°C; humidity, 60±10%; and light, 8 to 20 hours). Experiments were initiated at the same time each day to prevent any diurnal variation in blood pressure (BP). Mean arterial pressure (MAP) was measured by the direct intracarotid method with the use of a BP analyzer (Micromed Inc). MAP was recorded every 5 min and averaged for each of the control and experimental periods.
Pharmacological Agents
Ang III (des-Asp1)-angiotensin II (Bachem), an AT1R and AT2R agonist, was used for these studies (Ki 10.5×10−9 mol/L and 2.2×10−9 mol/L for AT1Rs and AT2Rs, respectively19). Candesartan (CAND), a specific, potent insurmountable inhibitor of AT1R (IC50 >1×10−5 mol/L and 2.9×10−8 mol/L for AT2Rs and AT1Rs, respectively), was used for systemic AT1R blockade. PD (Parke-Davis), a specific AT2R antagonist (IC50 2×10−8 mol/L and >1×10−4 mol/L for AT2Rs and AT1Rs, respectively), was employed interstitially to block the AT2R.
Effects on UNaV of Unilateral RI Ang III in the Presence of Systemic AT1R Blockade With and Without RI AT2R Blockade
WKY and SHR rats (n=6 in each group) were studied on normal Na+ intake with both kidneys intact. There were a total of 2 groups for WKY and 1 group for SHR. In all of the rats, the right kidney served as the control kidney and was infused with 5% dextrose in water (D5W) directly into the RI space during both control (30 min) and experimental collection periods (30 min each). In the first group, osmotic minipumps were implanted into the interscapular region with the animals under short-term anesthesia with ketamine (100 mg/mL) and xylazine (20 mg/mL) for systemic CAND (0.01 mg/kg per min) infusion 24 h before and during the experiment. Ang III (3.5, 7, 14, and 28 nmol/kg per min) was then infused cumulatively (each concentration for 30 min) into the RI space of the left (experimental) kidney after a 30 min control infusion of D5W (2.5 μL/min). In the second group, systemic CAND infusion was achieved in the same manner as in the first group, but Ang III and PD (10 μg/kg per minute) were co-infused into the RI space of the left kidney. In all rats, both ureters were cannulated individually to collect urine for quantification of UNaV for the control and 4 experimental periods from each kidney.
In Vivo Kidney Perfusion and Fixation Procedure
WKY and SHR kidneys were subject to the following fixation protocol after receiving either vehicle (D5W) or RI Ang III infusions. With RI infusions continuing, the rat heart left ventricular cavity was cannulated and the animal was perfused with 40 ml of 4% sucrose in Dulbecco's phosphate-buffered saline with calcium and magnesium chloride (DPBS++) followed by perfusion with 40 ml of 4% paraformaldehyde (PFA) in DPBS++. Sections of cortex were removed and placed in 4% PFA for 2 h at room temperature. Cortical sections were rinsed and immersed in 100 mM Tris-HCl for 30 min, before storage in 30% sucrose in DPBS++ overnight at 4°C. Cortical specimens were embedded in Tissue Tek OCT Compound freezing medium, frozen on liquid nitrogen and stored at −80°C. Cryostat thin sections (8 μm) were placed on Probe On Plus positively charged microscope slides (Fisher Scientific, Pittsburg, PA) and a PAP pen was used to draw a hydrophobic ring around the sections for immediate staining.
Immunofluorescence (IF) microscopy
After specimens had been spotted onto slides and washed with triphosphate- buffered saline (TBS), they were permeabilized with 0.2 % triton x-100 in TBS for 5 min. The sections were washed several times with TBS with 0.02% Tween 20 (TBST), and then blocked in 1% milk in TBST for 1 h. The kidney sections were incubated with anti-AT2R primary antibody at 1:100 dilution in 1% milk overnight at 4°C. ALEXA 647 conjugated donkey anti-rabbit secondary antibody diluted at 1:500 was then added for 90 min at room temperature. In order to identify RPTCs, the preparation was stained further with Texas-Red phalloidin (1:200) which labels actin containing structures including RPTC AMs. Hoescht (10 mg/mL stock) was diluted at 1:2500 and added to identify nuclei. Following several TBST washes, Fluoromount was applied before covering with a glass coverslip.
Confocal microscopy and quantification of immunofluorescence signals
Confocal microscopy recordings were performed using a Leica TCS SP inverted confocal scanning laser microscope with excitation at 490 and 647 and detection at 510 and 660 nm. Images were captured using identical capture parameters for each section on an Olympus IX81 Spinning Disk Confocal Microscope using a 60X 1.2 NA UIS2 water immersion objective and a Hammamatsu 9100-02 EMCCD camera with Slidebook 4.2 software. Images were exported as 16 bit tiffs and analyzed using MacBiophotonics ImageJ v1.38m and the Sync Measure 3D pluggin written by Joachim Walter. In order to generate line intensity plots, the AT2R-stained slide and the corresponding phalloidin-stained slide were opened, and the Sync Measure 3D pluggin was run. The line tool was used to draw a line from the tip of the phalloidin stain at the brush border of one cell to the phalloidin-stained basolateral membrane at the base of the same cell, and the AT2R immunofluorescence intensity plot along this trajectory was measured.
Membrane Preparations and Western Blot Analysis
After the in vivo RI D5W or Ang III infusions, RPTC total membranes and AMs were isolated as previously published8. Briefly, RPTCs were obtained after whole body perfusion with 40 ml of 4°C DPBS without Ca++ or Mg++ (DPBS−−), to halt endosomal movement and slow cellular respiration. Immediately following the perfusion, the kidney cortex was removed and minced into 1 mm3 pieces. The specimens were rinsed in 20 mL of 4°C DPBS−− containing 0.5 mmol glycol tetraacetic acid (EGTA) at 1200 rpm to open the tight junctions between the RPTCs, and then rinsed again in 20 mL of 4°C DPBS++ at 1200 rpm to remove the EGTA. The samples were digested in a collagenase solution (2 mg/mL in DPBS++; Boehringer Manheim Biochemicals) at room temperature for 3.5 h in a spinner flask that provided gentle agitation. Following the digestion, 15 mL of DPBS++ was added to the collagenase mixture, which was then transferred through two successive sieves, 212 μm and 106 μm, respectively (U.S.A Standard Sieve Series). The supernatant was collected and spun at 1200 rpm for 10 min in order to isolate RPTCs. Total cell membranes of the RPTCs were subsequently isolated by the method of Nagamatsu et al.20 AMs of RPTCs were isolated by first lysing the cells in detergent-free lysis buffer and performing a biotinylated lectin pull-down assay as follows. After the tissue was homogenized, 1 mg of total protein was incubated with 20 μg of biotinylated Lotus tetragonolobus agglutinin (LTA) lectin (Vector Laboratories) in 10-mL volume for 2 hours at room temperature. A 50% vol/vol slurry (40 μL) of Ultralink Neutravidin beads (Pierce Laboratory) was then added and incubated for 30 minutes. The beads were then pelleted and thoroughly washed using a microcentrifuge spin cup filter. The LTA affinity attached membranes were eluted by incubating the beads in the spin cup filter with 40 μL of 50°C sample buffer and 20 μL loaded per lane on a gel for Western blotting. After preparation, total RPTC membranes and AMs were incubated with rabbit AT2R polyclonal antibody (1:100 dilution, H-143 Santa Cruz) and villin monoclonal antibody (1:2500 dilution, Immunotech), which is enriched in RPTCs8. A review of the literature suggests that the H-143 AT2R antibody from Santa Cruz is specific to the AT2R, and does not cross react with the AT1R [see product citations for AT2R (H-143) catalog number sc-9040 (Santa Cruz Biotechnology, Inc.)]. In addition, a database search, e.g., Protein Blast indicates that the epitope recognized by H-143 is unique to the AT2R. Membranes were subsequently incubated with infrared secondary antibodies (anti-rabbit IRDye 680nm and anti-mouse IRDye 800nm, each at 1:15,000, Licor Biosciences). Immunoreactivity and quantitative assessment of band densities was performed using the Odyssey Infrared Imaging System (Licor Biosciences). Results were reported as a ratio of AT2R/villin expression.
Statistical Analysis
Comparisons among vehicle, Ang III, and Ang III plus PD were estimated by ANOVA, including a repeated-measures term, by using the general linear models procedure of the Statistical Analysis System. Multiple comparisons of individual pairs of effect means were conducted by the use of least-square means pooled variance. Data are expressed as mean ±1 SE. Statistical significance was identified at a level of P<0.05.
RESULTS
Effects of RI Ang III Infusion and Ang III Plus PD Infusion on UNaV and MAP in Uninephrectomized WKY and SHR Rats
As demonstrated in Figure 1, in WKY, RI Ang III infusion (in the presence of systemic AT1R blockade) increased UNaV from a baseline of 0.043±0.006 to 0.11±0.025 μmol/min (P<0.05) during 3.5 nmol/mg/kg Ang III infusion, to 0.23±0.069 μmol/min (P<0.05) during 7.0 nmol/mg/kg Ang III infusion, to 0.18±0.041 μmol/min (P<0.01) during 14 nmol/mg/kg Ang III infusion, and to 0.19±0.060 μmol/min (P<0.05) during 28 nmol/mg/kg Ang III infusion. PD co-infusion abolished the natriuretic responses to RI Ang III (P<0.05 or <0.01 from Ang III infusion). In SHR, however, identical infusions of Ang III failed to increase UNaV (baseline 0.033±0.016 to 0.031±0.009 μmol/min after 2 h of Ang III infusion, P=NS). Infusion of vehicle (D5W) resulted in no change in UNaV in WKY or SHR during control or experimental periods. As illustrated in Figure 2, RI Ang III infusion did not significantly alter MAP from baseline in WKY or SHR. Similarly, co-infusion of PD with Ang III did not influence MAP in WKY.
FIGURE 1.

Urine Na+ excretion rate (UNaV) in the presence of CAND in WKY after renal interstitial (RI) infusion of D5W ( ), Ang III (
), Ang III ( ), Ang III + PD (
), Ang III + PD ( ), and in SHR in response to D5W (
), and in SHR in response to D5W ( ) or Ang III (
) or Ang III ( ), N=6 per group. During the control period, only D5W was infused in all groups. Data represent mean ±1SE; *P<0.05, **P<0.01 from respective control period, +P<0.05, ++P<0.01, XP<0.05, and XXP<0.01 from Ang III-infused kidney in WKY.
), N=6 per group. During the control period, only D5W was infused in all groups. Data represent mean ±1SE; *P<0.05, **P<0.01 from respective control period, +P<0.05, ++P<0.01, XP<0.05, and XXP<0.01 from Ang III-infused kidney in WKY.
FIGURE 2.

MAP responses in the presence of CAND during the control and experimental periods in WKY during RI infusion of Ang III ( ), Ang III + PD (
), Ang III + PD ( ), and in SHR during RI Ang III (
), and in SHR during RI Ang III ( ) infusion, N=6 per group. During the control period, only D5W was infused in all groups of animals. Data represent mean ±1 SE.
) infusion, N=6 per group. During the control period, only D5W was infused in all groups of animals. Data represent mean ±1 SE.
Confocal Microscopy Analysis of RPTC AT2R Localization in Response to In Vivo RI Ang III Infusion in the Presence of Systemic AT1R Blockade in WKY and SHR
Figure 3 demonstrates high power (600X) confocal micrographs of WKY (Panel A) and SHR (Panel B) renal cortex labeled with antibodies to phalloidin (red), to AT2Rs (green), and Hoechst nuclear stain (blue) after vehicle, D5W infusion (control) (Panel A, Micrograph 1) and at the end of 2 h of in vivo Ang III infusion (Panel A, Micrograph 2). UNaV increased significantly from 0.032 to 0.192 μmol/min μmol/min after 2 h of Ang III infusion in WKY, but failed to increase in SHR (baseline 0.034 to 0.032 after 2 h of Ang III infusion). As indicated by the colocalization of AT2R and phalloidin in WKY (Panel A, Micrograph 2; yellow color), the Ang III-infused kidney demonstrated markedly increased AM expression of AT2Rs in RPTCs. However, as shown in Panel B, Micrograph 2, RI Ang III infusion did not result in increased apical membrane AT2R localization in SHR. Figure 4 quantifies AT2R fluorescence intensity as a function of its distance from the apical tip of RPTCs. Compared with SHR, Ang III-infused kidneys from WKY demonstrated greater RPTC AM fluorescence intensity (5385 ± 725 vs. 919 ± 35, p <.0001, quantified as the average of AT2R fluorescence intensity 0-2 μm from the apical tip) and reduced cytosolic AT2R fluorescence intensity (1878 ± 311 vs. 1044 ± 24, p<0.001, average of AT2R fluorescence intensity 2-6 μm from the apical tip).
FIGURE 3.
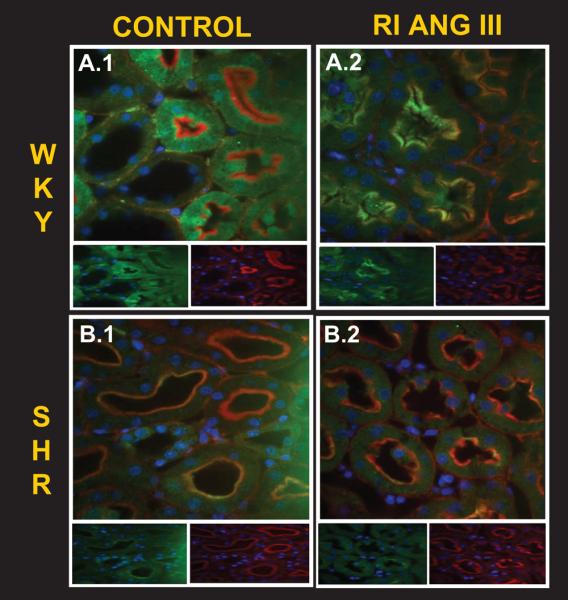
Confocal micrographs (600x) of renal cortical sections (8μm) obtained from control (D5W-) and Ang III-infused WKY (A) and SHR (B) kidneys, in the presence of CAND. In vivo, Ang III induced natriuresis in WKY, but not in SHR. On confocal microscopy of the same Ang III-infused kidneys, RPTC AT2R (green), co-localized with phalloidin (red), a marker of proximal tubule brush border, to produce a gold color in WKY (A.2). In SHR (B), the RPTC AT2R (green) failed to localize in the brush border (red) in response to renal Ang III infusion. Insets show individual AT2R (left) and phalloidin (right) staining for each condition.
FIGURE 4.

AT2R expression (relative fluorescence units) in thin sections of WKY (black) or SHR (gray) kidneys infused with RI Ang III (in the presence of CAND), as a function of the distance from the tip of the AM of RPTCs. Each data point represents mean ± SE of 9 independent measurements.
RPTC Total Cell and AM AT2R Expression In Response to RI Vehicle or Ang III Infusion In WKY and SHR
As demonstrated by Western blot analysis in Figure 5A, RPTC total cell and AM AT2R protein expression was similar between WKY and SHR after 2.5 h of RI vehicle infusion. However, compared to RI D5W infusion, RI Ang III infusion in WKY resulted in a 30% increase in AM (P<0.01), but not total RPTC membrane AT2R protein expression (Figure 5B). In SHR, AM AT2R protein expression failed to increase from baseline in response to RI Ang III infusion (Figure 5C).
FIGURE 5.

Western blot analysis of RPTC total cell and AM AT2R protein expression in response to RI vehicle (A) or Ang III infusion in WKY (B) and SHR (C), N=6 per group. Panel A: Total RPTC and AM AT2R expression in vehicle-infused WKY ( ) and SHR (
) and SHR ( ). Panel B: Total RPTC and AM AT2R expression in vehicle-infused (
). Panel B: Total RPTC and AM AT2R expression in vehicle-infused ( ) or Ang III-infused (
) or Ang III-infused ( ) WKY. Panel C: Total RPTC and AM AT2R expression in vehicle-infused (
) WKY. Panel C: Total RPTC and AM AT2R expression in vehicle-infused ( ) or Ang III-infused (
) or Ang III-infused ( ) SHR. Data represent mean ±1SE; **P<0.01, compared to vehicle-infused WKY.
) SHR. Data represent mean ±1SE; **P<0.01, compared to vehicle-infused WKY.
DISCUSSION
The present studies demonstrate that in normal WKY rats, RI Ang III infusion, in the presence of systemic AT1R blockade, results in AT2R-mediated natriuresis that is accompanied by increased AM but not total RPTC membrane AT2R expression. Identical Ang III infusions fail to stimulate natriuresis or alter AT2R localization or expression in SHR, suggesting that defects in AT2R function and/or trafficking may be important in the excess Na+ retention and ultimate development of hypertension in these animals.
Previous studies have shown that intrarenal Ang III infusion, in the presence of systemic AT1R blockade, results in AT2R-mediated natriuresis in Sprague-Dawley rats on normal Na+ intake3. These results are not specific for the Sprague-Dawley strain, however, since the present study demonstrates similar natriuresis to identical RI Ang III infusions in WKY. Indeed, in Sprague-Dawley rats, natriuresis to RI Ang III can be augmented by inhibiting the activity of the enzyme responsible for metabolizing Ang III (aminopeptidase N) and can be abolished by inhibiting the enzyme responsible for the formation of Ang III (aminopeptidase A)4. Thus, RI Ang III, and not Ang II, was employed to elicit natriuretic responses in WKY rats in this study. It is probable, and other authors have also proposed21, that Ang III, and not Ang II, is the preferred agonist of AT2Rs. Because the natriuresis in response to Ang III infusion in WKY was abolished to control levels with PD, the observed natriuresis was likely mediated by renal AT2Rs.
The mechanisms by which renal AT2Rs mediate natriuresis have not been established. However, it is well known that the regulation of cell surface expression of several membrane proteins by different hormones is crucial in the regulation of various aspects of whole body homeostasis22-24. In the kidney, the regulation of the abundance of the water transporter aquaporin-2 at the AM of collecting duct cells by vasopressin determines water excretion and thus plays a crucial role in the control of water homeostasis25. Likewise, it has been established that the natriuretic D1R translocates to the cell surface in response to D1R activation in cultured kidney cells, kidney slice preparations and isolated proximal tubules26-28. This event requires signaling through cAMP29 and is dependent upon intact microtubules and vesicular fusion proteins28. The physiological significance of RPTC D1R trafficking is not known, but since natriuretic responses to D1R activation are mediated, at least in part, by AT2R activation8, the present study investigated the trafficking patterns of renal AT2Rs under natriuretic stimulation.
At baseline, both WKY and SHR have been reported to demonstrate similar AT2R mRNA expression levels in the whole kidney and mesenteric vessels30-32. However, to our knowledge, this is the first report that documents similar RPTC AT2R protein expression levels after 24 hours of systemic AT1R blockade between WKY and SHR, suggesting that defects in sodium excretion in response to Ang III in SHR are not accounted for by decreased expression of natriuretic AT2Rs before infusion. The significance of characterizing RPTC AT2R expression levels is underscored by recent reports suggesting that BP regulation at baseline, and in response to Ang II infusion, is primarily determined by natriuretic responses in the renal proximal, and not distal tubules [Gurley GB, Aleen AM, Haase VH, Snouwaert JN, Koller BH, Le TH, and Coffman TM. AT1Rs in the Proximal Tubule of the Kidney Are Essential for Blood Pressure Regulation, AHA Council For High Blood Pressure Research Annual Meeting, 2008; Abstract 062].
In the present study, RPTC AM but not total cell AT2R expression was increased in response to Ang III-induced natriuresis in WKY. The lack of increase in total RPTC AT2R expression suggests that the trafficking of RPTC AT2Rs may be regulated under conditions that promote natriuresis. Whether AT2R translocation is required for natriuresis is presently unknown, but the absence of natriuresis or increased AM AT2R expression in SHR strongly suggests that the intracellular trafficking of RPTC AT2Rs may help to determine natriuretic responses. In fact, previous studies have also documented increased AM but not total RPTC AT2R protein expression in response to D1R-induced natriuresis8. Given similar baseline RPTC AT2R protein expression levels, the failure of SHR to increase AM AT2R expression in response to identical natriuretic stimuli indicates that functional abnormalities in RPTC AT2Rs may contribute to the excess sodium retention that occurs in these animals. Further studies will investigate the mechanisms of renal AT2R trafficking in normal rodents, and determine whether defects in AT2R signaling or trafficking machinery predispose SHR to defective Na+ excretion.
Acknowledgments
Sources of Funding: AHA Fellow-To-Faculty Award 0675020N (SHP)
Footnotes
PERSPECTIVES
Renal AT2Rs mediate sodium excretion in normal animals in response to RI Ang III infusion. The natriuresis is accompanied by increased RPTC AM AT2R distribution in WKY, but not in SHR. In light of the major critical importance of excess sodium retention in the development of hypertension in SHR, the present studies provide a potential mechanistic explanation for the pathogenesis of hypertension in SHR.
Disclosures: None
REFERENCES
- 1.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005;115:1092–1099. doi: 10.1172/JCI200523378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Padia SH, Howell NL, Siragy HM, Carey RM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension. 2006;47:537–544. doi: 10.1161/01.HYP.0000196950.48596.21. [DOI] [PubMed] [Google Scholar]
- 4.Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension. 2008;51:460–465. doi: 10.1161/HYPERTENSIONAHA.107.103242. [DOI] [PubMed] [Google Scholar]
- 5.Padia SH, Kemp BA, Howell NL, Siragy HM, Fournie-Zaluski MC, Roques BP, Carey RM. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension. 2007;49:625–630. doi: 10.1161/01.HYP.0000254833.85106.4d. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZQ, Siragy HM, Felder RA, Carey RM. Intrarenal dopamine production and distribution in the rat. Physiological control of sodium excretion. Hypertension. 1997;29:228–234. doi: 10.1161/01.hyp.29.1.228. [DOI] [PubMed] [Google Scholar]
- 7.Wang ZQ, Way D, Shimizu K, Fong F, Trigg L, McGrath BP. Beneficial acute effects of selective modulation of renal dopamine system by gamma-L-glutamyl-L-dopa in rabbits with congestive heart failure. J Cardiovasc Pharmacol. 1993;21:1004–1011. doi: 10.1097/00005344-199306000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Salomone LJ, Howell NL, McGrath HE, Kemp BA, Keller SR, Gildea JJ, Felder RA, Carey RM. Intrarenal dopamine D1-like receptor stimulation induces natriuresis via an angiotensin type-2 receptor mechanism. Hypertension. 2007;49:155–161. doi: 10.1161/01.HYP.0000251881.89610.ee. [DOI] [PubMed] [Google Scholar]
- 9.Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension. Man and rat. Circ Res. 1981;48:309–319. doi: 10.1161/01.res.48.3.309. [DOI] [PubMed] [Google Scholar]
- 10.Beierwaltes WH, Arendshorst WJ, Klemmer PJ. Electrolyte and water balance in young spontaneously hypertensive rats. Hypertension. 1982;4:908–915. doi: 10.1161/01.hyp.4.6.908. [DOI] [PubMed] [Google Scholar]
- 11.Biollaz J, Waeber B, Diezi J, Burnier M, Brunner HR. Lithium infusion to study sodium handling in unanesthetized hypertensive rats. Hypertension. 1986;8:117–121. doi: 10.1161/01.hyp.8.2.117. [DOI] [PubMed] [Google Scholar]
- 12.Christiansen RE, Roald AB, Tenstad O, Iversen BM. Renal hemodynamics during development of hypertension in young spontaneously hypertensive rats. Kidney Blood Press Res. 2002;25:322–328. doi: 10.1159/000066792. [DOI] [PubMed] [Google Scholar]
- 13.Firth JD, Raine AE, Ledingham JG. Sodium and lithium handling in the isolated hypertensive rat kidney. Clin Sci (Lond) 1989;76:335–341. doi: 10.1042/cs0760335. [DOI] [PubMed] [Google Scholar]
- 14.Heckmann U, Zidek W, Schurek HJ. Sodium reabsorption in the isolated perfused kidney of normotensive and spontaneously hypertensive rats. J Hypertens Suppl. 1989;7:S172–173. doi: 10.1097/00004872-198900076-00082. [DOI] [PubMed] [Google Scholar]
- 15.Mozaffari MS, Jirakulsomchok S, Shao ZH, Wyss JM. High-NaCl diets increase natriuretic and diuretic responses in salt-resistant but not salt-sensitive SHR. Am J Physiol. 1991;260:F890–897. doi: 10.1152/ajprenal.1991.260.6.F890. [DOI] [PubMed] [Google Scholar]
- 16.Nagaoka A, Kakihana M, Shibota M, Fujiwara K, Shimakawa K. Reduced sodium excretory ability in young spontaneously hypertensive rats. Jpn J Pharmacol. 1982;32:839–844. doi: 10.1254/jjp.32.839. [DOI] [PubMed] [Google Scholar]
- 17.Guyton AC, Coleman TG, Cowley AV, Jr., Scheel KW, Manning RD, Jr., Norman RA., Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1972;52:584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 18.Guyton AC, Coleman TG, Young DB, Lohmeier TE, DeClue JW. Salt balance and long-term blood pressure control. Annu Rev Med. 1980;31:15–27. doi: 10.1146/annurev.me.31.020180.000311. [DOI] [PubMed] [Google Scholar]
- 19.Wright JW, Harding JW. Important role for angiotensin III and IV in the brain renin-angiotensin system. Brain Res Brain Res Rev. 1997;25:96–124. doi: 10.1016/s0165-0173(97)00019-2. [DOI] [PubMed] [Google Scholar]
- 20.Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE, Bell GI. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem. 1992;267:467–472. [PubMed] [Google Scholar]
- 21.van Esch JH, Oosterveer CR, Batenburg WW, van Veghel R, Danser AH. Effects of angiotensin II and its metabolites in the rat coronary vascular bed: Is angiotensin III the preferred ligand of the angiotensin AT(2) receptor? Eur J Pharmacol. 2008;588:286–293. doi: 10.1016/j.ejphar.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 22.Bradbury NA, Bridges RJ. Role of membrane trafficking in plasma membrane solute transport. Am J Physiol. 1994;267:C1–24. doi: 10.1152/ajpcell.1994.267.1.C1. [DOI] [PubMed] [Google Scholar]
- 23.Jessen N, Goodyear LJ. Contraction signaling to glucose transport in skeletal muscle. J Appl Physiol. 2005;99:330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- 24.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology. 2005;146:5063–5070. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- 25.Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch. 2008 doi: 10.1007/s00424-008-0498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brismar H, Asghar M, Carey RM, Greengard P, Aperia A. Dopamine-induced recruitment of dopamine D1 receptors to the plasma membrane. Proc Natl Acad Sci U S A. 1998;95:5573–5578. doi: 10.1073/pnas.95.10.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holtback U, Brismar H, DiBona GF, Fu M, Greengard P, Aperia A. Receptor recruitment: a mechanism for interactions between G protein-coupled receptors. Proc Natl Acad Sci U S A. 1999;96:7271–7275. doi: 10.1073/pnas.96.13.7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruse MS, Adachi S, Scott L, Holtback U, Greengard P, Aperia A, Brismar H. Recruitment of renal dopamine 1 receptors requires an intact microtubulin network. Pflugers Arch. 2003;445:534–539. doi: 10.1007/s00424-002-0899-5. [DOI] [PubMed] [Google Scholar]
- 29.Trivedi M, Narkar VA, Hussain T, Lokhandwala MF. Dopamine recruits D1A receptors to Na-K-ATPase-rich caveolar plasma membranes in rat renal proximal tubules. Am J Physiol Renal Physiol. 2004;287:F921–931. doi: 10.1152/ajprenal.00023.2004. [DOI] [PubMed] [Google Scholar]
- 30.Correa FM, Viswanathan M, Ciuffo GM, Tsutsumi K, Saavedra JM. Kidney angiotensin II receptors and converting enzyme in neonatal and adult Wistar-Kyoto and spontaneously hypertensive rats. Peptides. 1995;16:19–24. doi: 10.1016/0196-9781(94)00150-5. [DOI] [PubMed] [Google Scholar]
- 31.Onorati F, Forte A, Mastroroberto P, Sante P, Esposito S, Pezzo F, Agozzino L, Cipollaro M, Cascino A, Renzulli A. Hypertension induces compensatory arterial remodeling following arteriotomy. J Surg Res. 2007;143:300–310. doi: 10.1016/j.jss.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 32.Touyz RM, Endemann D, He G, Li JS, Schiffrin EL. Role of AT2 receptors in angiotensin II-stimulated contraction of small mesenteric arteries in young SHR. Hypertension. 1999;33:366–372. doi: 10.1161/01.hyp.33.1.366. [DOI] [PubMed] [Google Scholar]